
Vaccine Increases the Diversity and Activation of Intratumoral T Cells in the Context of Combination Immunotherapy

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Mice
2.3. Tumor Inoculation, Treatment Schedule, and Metastasis Assay
2.4. Depletion Studies
2.5. Flow Cytometry
2.6. ELISPOT Assays
2.7. Real-Time PCR, Nanostring and TCR Analysis
2.8. OPAL Immunofluorescence
2.9. Statistical Methods
3. Results
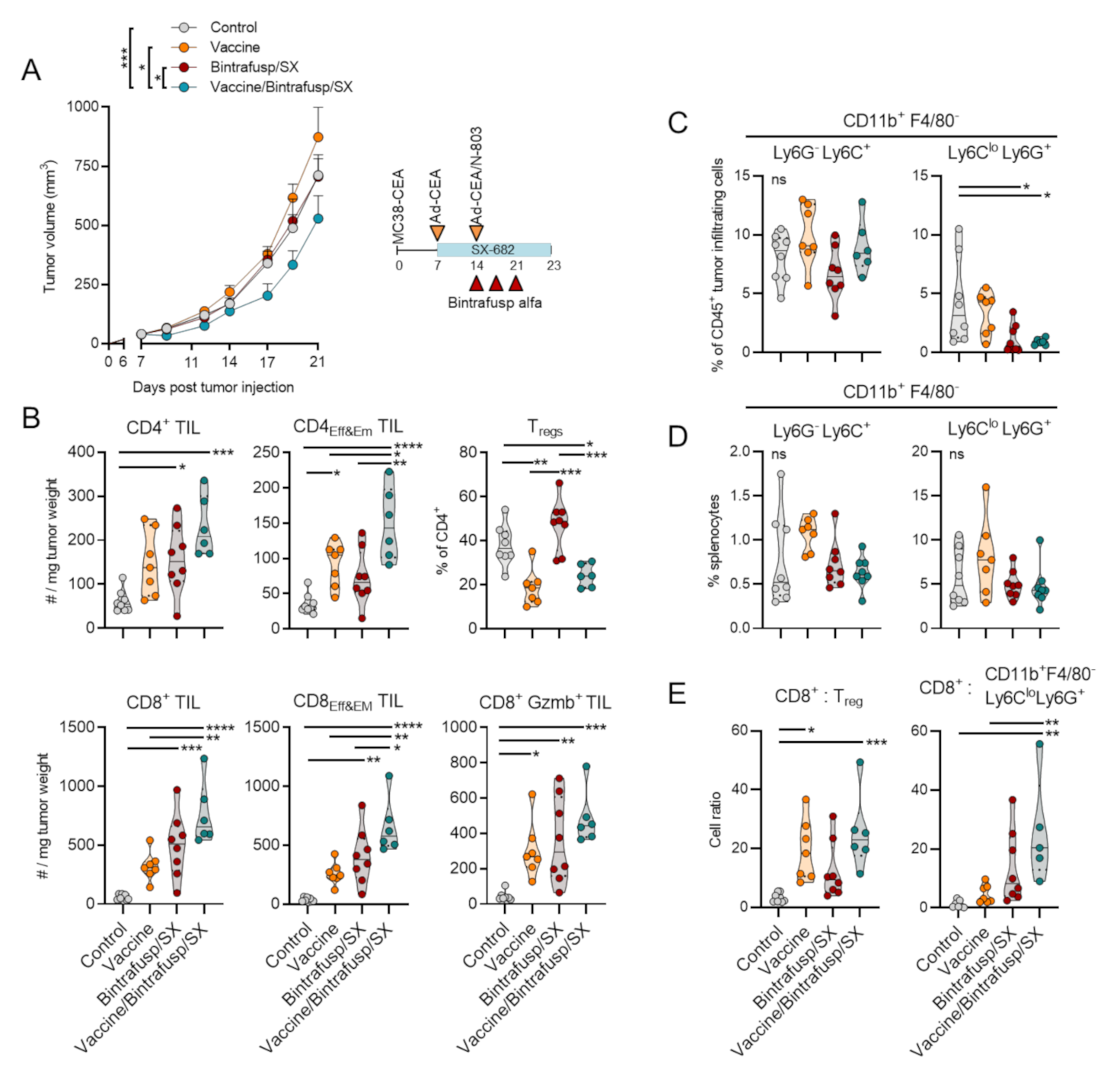
3.1. Addition of Vaccine to Checkpoint Blockade-Based Therapy Enhances Immune T-Cell Infiltration and Promotes a Th1 Tumor-Infiltrating Lymphocyte (TIL) Phenotype
3.2. Addition of Vaccine to Checkpoint Blockade-Based Therapy Enhances Immune T-Cell Activation and TCR Diversity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Horn, L.A.; Fousek, K.; Palena, C. Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 2020, 6, 432–441. [Google Scholar] [CrossRef]
- Drake, C.G. Combination immunotherapy approaches. Ann. Oncol. 2012, 23, viii41–viii46. [Google Scholar] [CrossRef]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193e7. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, T.F.; Corrales, L.; Williams, J.; Horton, B.; Sivan, A.; Spranger, S. Cancer Immunotherapy Targets Based on Understanding the T Cell-Inflamed Versus Non-T Cell-Inflamed Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283ra52. [Google Scholar] [CrossRef] [Green Version]
- Ali, O.A.; Lewin, S.A.; Dranoff, G.; Mooney, D.J. Vaccines Combined with Immune Checkpoint Antibodies Promote Cytotoxic T-cell Activity and Tumor Eradication. Cancer Immunol. Res. 2016, 4, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.L.; Benz, S.C.; Hicks, K.C.; Nguyen, A.; Gameiro, S.R.; Palena, C.; Sanborn, J.Z.; Su, Z.; Ordentlich, P.; Rohlin, L.; et al. Efficient Tumor Clearance and Diversified Immunity through Neoepitope Vaccines and Combinatorial Immunotherapy. Cancer Immunol. Res. 2019, 7, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Rudqvist, N.-P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.-W.; Emerson, R.O.; Robins, H.S.; Schneck, J.; Formenti, S.C.; DeMaria, S. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol. Res. 2018, 6, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Collins, J.M.; Donahue, R.N.; Tsai, Y.; Manu, M.; Palena, C.; Gatti-Mays, M.E.; Marté, J.L.; Madan, R.A.; Karzai, F.; Heery, C.R.; et al. Phase I Trial of a Modified Vaccinia Ankara Priming Vaccine Followed by a Fowlpox Virus Boosting Vaccine Modified to Express Brachyury and Costimulatory Molecules in Advanced Solid Tumors. Oncology 2019, 25, 560–e1006. [Google Scholar] [CrossRef] [Green Version]
- Gatti-Mays, M.E.; Redman, J.M.; Donahue, R.N.; Palena, C.; Madan, R.A.; Karzai, F.; Bilusic, M.; Sater, H.A.; Marté, J.L.; Cordes, L.M.; et al. A Phase I Trial Using a Multitargeted Recombinant Adenovirus 5 (CEA/MUC1/Brachyury)-Based Immunotherapy Vaccine Regimen in Patients with Advanced Cancer. Oncology 2019, 25, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatti-Mays, M.E.; Strauss, J.; Donahue, R.N.; Palena, C.; Del Rivero, J.; Redman, J.M.; Madan, R.A.; Marté, J.L.; Cordes, L.M.; Lamping, E.; et al. A Phase I Dose-Escalation Trial of BN-CV301, a Recombinant Poxviral Vaccine Targeting MUC1 and CEA with Costimulatory Molecules. Clin. Cancer Res. 2019, 25, 4933–4944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, H.; Gameiro, S.R.; Jochems, C.; Donahue, R.N.; Strauss, J.; Gulley, J.L.; Palena, C.; Schlom, J. Dual targeting of TGF-β and PD-L1 via a bifunctional anti-PD-L1/TGF-βRII agent: Status of preclinical and clinical advances. J. Immunother. Cancer 2020, 8, e000433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, J.; E Gatti-Mays, M.; Cho, B.C.; Hill, A.; Salas, S.; McClay, E.; Redman, J.M.; A Sater, H.; Donahue, R.N.; Jochems, C.; et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in patients with human papillomavirus-associated malignancies. J. Immunother. Cancer 2020, 8, e001395. [Google Scholar] [CrossRef] [PubMed]
- A Horn, L.; Riskin, J.; A Hempel, H.; Fousek, K.; Lind, H.; Hamilton, D.H.; McCampbell, K.K.; Maeda, D.Y.; A Zebala, J.; Su, Z.; et al. Simultaneous inhibition of CXCR1/2, TGF-β, and PD-L1 remodels the tumor and its microenvironment to drive antitumor immunity. J. Immunother. Cancer 2019, 8, e000326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, K.P.; Malamas, A.S.; Padget, M.R.; Solocinski, K.; Wolfson, B.; Fujii, R.; Sater, H.A.; Schlom, J.; Hodge, J.W. Therapy of Established Tumors with Rationally Designed Multiple Agents Targeting Diverse Immune–Tumor Interactions: Engage, Expand, Enable. Cancer Immunol. Res. 2021, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.S.; Kwilas, A.R.; Xu, W.; Alter, S.; Jeng, E.K.; Wong, H.C.; Schlom, J.; Hodge, J.W. IL-15 superagonist/IL-15RαSushi-Fc fusion complex (IL-15SA/IL-15RαSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget 2016, 7, 16130–16145. [Google Scholar] [CrossRef]
- Wrangle, J.M.; Velcheti, V.; Patel, M.R.; Garrett-Mayer, E.; Hill, E.G.; Ravenel, J.G.; Miller, J.S.; Farhad, M.; Anderton, K.; Lindsey, K.; et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: A non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018, 19, 694–704. [Google Scholar] [CrossRef]
- Robbins, P.F.; A Kantor, J.; Salgaller, M.; Hand, P.H.; Fernsten, P.D.; Schlom, J. Transduction and expression of the human carcinoembryonic antigen gene in a murine colon carcinoma cell line. Cancer Res. 1991, 51, 3657–3662. [Google Scholar]
- Ardiani, A.; Gameiro, S.R.; Palena, C.; Hamilton, D.H.; Kwilas, A.; King, T.H.; Schlom, J.; Hodge, J.W. Vaccine-Mediated Immunotherapy Directed against a Transcription Factor Driving the Metastatic Process. Cancer Res. 2014, 74, 1945–1957. [Google Scholar] [CrossRef] [Green Version]
- Clarke, P.; Mann, J.; Simpson, J.F.; Rickard-Dickson, K.; Primus, F.J. Mice transgenic for human carcinoembryonic antigen as a model for immunotherapy. Cancer Res. 1998, 58, 1469–1477. [Google Scholar] [PubMed]
- Hance, K.W.; Zeytin, H.E.; Greiner, J.W. Mouse models expressing human carcinoembryonic antigen (CEA) as a transgene: Evaluation of CEA-based cancer vaccines. Mutat. Res. Mol. Mech. Mutagen. 2005, 576, 132–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudson, K.M.; Hicks, K.C.; Luo, X.; Chen, J.-Q.; Schlom, J.; Gameiro, S.R. M7824, a novel bifunctional anti-PD-L1/TGFβ Trap fusion protein, promotes anti-tumor efficacy as monotherapy and in combination with vaccine. OncoImmunology 2018, 7, e1426519. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10, eaan5488. [Google Scholar] [CrossRef] [Green Version]
- David, J.M.; Dominguez, C.; McCampbell, K.K.; Gulley, J.L.; Schlom, J.; Palena, C. A novel bifunctional anti-PD-L1/TGF-β Trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. OncoImmunology 2017, 6, e1349589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, R.I.; Castillo, M.D.; Litzinger, M.; Hamilton, D.H.; Palena, C. IL-8 Signaling Plays a Critical Role in the Epithelial–Mesenchymal Transition of Human Carcinoma Cells. Cancer Res. 2011, 71, 5296–5306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fousek, K.; Horn, L.A.; Palena, C. Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol. Ther. 2021, 219, 107692. [Google Scholar] [CrossRef]
- Yuen, K.C.; Liu, L.-F.; Gupta, V.; Madireddi, S.; Keerthivasan, S.; Li, C.; Rishipathak, D.; Williams, P.; Kadel, E.E.; Koeppen, H.; et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat. Med. 2020, 26, 693–698. [Google Scholar] [CrossRef]
- Schalper, K.A.; Carleton, M.; Zhou, M.; Chen, T.; Feng, Y.; Huang, S.-P.; Walsh, A.M.; Baxi, V.; Pandya, D.; Baradet, T.; et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat. Med. 2020, 26, 688–692. [Google Scholar] [CrossRef]
- Sun, L.; Clavijo, P.E.; Robbins, Y.; Patel, P.; Friedman, J.; Greene, S.; Das, R.; Silvin, C.; Van Waes, C.; Horn, L.A.; et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Greene, S.; Robbins, Y.; Mydlarz, W.K.; Huynh, A.P.; Schmitt, N.C.; Friedman, J.; Horn, L.A.; Palena, C.; Schlom, J.; Maeda, D.Y.; et al. Inhibition of MDSC Trafficking with SX-682, a CXCR1/2 Inhibitor, Enhances NK-Cell Immunotherapy in Head and Neck Cancer Models. Clin. Cancer Res. 2020, 26, 1420–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heery, C.R.; Palena, C.; McMahon, S.; Donahue, R.N.; Lepone, L.M.; Grenga, I.; Dirmeier, U.; Cordes, L.; Marté, J.; Dahut, W.; et al. Phase I Study of a Poxviral TRICOM-Based Vaccine Directed Against the Transcription Factor Brachyury. Clin. Cancer Res. 2017, 23, 6833–6845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duraiswamy, J.; Freeman, G.J.; Coukos, G. Therapeutic PD-1 Pathway Blockade Augments with Other Modalities of Immunotherapy T-Cell Function to Prevent Immune Decline in Ovarian Cancer. Cancer Res. 2013, 73, 6900–6912. [Google Scholar] [CrossRef] [Green Version]
- Otsuru, T.; Kobayashi, S.; Wada, H.; Takahashi, T.; Gotoh, K.; Iwagami, Y.; Yamada, D.; Noda, T.; Asaoka, T.; Serada, S.; et al. Epithelial-mesenchymal transition via transforming growth factor betain pancreatic cancer is potentiated by the inflammatory glycoproteinleucine-rich alpha-2 glycoprotein. Cancer Sci. 2018, 110, 985–996. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yin, K.; Tian, J.; Xia, X.; Ma, J.; Tang, X.; Xu, H.; Wang, S. Granulocytic Myeloid-Derived Suppressor Cells Promote the Stemness of Colorectal Cancer Cells through Exosomal S100A9. Adv. Sci. 2019, 6, 1901278. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.; Wolf, D.; Bortone, D.S.; Ouyang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalba, M.; Evans, S.R.; Vidal-Vanaclocha, F.; Calvo, A. Role of TGF-β in metastatic colon cancer: It is finally time for targeted therapy. Cell Tissue Res. 2017, 370, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Redman, J.M.; Steinberg, S.M.; Gulley, J.L. Quick efficacy seeking trial (QuEST1): A novel combination immunotherapy study designed for rapid clinical signal assessment metastatic castration-resistant prostate cancer. J. Immunother. Cancer 2018, 6, 91. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Fold Change | Gene | Fold Change | Gene | Fold Change |
|---|---|---|---|---|---|
| Ido1 | 10.13 | Tnfrsf18 | 2.59 | Csf1 | 2.13 |
| Cd247 | 8.73 | Cxcr6 | 2.53 | Pou2f2 | 2.13 |
| Gzmk | 8.56 | Traf3 | 2.53 | Igf2r | 2.12 |
| Zap70 | 7.41 | Igf1r | 2.52 | Itgal | 2.11 |
| Cxcl3 | 6.93 | Prg2 | 2.52 | Notch1 | 2.11 |
| Cd163 | 6.77 | Cd8b1 | 2.51 | Pnma1 | 2.11 |
| Cd27 | 6.75 | Tnfrsf4 | 2.51 | Hc | 2.1 |
| Il6 | 6.75 | Il2rb | 2.5 | Cmah | 2.09 |
| Cd8a | 6.7 | Nfatc2 | 2.49 | Inpp5d | 2.09 |
| F2rl1 | 5.76 | Dmbt1 | 2.47 | Cxcl2 | 2.08 |
| Prf1 | 5.37 | CD209e | 2.46 | Smad3 | 2.07 |
| Pparg | 5.35 | Cxcl5 | 2.46 | Angpt1 | 2.06 |
| Gzmb | 5.21 | Ccl3 | 2.45 | Tfe3 | 2.05 |
| Cd5 | 5.16 | Itga4 | 2.43 | Fcer1a | 2.04 |
| Il2ra | 4.85 | Polr2a | 2.43 | Masp1 | 2.04 |
| Egr3 | 4.33 | Egr1 | 2.42 | Bst1 | 2.02 |
| Cd6 | 4.01 | Gbp5 | 2.42 | Erbb2 | 2.02 |
| Cd3e | 3.8 | Sap130 | 2.39 | Rel | 2.02 |
| Cma1 | 3.73 | Tlr9 | 2.36 | Tapbp | 2.02 |
| Cxcl9 | 3.59 | Nlrc5 | 2.35 | Tirap | 2.01 |
| Lcn2 | 3.56 | Il25 | 2.33 | Sdha | 2.01 |
| Il12rb2 | 3.42 | Pin1 | 2.33 | Cr2 | 2 |
| S100a8 | 3.42 | C8b | 2.3 | Cd7 | −2.01 |
| Il18r1 | 3.34 | Icos | 2.28 | Il17b | −2.03 |
| Ikzf1 | 3.18 | Lyve1 | 2.28 | Aire | −2.08 |
| Il12rb1 | 3.18 | Elk1 | 2.27 | Tnfrsf17 | −2.15 |
| Cd3g | 3.17 | Ep300 | 2.27 | Ms4a1 | −2.21 |
| Igll1 | 3.16 | Gbp2b | 2.23 | Cfd | −2.43 |
| Cx3cl1 | 2.86 | C4b | 2.22 | Il12a | −2.48 |
| Runx3 | 2.84 | Crp | 2.22 | Il22 | −2.5 |
| Itk | 2.83 | Nfatc3 | 2.22 | Klra1 | −2.54 |
| Cxcl1 | 2.82 | Cxcl13 | 2.21 | Tdo2 | −2.82 |
| Gata3 | 2.8 | Atm | 2.2 | Ifna4 | −3.04 |
| Lrp1 | 2.79 | Il6ra | 2.2 | Xcl1 | −3.17 |
| Il13ra2 | 2.76 | Tnfrsf11b | 2.2 | Chit1 | −3.7 |
| Camp | 2.67 | Fasl | 2.19 | Il17rb | −3.94 |
| Marco | 2.67 | Jun | 2.19 | Epcam | −4.85 |
| Klrc1 | 2.65 | Ddx58 | 2.18 | Tnfrsf9 | −5.48 |
| Pdcd1 | 2.63 | Il18rap | 2.15 | ||
| Crebbp | 2.62 | Tigit | 2.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horn, L.A.; Fousek, K.; Hamilton, D.H.; Hodge, J.W.; Zebala, J.A.; Maeda, D.Y.; Schlom, J.; Palena, C. Vaccine Increases the Diversity and Activation of Intratumoral T Cells in the Context of Combination Immunotherapy. Cancers 2021, 13, 968. https://doi.org/10.3390/cancers13050968
Horn LA, Fousek K, Hamilton DH, Hodge JW, Zebala JA, Maeda DY, Schlom J, Palena C. Vaccine Increases the Diversity and Activation of Intratumoral T Cells in the Context of Combination Immunotherapy. Cancers. 2021; 13(5):968. https://doi.org/10.3390/cancers13050968
Chicago/Turabian StyleHorn, Lucas A., Kristen Fousek, Duane H. Hamilton, James W. Hodge, John A. Zebala, Dean Y. Maeda, Jeffrey Schlom, and Claudia Palena. 2021. "Vaccine Increases the Diversity and Activation of Intratumoral T Cells in the Context of Combination Immunotherapy" Cancers 13, no. 5: 968. https://doi.org/10.3390/cancers13050968