
Hybrid Minigene Assay: An Efficient Tool to Characterize mRNA Splicing Profiles of NF1 Variants

 ,
,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
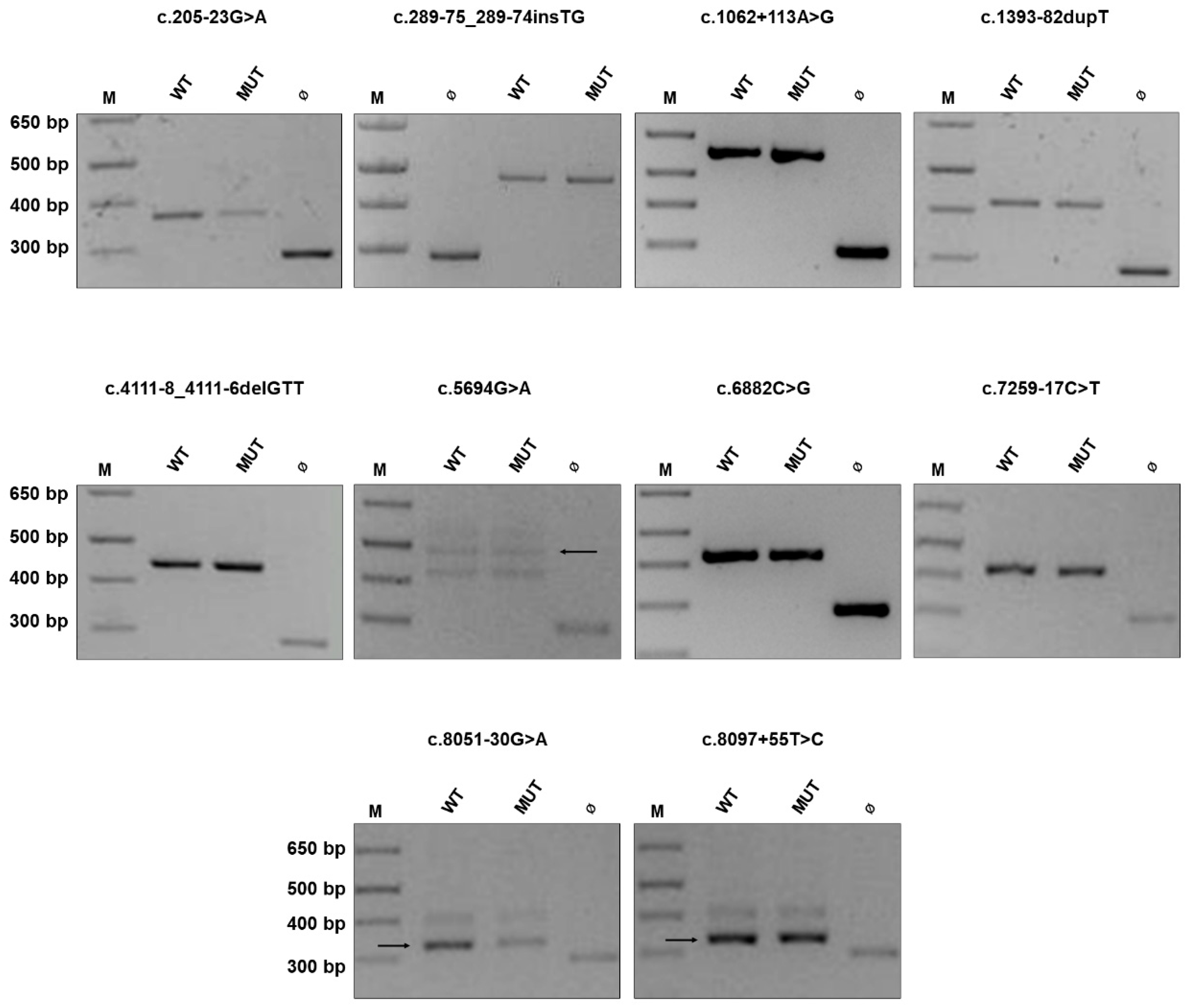
2.1. NF1 Benign Variants
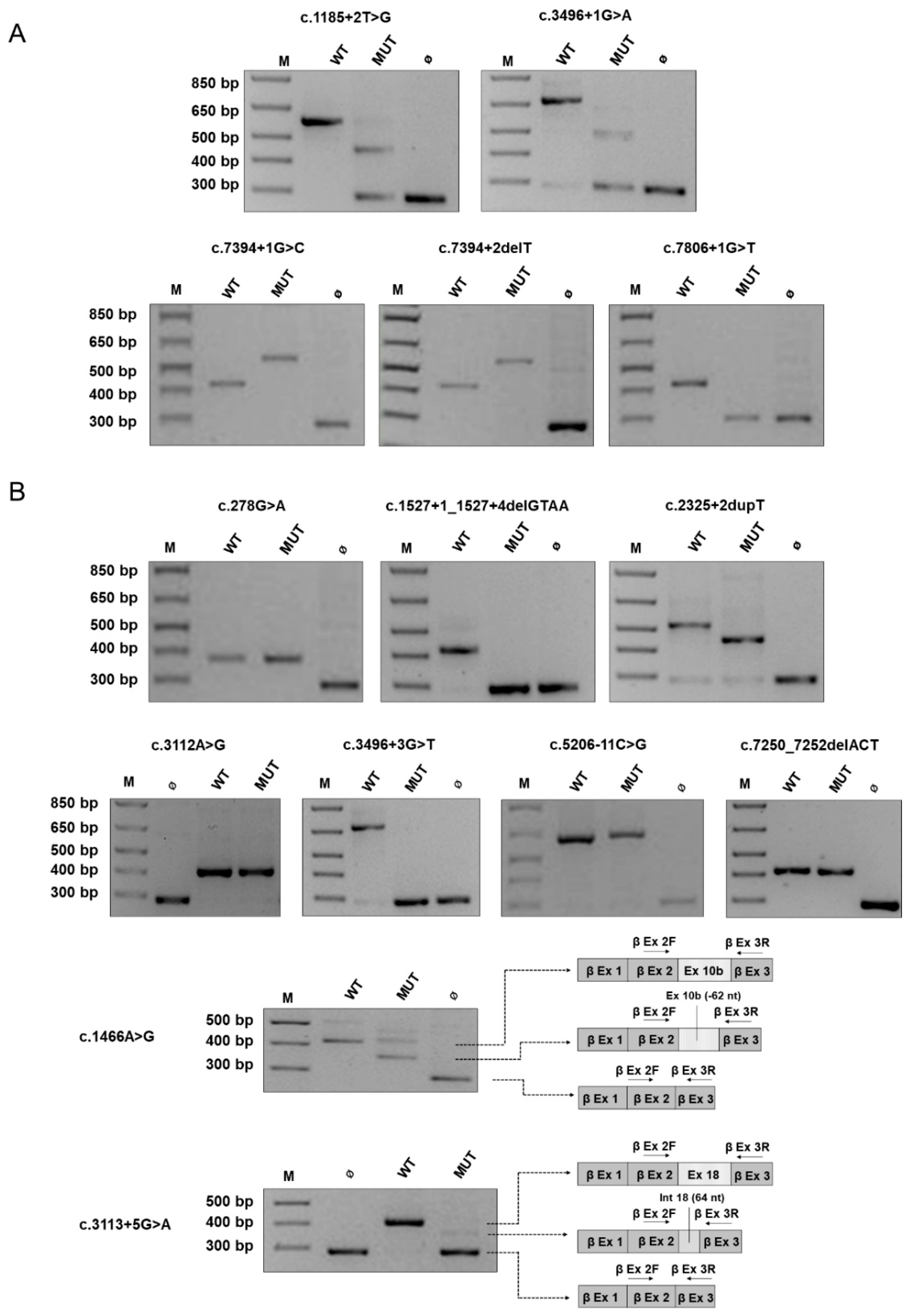
2.2. Canonical and Non-Canonical NF1 Variants
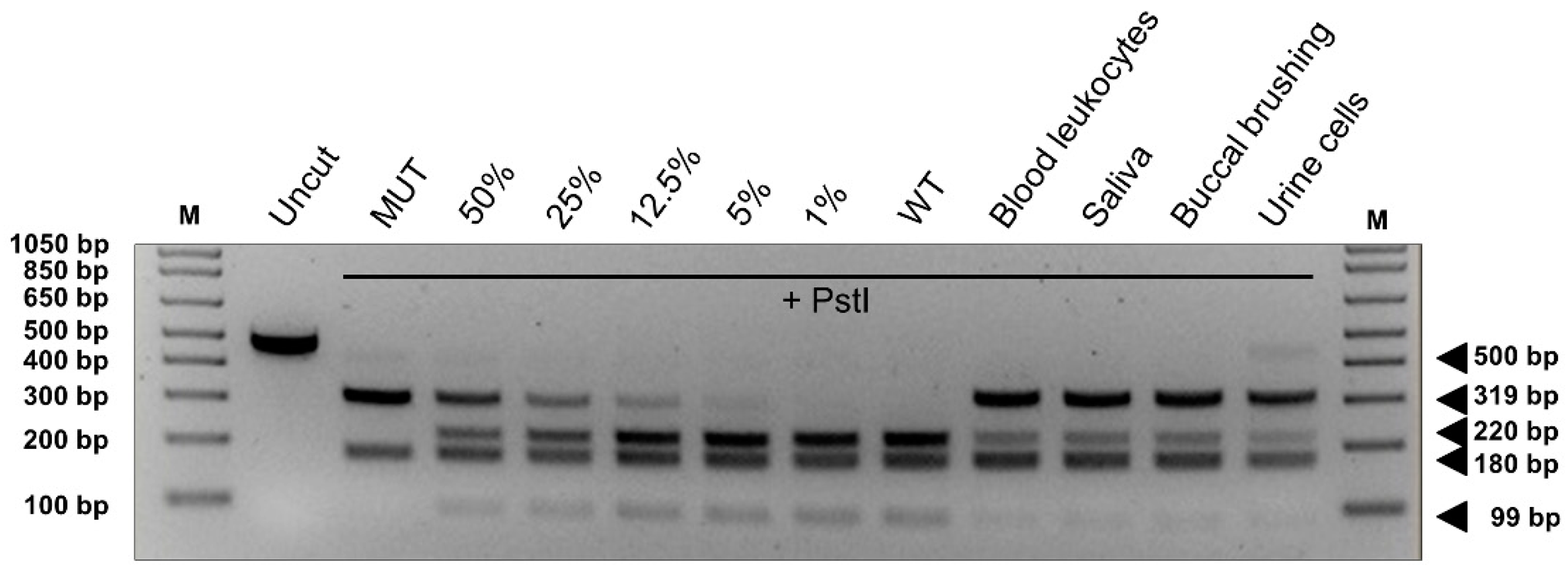
2.3. NF1 Variants Associated with the Residual Production of Wild-Type Transcript
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Molecular Analysis of the NF1 Gene
4.3. Bioinformatic Analysis of Variants
4.4. Construction and Expression of the Minigenes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clementi, M.; Barbujani, G.; Turolla, L.; Tenconi, R. Neurofibromatosis-1: A maximum likelihood estimation of mutation rate. Hum. Genet. 1990, 84, 116–118. [Google Scholar] [CrossRef]
- Jett, K.; Friedman, J.M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 2010, 12, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, Y.S.; McPherson, J.R.; Ong, C.K.; Rozen, S.G.; Teh, B.T.; Lee, A.S.; Callen, D.F. The NF1 gene revisited-from bench to bedside. Oncotarget 2014, 5, 5873–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallee, B.; Benedetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef]
- Cawthon, R.M.; Weiss, R.; Xu, G.F.; Viskochil, D.; Culver, M.; Stevens, J.; Robertson, M.; Dunn, D.; Gesteland, R.; O’Connell, P.; et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 1990, 62, 193–201. [Google Scholar] [CrossRef]
- Wallace, M.R.; Marchuk, D.A.; Andersen, L.B.; Letcher, R.; Odeh, H.M.; Saulino, A.M.; Fountain, J.W.; Brereton, A.; Nicholson, J.; Mitchell, A.L.; et al. Type 1 neurofibromatosis gene: Identification of a large transcript disrupted in three NF1 patients. Science 1990, 249, 181–186. [Google Scholar] [CrossRef]
- Vandenbroucke, I.; Callens, T.; De Paepe, A.; Messiaen, L. Complex splicing pattern generates great diversity in human NF1 transcripts. BMC Genom. 2002, 3, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottillo, I.; De Luca, A.; Schirinzi, A.; Guida, V.; Torrente, I.; Calvieri, S.; Gervasini, C.; Larizza, L.; Pizzuti, A.; Dallapiccola, B. Functional analysis of splicing mutations in exon 7 of NF1 gene. BMC Med. Genet. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.B.; Ballester, R.; Marchuk, D.A.; Chang, E.; Gutmann, D.H.; Saulino, A.M.; Camonis, J.; Wigler, M.; Collins, F.S. A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol. Cell. Biol. 1993, 13, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988, 45, 575–578. [Google Scholar]
- Brems, H.; Chmara, M.; Sahbatou, M.; Denayer, E.; Taniguchi, K.; Kato, R.; Somers, R.; Messiaen, L.; De Schepper, S.; Fryns, J.P.; et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet. 2007, 39, 1120–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, S. Neurofibromatosis: Emerging phenotypes, mechanisms and management. Clin. Med. 2008, 8, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Korf, B.R. Spinal neurofibromatosis and phenotypic heterogeneity in NF1. Clin. Genet. 2015, 87, 399–400. [Google Scholar] [CrossRef] [PubMed]
- DeBella, K.; Szudek, J.; Friedman, J.M. Use of the National Institutes of Health Criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000, 105, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Ars, E.; Serra, E.; Garcia, J.; Kruyer, H.; Gaona, A.; Lazaro, C.; Estivill, X. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum. Mol. Genet. 2000, 9, 237–247. [Google Scholar] [CrossRef] [Green Version]
- De Raedt, T.; Brems, H.; Lopez-Correa, C.; Vermeesch, J.R.; Marynen, P.; Legius, E. Genomic organization and evolution of the NF1 microdeletion region. Genomics 2004, 84, 346–360. [Google Scholar] [CrossRef]
- Leppig, K.A.; Kaplan, P.; Viskochil, D.; Weaver, M.; Ortenberg, J.; Stephens, K. Familial neurofibromatosis 1 microdeletions: Cosegregation with distinct facial phenotype and early onset of cutaneous neurofibromata. Am. J. Med. Genet. 1997, 73, 197–204. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Mautner, V.F.; Cooper, D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017, 136, 349–376. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Koczkowska, M.; Callens, T.; Gomes, A.; Sharp, A.; Chen, Y.; Hicks, A.D.; Aylsworth, A.S.; Azizi, A.A.; Basel, D.G.; Bellus, G.; et al. Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): An update of genotype-phenotype correlation. Genet. Med. 2019, 21, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Pinna, V.; Lanari, V.; Daniele, P.; Consoli, F.; Agolini, E.; Margiotti, K.; Bottillo, I.; Torrente, I.; Bruselles, A.; Fusilli, C.; et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 2015, 23, 1068–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.J.; Liu, Y.; Cochran, M.; Abbott, M.A.; Atkin, J.; et al. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum. Mutat. 2015, 36, 1052–1063. [Google Scholar] [CrossRef] [Green Version]
- Trevisson, E.; Morbidoni, V.; Forzan, M.; Daolio, C.; Fumini, V.; Parrozzani, R.; Cassina, M.; Midena, E.; Salviati, L.; Clementi, M. The Arg1038Gly missense variant in the NF1 gene causes a mild phenotype without neurofibromas. Mol. Genet. Genom. Med. 2019, 7, e616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844-848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar] [CrossRef] [Green Version]
- Koczkowska, M.; Callens, T.; Chen, Y.; Gomes, A.; Hicks, A.D.; Sharp, A.; Johns, E.; Uhas, K.A.; Armstrong, L.; Bosanko, K.A.; et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: Genotype-phenotype study in neurofibromatosis type 1. Hum. Mutat. 2020, 41, 299–315. [Google Scholar] [CrossRef]
- Kaufmann, D. Neurofibromatoses; Karger: Basel, Switzerland; New York, NY, USA, 2008; p. 192. [Google Scholar]
- Evans, D.G.; Bowers, N.; Burkitt-Wright, E.; Miles, E.; Garg, S.; Scott-Kitching, V.; Penman-Splitt, M.; Dobbie, A.; Howard, E.; Ealing, J.; et al. Comprehensive RNA Analysis of the NF1 Gene in Classically Affected NF1 Affected Individuals Meeting NIH Criteria has High Sensitivity and Mutation Negative Testing is Reassuring in Isolated Cases With Pigmentary Features Only. EBioMedicine 2016, 7, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, K.; Eckart, M.; Rehder, H.; Fonatsch, C. Illegitimate splicing of the NF1 gene in healthy individuals mimics mutation-induced splicing alterations in NF1 patients. Hum. Genet. 2000, 106, 311–313. [Google Scholar] [CrossRef]
- Valero, M.C.; Martin, Y.; Hernandez-Imaz, E.; Marina Hernandez, A.; Melean, G.; Valero, A.M.; Javier Rodriguez-Alvarez, F.; Telleria, D.; Hernandez-Chico, C. A highly sensitive genetic protocol to detect NF1 mutations. J. Mol. Diagn. 2011, 13, 113–122. [Google Scholar] [CrossRef]
- Maruoka, R.; Takenouchi, T.; Torii, C.; Shimizu, A.; Misu, K.; Higasa, K.; Matsuda, F.; Ota, A.; Tanito, K.; Kuramochi, A.; et al. The use of next-generation sequencing in molecular diagnosis of neurofibromatosis type 1: A validation study. Genet. Test Mol. Biomark. 2014, 18, 722–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasmant, E.; Parfait, B.; Luscan, A.; Goussard, P.; Briand-Suleau, A.; Laurendeau, I.; Fouveaut, C.; Leroy, C.; Montadert, A.; Wolkenstein, P.; et al. Neurofibromatosis type 1 molecular diagnosis: What can NGS do for you when you have a large gene with loss of function mutations? Eur. J. Hum. Genet. 2015, 23, 596–601. [Google Scholar] [CrossRef] [Green Version]
- Cunha, K.S.; Oliveira, N.S.; Fausto, A.K.; de Souza, C.C.; Gros, A.; Bandres, T.; Idrissi, Y.; Merlio, J.P.; de Moura Neto, R.S.; Silva, R.; et al. Hybridization Capture-Based Next-Generation Sequencing to Evaluate Coding Sequence and Deep Intronic Mutations in the NF1 Gene. Genes 2016, 7, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellanos, E.; Rosas, I.; Negro, A.; Gel, B.; Alibes, A.; Baena, N.; Pineda, M.; Pi, G.; Pintos, G.; Salvador, H.; et al. Mutational spectrum by phenotype: Panel-based NGS testing of patients with clinical suspicion of RASopathy and children with multiple cafe-au-lait macules. Clin. Genet. 2020, 97, 264–275. [Google Scholar] [CrossRef]
- Wimmer, K.; Roca, X.; Beiglbock, H.; Callens, T.; Etzler, J.; Rao, A.R.; Krainer, A.R.; Fonatsch, C.; Messiaen, L. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5’ splice-site disruption. Hum. Mutat. 2007, 28, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Pros, E.; Gomez, C.; Martin, T.; Fabregas, P.; Serra, E.; Lazaro, C. Nature and mRNA effect of 282 different NF1 point mutations: Focus on splicing alterations. Hum. Mutat. 2008, 29, E173–E193. [Google Scholar] [CrossRef]
- Zatkova, A.; Messiaen, L.; Vandenbroucke, I.; Wieser, R.; Fonatsch, C.; Krainer, A.R.; Wimmer, K. Disruption of exonic splicing enhancer elements is the principal cause of exon skipping associated with seven nonsense or missense alleles of NF1. Hum. Mutat. 2004, 24, 491–501. [Google Scholar] [CrossRef]
- Trevisson, E.; Salviati, L.; Baldoin, M.C.; Toldo, I.; Casarin, A.; Sacconi, S.; Cesaro, L.; Basso, G.; Burlina, A.B. Argininosuccinate lyase deficiency: Mutational spectrum in Italian patients and identification of a novel ASL pseudogene. Hum. Mutat. 2007, 28, 694–702. [Google Scholar] [CrossRef]
- Forzan, M.; Salviati, L.; Pertegato, V.; Casarin, A.; Bruson, A.; Trevisson, E.; Di Gianantonio, E.; Clementi, M. Is CFTR 621 + 3 A > G a cystic fibrosis causing mutation? J. Hum. Genet. 2010, 55, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, G.; Casarin, A.; Trevisson, E.; Dona, M.; Cassina, M.; Graziano, C.; Picci, L.; Clementi, M.; Salviati, L. Validation of CFTR intronic variants identified during cystic fibrosis population screening by a minigene splicing assay. Clin. Chem. Lab. Med. 2015, 53, 1719–1723. [Google Scholar] [CrossRef]
- Cassina, M.; Cerqua, C.; Rossi, S.; Salviati, L.; Martini, A.; Clementi, M.; Trevisson, E. A synonymous splicing mutation in the SF3B4 gene segregates in a family with highly variable Nager syndrome. Eur. J. Hum. Genet. 2017, 25, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Bianchessi, D.; Morosini, S.; Saletti, V.; Ibba, M.C.; Natacci, F.; Esposito, S.; Cesaretti, C.; Riva, D.; Finocchiaro, G.; Eoli, M. 126 novel mutations in Italian patients with neurofibromatosis type 1. Mol. Genet. Genomic. Med. 2015, 3, 513–525. [Google Scholar] [CrossRef]
- Mattocks, C.; Baralle, D.; Tarpey, P.; ffrench-Constant, C.; Bobrow, M.; Whittaker, J. Automated comparative sequence analysis identifies mutations in 89% of NF1 patients and confirms a mutation cluster in exons 11–17 distinct from the GAP related domain. J. Med. Genet. 2004, 41, e48. [Google Scholar] [CrossRef] [Green Version]
- Fahsold, R.; Hoffmeyer, S.; Mischung, C.; Gille, C.; Ehlers, C.; Kucukceylan, N.; Abdel-Nour, M.; Gewies, A.; Peters, H.; Kaufmann, D.; et al. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am. J. Hum. Genet. 2000, 66, 790–818. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ars, E.; Kruyer, H.; Morell, M.; Pros, E.; Serra, E.; Ravella, A.; Estivill, X.; Lazaro, C. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J. Med. Genet. 2003, 40, e82. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Montmain, G.; Ruano, E.; Upadhyaya, M.; Dudley, S.; Liskay, R.M.; Thibodeau, S.N.; Puisieux, A. Neurofibromatosis type 1 gene as a mutational target in a mismatch repair-deficient cell type. Hum. Genet. 2003, 112, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Van Roy, N.; Speleman, F.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum. Mutat. 2000, 15, 541–555. [Google Scholar] [CrossRef]
- Zhu, G.; Zheng, Y.; Liu, Y.; Yan, A.; Hu, Z.; Yang, Y.; Xiang, S.; Li, L.; Chen, W.; Peng, Y.; et al. Identification and characterization of NF1 and non-NF1 congenital pseudarthrosis of the tibia based on germline NF1 variants: Genetic and clinical analysis of 75 patients. Orphanet. J. Rare Dis. 2019, 14, 221. [Google Scholar] [CrossRef]
- Laycock-van Spyk, S.; Thomas, N.; Cooper, D.N.; Upadhyaya, M. Neurofibromatosis type 1-associated tumours: Their somatic mutational spectrum and pathogenesis. Hum. Genom. 2011, 5, 623–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messiaen, L.M.; Callens, T.; Roux, K.J.; Mortier, G.R.; De Paepe, A.; Abramowicz, M.; Pericak-Vance, M.A.; Vance, J.M.; Wallace, M.R. Exon 10b of the NF1 gene represents a mutational hotspot and harbors a recurrent missense mutation Y489C associated with aberrant splicing. Genet. Med. 1999, 1, 248–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wimmer, K.; Schamschula, E.; Wernstedt, A.; Traunfellner, P.; Amberger, A.; Zschocke, J.; Kroisel, P.; Chen, Y.; Callens, T.; Messiaen, L. AG-exclusion zone revisited: Lessons to learn from 91 intronic NF1 3’ splice site mutations outside the canonical AG-dinucleotides. Hum Mutat. 2020, 41, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Bausch, B.; Borozdin, W.; Mautner, V.F.; Hoffmann, M.M.; Boehm, D.; Robledo, M.; Cascon, A.; Harenberg, T.; Schiavi, F.; Pawlu, C.; et al. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. J. Clin. Endocrinol. Metab. 2007, 92, 2784–2792. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.M.; Sohn, Y.B.; Jeong, S.Y.; Kim, H.J.; Messiaen, L.M. Mutation spectrum of NF1 and clinical characteristics in 78 Korean patients with neurofibromatosis type 1. Pediatr. Neurol. 2013, 48, 447–453. [Google Scholar] [CrossRef]
- Steffensen, A.Y.; Dandanell, M.; Jonson, L.; Ejlertsen, B.; Gerdes, A.M.; Nielsen, F.C.; Hansen, T. Functional characterization of BRCA1 gene variants by mini-gene splicing assay. Eur. J. Hum. Genet. 2014, 22, 1362–1368. [Google Scholar] [CrossRef]
- Fraile-Bethencourt, E.; Diez-Gomez, B.; Velasquez-Zapata, V.; Acedo, A.; Sanz, D.J.; Velasco, E.A. Functional classification of DNA variants by hybrid minigenes: Identification of 30 spliceogenic variants of BRCA2 exons 17 and 18. PLoS Genet. 2017, 13, e1006691. [Google Scholar] [CrossRef]
- Aissat, A.; de Becdelievre, A.; Golmard, L.; Vasseur, C.; Costa, C.; Chaoui, A.; Martin, N.; Costes, B.; Goossens, M.; Girodon, E.; et al. Combined computational-experimental analyses of CFTR exon strength uncover predictability of exon-skipping level. Hum. Mutat. 2013, 34, 873–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villate, O.; Ibarluzea, N.; Fraile-Bethencourt, E.; Valenzuela, A.; Velasco, E.A.; Grozeva, D.; Raymond, F.L.; Botella, M.P.; Tejada, M.I. Functional Analyses of a Novel Splice Variant in the CHD7 Gene, Found by Next Generation Sequencing, Confirm Its Pathogenicity in a Spanish Patient and Diagnose Him with CHARGE Syndrome. Front Genet. 2018, 9, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubeuf, H.; Caputo, S.M.; Sullivan, T.; Rondeaux, J.; Krieger, S.; Caux-Moncoutier, V.; Hauchard, J.; Castelain, G.; Fievet, A.; Meulemans, L.; et al. Calibration of Pathogenicity Due to Variant-Induced Leaky Splicing Defects by Using BRCA2 Exon 3 as a Model System. Cancer Res. 2020, 80, 3593–3605. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Takeda, J.I.; Masuda, A. Rules and tools to predict the splicing effects of exonic and intronic mutations. Wiley Interdiscip Rev RNA 2018, 9, e1551. [Google Scholar] [CrossRef]
- Takahara, K.; Schwarze, U.; Imamura, Y.; Hoffman, G.G.; Toriello, H.; Smith, L.T.; Byers, P.H.; Greenspan, D.S. Order of intron removal influences multiple splice outcomes, including a two-exon skip, in a COL5A1 acceptor-site mutation that results in abnormal pro-alpha 1(V) N-propeptides and Ehlers-Danlos syndrome type I. Am. J. Hum. Genet. 2002, 71, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, T.; Fukao, T.; Murase, K.; Sakaguchi, N.; Harding, C.O.; Kondo, N. Molecular basis of two-exon skipping (exons 12 and 13) by c.1248 + 5g > a in OXCT1 gene: Study on intermediates of OXCT1 transcripts in fibroblasts. Hum. Mutat. 2013, 34, 473–480. [Google Scholar] [CrossRef]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef]
- Assunto, A.; Ferrara, U.; De Luca, A.; Pivonello, C.; Lombardo, L.; Piscitelli, A.; Tortora, C.; Pinna, V.; Daniele, P.; Pivonello, R.; et al. Isoform-specific NF1 mRNA levels correlate with disease severity in Neurofibromatosis type 1. Orphanet. J. Rare Dis. 2019, 14, 261. [Google Scholar] [CrossRef]
- Fernandez-Rodriguez, J.; Castellsague, J.; Benito, L.; Benavente, Y.; Capella, G.; Blanco, I.; Serra, E.; Lazaro, C. A mild neurofibromatosis type 1 phenotype produced by the combination of the benign nature of a leaky NF1-splice mutation and the presence of a complex mosaicism. Hum. Mutat. 2011, 32, 705–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferner, R.E.; Huson, S.M.; Thomas, N.; Moss, C.; Willshaw, H.; Evans, D.G.; Upadhyaya, M.; Towers, R.; Gleeson, M.; Steiger, C.; et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J. Med. Genet. 2007, 44, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heim, R.A.; Kam-Morgan, L.N.; Binnie, C.G.; Corns, D.D.; Cayouette, M.C.; Farber, R.A.; Aylsworth, A.S.; Silverman, L.M.; Luce, M.C. Distribution of 13 truncating mutations in the neurofibromatosis 1 gene. Hum. Mol. Genet. 1995, 4, 975–981. [Google Scholar] [CrossRef] [Green Version]
- Martella, M.; Salviati, L.; Casarin, A.; Trevisson, E.; Opocher, G.; Polli, R.; Gross, D.; Murgia, A. Molecular analysis of two uncharacterized sequence variants of the VHL gene. J. Hum. Genet. 2006, 51, 964–968. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant [HGVS) | Familial/Sporadic Disease | Segregation Analysis |
|---|---|---|
| Benign Variant | ||
| c.205-23G > A (c.974delT) | F (mother, brother, sister) | n.a. |
| c.289-75_289-74insTG | S 1 | Variant inherited from the unaffected mother |
| c.1062 + 113A > G (c.7682_7683delAG) | S | n.a. |
| c.1393-82dupT 2 | F (mother, sister, maternal aunt) | Variant identified in mother, sister, unaffected maternal aunt and grandfather) |
| c.4111-8_4111-6delGTT | S | Variant inherited from the unaffected father |
| c.5694G > A p.(Glu1898=) (288 + 1delG) | S | n.a. |
| c.6882C > G 2 p.(Leu2294=) | F (father, paternal grandmother and aunt) | n.a. |
| c.7259-17C > T (exon 39 deletion) | S | n.a. |
| c.8051-30G > A | S | Variant inherited from the unaffected mother |
| c.8097 + 55T > C (exon 14 deletion) | S | n.a. |
| Canonical Variant | ||
| c.1185 + 2T > G | U | n.a. |
| c.3496 + 1G > A | S | De novo |
| c.7394 + 1G > C | S | De novo |
| c.7394 + 2delT | S | De novo |
| c.7806 + 1G > T | S | De novo |
| Non-canonical Variant | ||
| c.278G > A p.(Cys93Tyr) | F (mother, brother, son) | Variant identified in affected son (mother and brother not tested) |
| c.1466A > G 3 p.(Tyr489Cys) | S | De novo |
| c.1527 + 1delGTAA | F (mother, brother, maternal uncle and grandmother) | Variant identified in affected mother and brother (other family member not tested) |
| c.1722-3C > T | S | De novo |
| c.1722-3C > G 4 | - | - |
| c.1722-3C > A 4 | - | - |
| c.2325 + 2dupT | U | n.a. |
| c.3112A > G 5 p.(Arg1038Gly) | Case 1: F (mother, maternal uncle and 1st cousin; son, daughters); Case 2: S | Case 1: Variant identified in affected mother, 1st maternal cousin, son, daughters (maternal uncle not tested); Case 2: De novo |
| c.3113 + 5G > A | U | n.a. |
| c.3496 + 3G > T | U | n.a. |
| c.3496 + 5G > A | S | De novo |
| c.4538_4540delGAC p.(Arg1513del) | S | Variant inherited from the unaffected mother |
| c.5206-11C > G | F (mother) | n.a. |
| c.7250_7252delACT p.(Tyr2417del) | S | De novo |
| Genomic Coordinate | cDNA Change | Effect on RNA 1 | Effect on Protein | Location | GnomAD Allele Frequency 2 | TOPMED Allele Frequency | Human Splicing Finder | Mutation Taster | CADD Score | LOVD, HGMD, ClinVar 3 | References | Splicing Alterations on Minigene | ACMG Class 3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 29486005-G-A | c.205-23G > A | intron 2 (intron 2) | 1/249602 (0.000004006) (E) | – | No significant impact on splicing signals. | Acceptor site marginally increased | 14.08 | - - - | – | No | B | ||
| 29490130--TG | c.289-75_289-74insTG | No 1 | intron 3 (intron 3) | – | – | No significant impact on splicing signals. | Acceptor site marginally increased; acceptor site increased. | 12.91 | - - - | – | No | B | |
| 29527726-A-G | c.1062 + 113A > G | intron 7 (intron 9) | 7/31406 (0.00022) (G) | 35/125568 (0.000279) | New donor splice site: Activation of a cryptic donor site. Potential alteration of splicing. | No significant impact on splicing signals. | 0.315 | P CS1512923 - | [41] | No | B | ||
| 29541383--T | c.1393-82dupT | intron 10a (intron 12) | 2/31276 (0.00006) (G) | 9/125568 (0.000072) | No significant impact on splicing signals. | No significant impact on splicing signals. | 3.171 | - - - | – | No | B | ||
| 29585354-GTT- | c.4111-8_4111-6delGTT | intron 23a (intron 30) | 116/251202 (0.00046) (E) | 67/125568 (0.000534) | No significant impact on splicing signals. | Acceptor site increased; acceptor site marginally decreased. | 14.57 | B - LB | [42] | No | B | ||
| 29657461-G-A | c.5694G > A | p.(Glu1898=) | exon 30 (exon 38) | 35/251248 (0.000139) (E) | 9/125568 (0.000072) | Alteration of auxiliary sequences: Significant alteration of exonic splicing enhancers/silencers (ESE/ESS) motifs ratio. | Donor site marginally increased; donor site gained. | 11.45 | B - LB/VUS | – | No | B | |
| 29667546-C-G | c.6882C > G | p.(Leu2294=) | exon 38 (exon 46) | 1/251486 (0.000004) (E) | 3/125568 (0.000024) | No significant impact on splicing signals. | Acceptor site marginally increased; donor site gained. | 10.03 | - - LB | – | No | B | |
| 29677184-C-T | c.7259-17C > T | intron 40 (intron 48) | 603/251342 (0.002399) (E) | 940/125568 (0.007486) | No significant impact on splicing signals. | No significant impact on splicing signals. | 3.685 | B CS00088 B | [43] | No | B | ||
| 29685957-G-A | c.8051-30G > A | intron 46 (intron 54) | 37/250938 (0.0001474) (E) | 22/125568 (0.000175) | Alteration of auxiliary sequences: Significant alteration of ESE/ESS motifs ratio | No significant impact on splicing signals. | 3.438 | LB - - | – | No | B | ||
| 29686088-T-C | c.8097 + 55T > C | intron 47 (intron 55) | 16/31406 (0.00051) (G) | 66/125568 (0.000526) | Alteration of auxiliary sequences: Significant alteration of ESE/ESS motifs ratio. | No significant impact on splicing signals. | 3.611 | - - - | – | No | B |
| Genomic Coordinate | cDNA Change | Effect on RNA 1 | Effect on Protein | Location | GnomAD Allele Frequency 2 | TOPMED Allele Frequency | Human Splicing Finder | Mutation Taster | CADD Score | LOVD, HGMD, ClinVar 3 | References | Splicing Alterations on Minigene | ACMG Class 3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Canonical variants | |||||||||||||
| 29528179-T-G | c.1185 + 2T > G | intron 8 (intron 10) | – | – | Alteration of the WT donor site, most probably affecting splicing. | Alteration within used splice site, likely to disturb normal splicing; donor site lost, donor site marginally increased, donor site gained. | 34 | P - P | – | Skipping of exon 8 (10) alone and of both exon 7 (9) and 8 (10) | P | ||
| 29559900-G-A | c.3496 + 1G > A | Skipping of exon 20: r.3315_3496del182 [34] | intron 20 (intron 26) | – | – | Alteration of the WT donor site, most probably affecting splicing. | Alteration within used splice site, likely to disturb normal splicing; donor site lost; acceptor site marginally increased; donor site increased; donor gained. | 33 | - CS072245 - | [34] | Skipping of exon 20 (26) alone and of both exon 20 (26) and 21 (27) | P | |
| 29677337-G-C | c.7394 + 1G > C | intron 41 (intron 49) | – | – | Alteration of the WT donor site, most probably affecting splicing. | Alteration within used splice site, likely to disturb normal splicing; donor site lost; acceptor site increased; donor marginally increased. | 35 | - - - | – | Intronic retention of 126 nucleotides | P | ||
| 29677338-T- | c.7394 + 2delT | intron 41 (intron 49) | – | – | Alteration of the WT donor site, most probably affecting splicing | Alteration within used splice site, likely to disturb normal splicing; donor site lost; acceptor site marginally increased; acceptor site increased; donor site increased: acceptor site gained; donor site gained. | 32 | - - - | – | Intronic retention of 126 nucleotides | P | ||
| 29684109-G-T | c.7806 + 1G > T | r.7676_7806del131 [35,45] | p.(Glu2559Glyfs*10) | intron 44 (intron 52) | – | – | Alteration of the WT donor site, most probably affecting splicing | Alteration within used splice site, likely to disturb normal splicing; donor site lost; acceptor site increased; acceptor site marginally increased; acceptor site gained. | 34 | P CS031796 - | [35,45] | Skipping of exon 44 (52) | P |
| Non-canonical variants | |||||||||||||
| 29486101-G-A | c.278G > A | no effect on splicing: r.278g > a [46] | p.(Cys93Tyr) | exon 3 (exon 3) | – | 1/125568 (0.000008) | No significant impact on splicing signals. | Donor site marginally increased. | 29.5 | LP CM001252 P/VUS | [46,47] | No | LP |
| 29541542-A-G | c.1466A > G | r.(1466a > g, 1466_1527del62) 1 [35] | p.(Tyr489Cys, Tyr489*) | exon 10b (exon 13) | 3/250646 (0.000012) (E) | – | Significant alteration of ESE/ESS motifs ratio; activation of a cryptic donor site. Potential alteration of splicing. | Donor site increased. | 23.9 | P CM1111787 P | [15,35,48,49,50] | Loss of 62 nucleotides of exon 10b (13) downstream the mutation; production of the correctly spliced transcript | P |
| 29541604-GTAA- | c.1527 + 1_1527 + 4delGTAA | r.1393_1527del135 [35,45,50] | p.(Ser465_Cys509del) | intron 10b (intron 13) | – | – | Alteration of the WT donor site, most probably affecting splicing. | Alteration within used splice site, likely to disturb normal splicing; donor site lost, acceptor site gained. | 33 | - - LP | [35,45,50] | Skipping of exon 10b (13) | P |
| 29550459-C-T | c.1722-3C > T | intron 11 (intron 15) | 1/250978 (0.000004) (E) | – | Activation of a cryptic donor site. Potential alteration of splicing | Alteration within used splice site, likely to disturb normal splicing; acceptor site lost, acceptor site increased. | 20.2 | - - - | – | Skipping of exon 12a (16) and production of WT transcript | LP | ||
| 29550459-C-G | c.1722-3C > G | r.1721_1722ins1722-43_1722-1 [35] | p.(Ser574Argfs*28) | intron 11 (intron 15) | – | – | Alteration of the WT acceptor site, most probably affecting splicing | Alteration within used splice site, likely to disturb normal splicing; acceptor site lost, donor site gained. | 25.1 | P CS000051 - | [15,35] | Skipping of exon 12a (16) and production of a transcript retaining 43 nucleotides of intron 11 (15) | LP |
| 29550459-C-A | c.1722-3C > A | intron 11 (intron 15) | – | – | Alteration of the WT acceptor site, most probably affecting splicing | Alteration within used splice site, likely to disturb normal splicing; acceptor site lost, donor site marginally increased, donor site gained. | 24.1 | - - P | – | Skipping of exon 12a (16) and production of a transcript retaining 43 nucleotides of intron 11 (15) | LP | ||
| 29554312--T | c.2325 + 2dupT | intron 14 (intron 19) | – | – | Alteration of the WT donor site, most probably affecting splicing; activation of a cryptic donor site. Potential alteration of splicing. | Donor site lost; donor site gained. | 23 | - - - | – | Skipping of exon 14 (19) | P | ||
| 29557399-A-G | c.3112A > G | r.3112a > g [23] | p.(Arg1038Gly) | exon 18 (exon 23) | – | – | Significant alteration of ESE/ESS motifs ratio. | Donor site lost; donor site marginally increased. | 29.1 | LP - - | [23] | No | P |
| 29557405-G-A | c.3113 + 5G > A | r.2991_3113del123 [34,35] | p.(Tyr998_Arg1038del) | intron 18 (intron 23) | – | – | Alteration of the WT donor site, most probably affecting splicing. | Donor site decreased; donor site marginally increased; donor site increased. | 23.1 | - CS072240 P | [34,35] | Skipping of exon 18 (23) and production of a transcript devoid of exon 18 (23) retaining 64 nucleotides of intron 18 (23) | P |
| 29559902-G-T | c.3496 + 3G > T | intron 20 (intron 26) | – | – | Alteration of the WT donor site, most probably affecting splicing. | Acceptor site marginally increased; donor site decreased; donor site increased. | 21.7 | - - - | – | Skipping of exon 20 (26) and 21 (27) | P | ||
| 29559904-G-A | c.3496 + 5G > A | r.3315_3496del182 1 | intron 20 (intron 26) | – | – | Alteration of the WT donor site, most probably affecting splicing. | Acceptor site marginally increased; donor site increased; donor site lost; donor site increased; donor site marginally increased; donor site gained. | 23.0 | - - VUS | – | Skipping of exon 20 (26), loss of the last 66 nucleotides of exon 20 (26) and production of WT transcript. | P | |
| 29588752-GAC- | c.4538_4540delGAC | p.(Arg1513del) | exon 27a (exon 34) | – | – | Significant alteration of ESE/ESS motifs ratio; activation of a cryptic acceptor site. Potential alteration of splicing. | Donor site increased. Disease causing at protein level. | 22.5 | - - - | – | Skipping of exon 27a (34); production of correctly spliced transcript | VUS | |
| 29654506-C-G | c.5206-11C > G | intron 28 (intron 36) | – | – | Alteration of the WT acceptor site, most probably affecting splicing; activation of a cryptic acceptor site. Potential alteration of splicing. | Acceptor site marginally increased; acceptor site increased. | 16.7 | - - - | – | Intronic retention of 10 nucleotides 4 | LP | ||
| 29676261-ACT- | c.7250_7252delACT | p.(Tyr2417del) | exon 40 (intron 48) | – | – | Alteration of the WT donor site, most probably affecting splicing; activation of a cryptic donor site. Activation of a cryptic acceptor site. Potential alteration of splicing. | No significant impact on splicing signals. Disease causing at protein level. | 19.9 | - - - | – | No | LP | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morbidoni, V.; Baschiera, E.; Forzan, M.; Fumini, V.; Ali, D.S.; Giorgi, G.; Buson, L.; Desbats, M.A.; Cassina, M.; Clementi, M.; et al. Hybrid Minigene Assay: An Efficient Tool to Characterize mRNA Splicing Profiles of NF1 Variants. Cancers 2021, 13, 999. https://doi.org/10.3390/cancers13050999
Morbidoni V, Baschiera E, Forzan M, Fumini V, Ali DS, Giorgi G, Buson L, Desbats MA, Cassina M, Clementi M, et al. Hybrid Minigene Assay: An Efficient Tool to Characterize mRNA Splicing Profiles of NF1 Variants. Cancers. 2021; 13(5):999. https://doi.org/10.3390/cancers13050999
Chicago/Turabian StyleMorbidoni, Valeria, Elisa Baschiera, Monica Forzan, Valentina Fumini, Dario Seif Ali, Gianpietro Giorgi, Lisa Buson, Maria Andrea Desbats, Matteo Cassina, Maurizio Clementi, and et al. 2021. "Hybrid Minigene Assay: An Efficient Tool to Characterize mRNA Splicing Profiles of NF1 Variants" Cancers 13, no. 5: 999. https://doi.org/10.3390/cancers13050999
APA StyleMorbidoni, V., Baschiera, E., Forzan, M., Fumini, V., Ali, D. S., Giorgi, G., Buson, L., Desbats, M. A., Cassina, M., Clementi, M., Salviati, L., & Trevisson, E. (2021). Hybrid Minigene Assay: An Efficient Tool to Characterize mRNA Splicing Profiles of NF1 Variants. Cancers, 13(5), 999. https://doi.org/10.3390/cancers13050999