1. Introduction
The incidence of pancreatic ductal adenocarcinoma (PDAC) has risen in the last 50 years and it is forecasted to be the second leading cause of cancer deaths within a decade. The dismal prognosis results from lack of specific early symptoms and early diagnostic methods. Patients usually present in the advanced stage of the disease and experience the failure of traditional anticancer therapies. Long-term disease-free survival is possible but the majority of diagnoses are unresectable cases, with an expected survival of less than 6 months. Modest curative benefits are offered only by early surgery with subsequent adjuvant therapy. Systemic recurrence after treatment suggests early metastasis in the course of pancreatic tumorigenesis, long before disease diagnosis [
1].
The therapeutic failure in PDAC can be attributed to specific tumor cell biology, as well as features of the tumor milieu, both contributing to the high malignancy of this cancer. The tumor microenvironment of PDAC indeed presents a formidable challenge to therapy. Considerable desmoplastic stroma can constitute up to 90% of the tumor volume and are believed to originate from cancer-associated fibroblasts (CAFs) [
2]. These are accountable for the chemoresistance of pancreatic tumors, by creating physical barriers that shield tumor cells from systemically injected therapeutic compounds. The heavy extracellular matrix within pancreatic cancer distorts tissue architecture and causes an abnormal structure of blood and lymphatic vasculature. The ensuing hypoxia is another hallmark of the PDAC microenvironment, associated with desmoplasia. It originates from desmoplasia-associated hypovascularization and, vice versa, favors desmoplastic progression by activating pancreatic stellate cells [
3,
4,
5]. Desmoplasia and hypoxia are also barriers to the infiltration of both regulatory and effector lymphocytes, as well as to T cell activation [
6,
7,
8]. Macrophages that are recruited adopt an immunosuppressive, pro-angiogenic M2-like state, block CD4+ T cell entry into the PDAC microenvironment, support PDAC progression, and thus are a marker of negative clinical prognosis [
5,
9,
10].
The repertoire of treatment strategies in PDAC has been expanding in the last decades to include novel approaches such as targeted drugs, non-coding RNAs (miRNAs) and immunotherapy [
11]. However, targeted chemotherapeutics have only moderately improved PDAC outcomes and have not altered 5-year survival. Gemcitabine and erlotinib, used to treat advanced disease, both yield only a modest clinical benefit. The development of chemoresistance and the pro-metastatic propensity of pancreatic cancer are related to the transition from epithelial to mesenchymal cells (EMT) [
12,
13]. Gene expression data have demonstrated the contribution of EMT cells to chemoresistance in PDAC [
13,
14].
Overall, the prevailing notion is that dense stroma, multiple genetic mutations, compensatory alternative pathways and aggressive metastatic spread cause PDAC chemoresistance, even to multidrug regimens. These factors are also at the root of the moderate success in targeting cancer-associated molecular pathways and the adverse effects linked to activated downstream effectors [
11].
The ineffective nature of existing treatments for PDAC has stimulated the search for innovative therapeutic strategies. Oncolytic virotherapy is becoming increasingly sought-after for the treatment of many different neoplasms, including pancreatic cancer. The therapeutic effectiveness of oncolytic viruses (OVs) is the result of direct oncolysis, virus spread to adjacent cancer cells, and the elicited anti-tumor immune response.
Oncolytic viruses (OVs) are replication-competent and selectively target tumor cells, but they are not able to bind and/or productively replicate in most normal somatic cells. OVs can be genetically engineered to express inserted foreign transgenes to induce cancer cell death in a fashion that can stimulate anti-tumor immune responses [
15]. OV replication in cancer cells yields infectious progeny that may further spread and reduce tumor burden [
16].
Myxoma virus (MYXV) is a poxvirus with attractive safety profile for oncolytic therapy. MYXV infects and produces symptoms only in rabbits. Although MYXV is nonpathogenic to humans and mice, the virus exhibits natural tropism for a wide spectrum of human cancers [
17]. MYXV has a 160-kb double-stranded DNA genome that has the capacity to accept numerous transgenes and is easy to manipulate. In this study, we armed oncolytic MYXV with the murine Light gene, encoding mouse tumor necrosis factor ligand superfamily member 14 (TNFSF14, synonym: LIGHT), which was designed to promote the influx of T lymphocytes into the tumor and thus intensify the immune response directed against the tumor. This ought to help stimulate the transformation of the “cold” tumor microenvironment, with a small number of effector immune cells, into a “hot” environment, with increased infiltration of other immune cells and cytokines. LIGHT is an inducible lymphotoxin which competes with HSV glycoprotein D for binding to a herpesvirus entry mediator (HVEM) expressed on T lymphocytes. LIGHT is expressed on activated T cells, natural killer (NK) cells, immature dendritic cells (DCs) and on the tumor and in the stroma [
18]. LIGHT is an immune stimulator that contributes to the anti-tumor immune response, and its expression in the TME is associated with improved overall survival and relapse-free survival [
19]. LIGHT–HVEM interaction has been shown to induce apoptosis of several tumor cell lines directly [
20]. Therefore, LIGHT is a promising biotherapeutic adjuvant agent for cancer immunotherapy with OVs [
21].
Systemic delivery represents the desired route to reach poorly accessible sites or disseminated metastatic lesions. IV administration of naked OVs is, however, a very inefficient strategy for delivery. Rapid elimination from the bloodstream by antiviral defense mechanisms is the major obstacle to achieving the effective transfer of the viral cargo to distant tumor sites. IV delivery of a viral construct to tumor sites can be improved by exploiting protective carrier cells, e.g., [
22]. Adipose-derived stem cells (ADSCs) of mesenchymal origin provide a unique cell carrier platform, where the virus can be at least partly protected from immune clearance pathways before delivery to the tumor site. In addition, ADSCs show natural chemotactic tropism for cancer tissues. However, in cases where IV delivery of cell-protected OV would not be effective (due to the “first pass” effect in the lungs) [
23], the transfer of oncovirus cargo to a defined and identifiable tumor site can be accomplished through locoregional injection. In this study, we investigated whether carrier cells could also be beneficial for the delivery of OVs to PDAC via the intraperitoneal (IP) route. IP administration remains an interesting alternative when targeting OVs to abdominal-area cancer targets. Comparison of the two modes of OV delivery using a peritoneal murine tumor model revealed [
24] that IP was associated with narrower biodistribution (a reduced frequency of virus detected in the kidney, lung and heart), decreased toxicity and greater therapeutic efficacy against peritoneal metastases. Tumor burden was more effectively reduced with IP, compared with IV administration. Median survival following IP administration was approximately twice that observed with IV administration.
Wennier et al. tested the use of unarmed recombinant vMyx-tdTr to treat disseminated pancreatic adenocarcinoma in the peritoneal cavity of immunocompetent mice. They showed that oncolytic therapy, followed by gemcitabine, could decisively improve animal survival [
25]. In our study, we assessed the usefulness of the novel armed MYXV construct (vMyx-mLIGHT-Fluc/tdTr) in experimental therapy on orthotopically-induced murine PDAC via IP delivery of the viral cargo using the mesenchymal ADSC platform.
2. Materials and Methods
2.1. Recombinant Viruses
Recombinant MYXV constructs (vMyx-EGFP, vMyx-EGFP/tdTr and vMyx-mLIGHT/FLuc/tdTr) derived from the wild-type Lausanne strain of myxoma virus (vMyx-WT) were used. The recombination cassettes were inserted in the intergenic region between the M135 and M136 open reading frames (ORFs). vMyx-EGFP expresses EGFP from the early/late promoter; the vMyx-EGFP/tdTr tandem system allows the expression of EGFP at both early and late infection stages (early/late promoter), whereas tdTr is expressed at the late infection stage (poxvirus p11 late promoter) [
26]. The vMyx-mLIGHT-FLuc/tdTr construct expresses mouse LIGHT and firefly luciferase at both the early and late infection stages (early/late promoter) and tdTr under a synthetic poxvirus late promoter. vMyx-mLIGHT-FLuc/tdTr was constructed by inserting a DNA cassette containing the coding sequences of murine LIGHT (TNFSF14, see
Supplementary Materials for the nucleotide sequence and
Figure S1), firefly luciferase and tdTr at an intergenic location between the M135 and M136 genes in the wild-type MYXV strain Lausanne genome. A recombinant plasmid was constructed using the Gateway System (Thermo Fisher Scientific, Waltham, MA, USA) [
17]. The sequence encoding murine LIGHT was PCR-amplified using gene-specific primers with the forward primer containing the synthetic early/late (sE/L) promoter sequence. The resultant sE/L-LIGHT fragment was PCR-ligated to another PCR fragment containing the MYXV M135 gene. The resultant attB1-M135-sE/L-LIGHT-attB4 PCR fragment was recombined into the pDONR221-P1P4 plasmid (Invitrogen, Waltham, MA, USA) using the BP clonase enzyme mix (Invitrogen). The construction of the plasmid having tdTr (attB4r-p11tdTr-attB3r) and another plasmid having FLuc and the MYXV M136 gene (attB3-sE/L-FLuc-M136-attB2) was described previously [
27]. All three plasmids, along with the pDEST40 destination plasmid (Invitrogen) were then subjected to an LR recombination reaction using LR clonase II (Invitrogen) to generate the final plasmid construct. The recombinant virus was then created using the method described previously for making recombinant MYXV [
17].
2.2. Virus Purification and Titration
MYXVs were produced in RK13 cells (at multiplicity of infection /MOI/ = 0.1). When the cytopathic effect was visible (ca. 72 h; ±80% confluency), cells were harvested, centrifuged (1500 rpm, 10 min, 4 °C), resuspended in 10 mM Tris–HCl (pH = 8), and subjected to three freeze/thaw cycles and cup sonication (5 × 1 min). Cell debris was removed by centrifugation. Homogenates containing the virus were layered onto the 36% sucrose cushion and ultracentrifuged (105 × g/1 h, 4 °C). The supernatant was removed, and the pellet resuspended in 10 mM Tris–HCl (pH = 8). The quantity of infectious viral particles was determined by titration on RK13 cells (4 × 105/well; 6-well plate). After 3–4 days, fluorescent foci were counted using an inverted microscope (Leica Microsystems, Mannheim Germany). Viral titer (focus-forming units per milliliter (FFU)/mL) was calculated as the number of foci multiplied by the dilution.
2.3. Cell Lines
Human (Panc–1, AsPC–1) and murine (Pan02) pancreatic ductal adenocarcinoma cell lines were used. The human lines were from ATCC; the murine line was a gift from GMF. Rabbit RK13 kidney epithelial cell line (ATCC) was used to propagate MYXV constructs. Cells were maintained in DMEM (RK13, Panc-1 and Pan02) or RPMI-1640 (AsPC–1) media, both supplemented with 10% FBS (EURx) and 1% penicillin-streptomycin (Sigma-Aldrich, Poznan, Poland) Cultures were grown in a humidified 5% CO2 incubator at 37 °C and were routinely tested for mycoplasma contamination.
2.4. ADSCs Isolation and Culture
Fragments of adipose tissue obtained on-site (MSC National Research Institute of Oncology) from human donors (
Table S1) were washed with PBS
− (Gibco™, ThermoFisher Scientific, Warsaw, Poland) containing 1% FBS (EURx) to remove contaminating hematopoietic cells. The tissue was cut into small pieces and digested using collagenase type I solution (200 U/mL, Gibco) and shaking (1 h/37 °C/180 rpm). Cell suspensions were centrifuged (1500 rpm/10 min), filtered through 70-μm and 40-μm strainers and seeded in culture flasks using MEM (Sigma-Aldrich, Poznan, Poland) supplemented with 10% human platelet lysate (Sigma), heparin (2 U/mL, Polfa), 1% non-essential amino acids (Gibco) and 1% penicillin–streptomycin (Sigma). After 72 h, cultures were washed with PBS
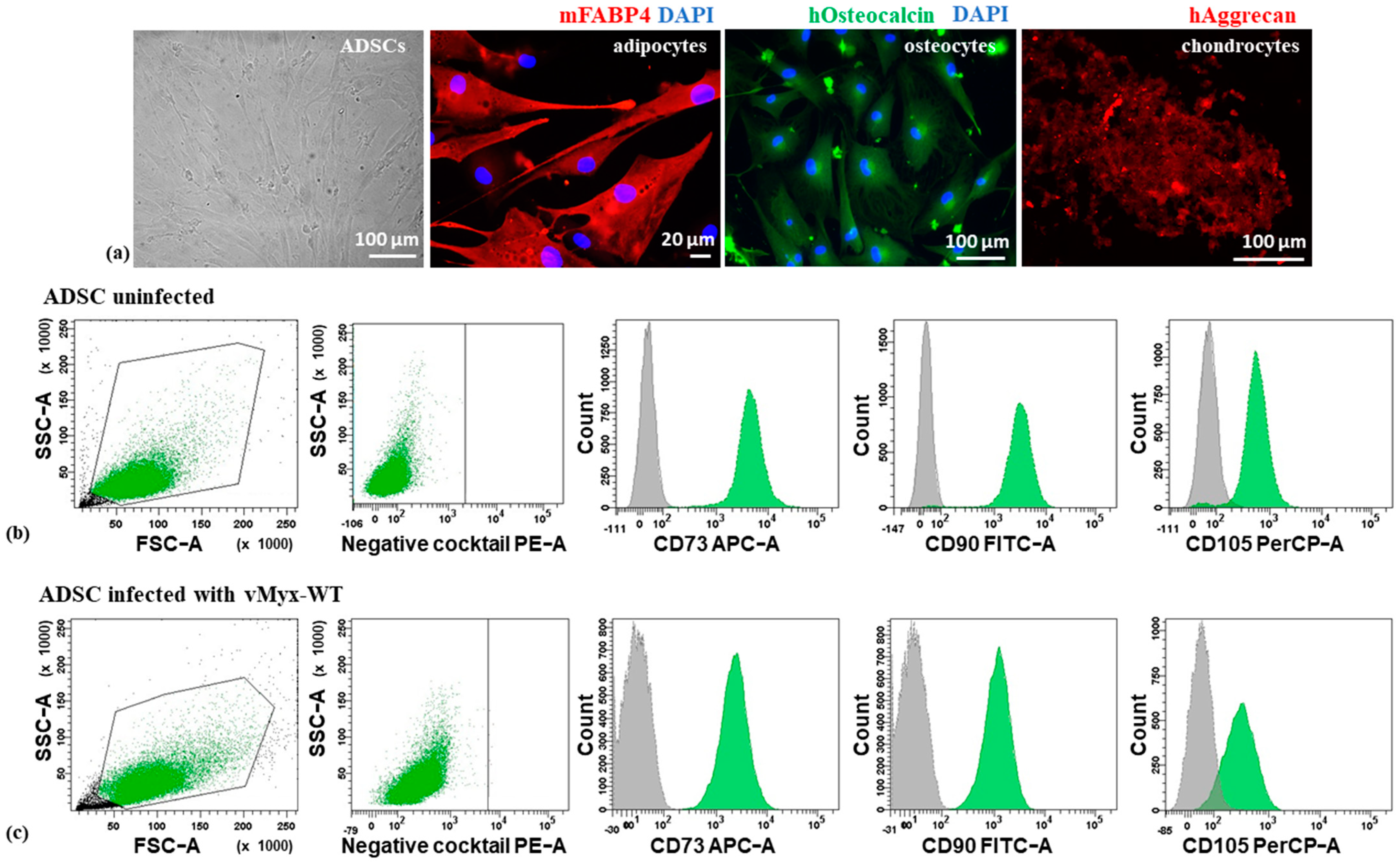
− to remove non-adherent cells and rinsed with fresh culture medium. Sub-confluent cultures were split at a 1:3 ratio. Differentiation into adipocytes, osteocytes and chondrocytes was analyzed (passage 3) using the Human Mesenchymal Stem Cell Functional Identification Kit (SC006, R&D Systems), containing Goat Anti-Mouse FABP-4 Antigen Affinity-purified Polyclonal Antibody (adipocyte marker), a Mouse Anti-Human Osteocalcin Monoclonal Antibody (osteocyte marker) and Goat Anti-Human Aggrecan Antigen Affinity-purified Polyclonal Antibody (chondrocyte marker). Cells were stained with Biotinylated Rabbit Anti-Goat IgG (immunoglobulin G) Antibody (H + L), Texas Red Streptavidin (Vector Laboratories, BA-5000 and SA-5006, respectively) or Goat Anti-Mouse Alexa Fluor Plus 488 secondary antibody (Thermo Fisher Scientific, No. A32723). Nuclei were counterstained with DAPI (Thermo Fisher Scientific, No. 62248). The morphology of adipose-derived stem cells was inspected using the Zeiss LSM 710 confocal microscope system.
2.5. Flow Cytometry Analysis of ADSC Phenotype
To confirm the phenotype of ADSCs, cultured cells were incubated with appropriate antibodies (20 min/RT), rinsed with and resuspended in Cell Wash Buffer (BD Biosciences). Analysis was performed using a BD FACS Canto II flow cytometer (Becton-Dickinson). The phenotype of both uninfected and infected ADSCs (vMyx-WT; MOI = 5) was analyzed. The positive cell population gates were set using isotype IgG controls. The presence of ADSC-associated surface markers (CD73, CD90, and CD105) and the co-occurring absence of blood cell-lineage-specific markers (CD11b, CD19, CD34, CD45 and HLA-DR) was examined using the Human MSC Analysis Kit (BD Biosciences, No. 562245).
2.6. Infectiveness of RK13, ADSC and Pancreatic Cell Lines to MYXV
ADSCs, RK13 and three pancreatic adenocarcinoma cell lines (murine Pan02, human Panc-1 and AsPC-1) were tested for susceptibility to MYXV infection. Cultured cells (2 × 105 cells/well; 6-well plate) infected with vMyx-EGFP (MOI = 5) were collected (24, 48 and 72 h p.i.), centrifuged (2000 rpm/2 min), washed twice and resuspended in PBS− (200 µL). Cell aliquots were incubated with 7-aminoactinomycin D (7-AAD; 5 µL) for 2 min to determine viability and analyzed for enhanced GFP expression using flow cytometry (BD FACS Canto II). 7-AAD emission was detected using a long-pass filter (670 nm), and a region for live cells was defined. Non-infected cells were used as a control.
2.7. Expression of Early and Late MYXV Genes
To visualize early and late gene expression, ADSCs, RK13 and three pancreatic adenocarcinoma cell lines (murine Pan02, human Panc-1 and AsPC-1) were infected with vMyx-EGFP/tdTr (MOI = 5). Cells were plated into 4-well chamber slides (5 × 104 cells/well). After 24 h p.i., cells were washed with PBS− and fixed in paraformaldehyde (4%) for 10 min at room temperature (RT). Cell nuclei were stained with DAPI Counterstain (Life Technologies™, ThermoFisher Scientific, Warsaw, Poland). Infection was evaluated using fluorescence microscopy (Zeiss LSM 710 confocal workstation).
2.8. Single-Step Growth Analysis of Viral Replication in Cell Cultures
Cultures (5 × 104 cells/well; 24-well plate) of ADSCs, RK13 and three pancreatic cancer cells lines (murine Pan02, human Panc-1 and AsPC-1) were infected (in triplicate) with vMyx-mLIGHT/Fluc/tdTr (MOI = 5). The inoculum was removed at 90 min post-infection and cells were further incubated (5% CO2, 37 °C) with fresh medium. Next, cells were trypsinized and collected (at 3-, 6-, 12- and 24-h time points). Following centrifugation (2000 rpm/2 min), cells were resuspended in 200 µL hypotonic swelling buffer (5 mL of 1M Tris-HCl (pH = 8.0) and 1 mL of 1M MgCl2) and frozen at −80 °C. Before titration, cells were thawed and sonicated (2 × 1 min) to disaggregate virus complexes. Samples from the examined time points were titrated back onto RK13 cells (4 × 105 cells/well; 6-well plate) by serial dilution. Foci were counted using an inverted fluorescent microscope (Leica Microsystems). Titers (FFU/mL) were calculated (number of foci multiplied by the dilution factor).
2.9. Cytotoxicity of MYXV for ADSCs, RK13 and Pancreatic Cancer Cell Lines
To assess cytotoxic effects of MYXV infection on ADSCs, RK13 and three pancreatic cancer cell lines (murine Pan02, human Panc-1 and AsPC-1) cell cultures (1 × 104 cells/well; 96-well plate) were infected with vMyx-mLIGHT/Fluc/tdTr at increasing MOIs (0.1, 1, 5 and 10). After 24 and 48 h, cell viability was evaluated using an MTS assay (CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay kit; Promega) and a Biotek plate reader (490 nm).
2.10. Animal Care
All animal procedures were performed in accordance with European Union (EU) law, after approval by the Local Ethics Committee, Medical University of Silesia, Katowice, Poland. Six-to-eight-week-old C57Bl/6NCrl female mice (total n = 235; Charles River Laboratories) were used. Animals (18–22 g) were housed in HEPA-filtered IVC System cages (Allentown Caging Equipment) under a controlled dark/light cycle (12 h/12 h) and were fed a pathogen-free standard diet (Altromin 1314) and water ad libitum. All efforts were made to minimize animal suffering.
2.11. Orthotopic Tumor Implantation
For orthotopic tumor implantation, C57Bl/6NCrl female mice (subtotal n = 163) were anesthetized with isoflurane (1–3% vol.) and injected with carprofen (5 mg/kg, ScanVet) into the nape of the neck. The surgical field was sterilized with iodine and a ca. 1-cm-long incision was made beside the splenic silhouette. With the entire pancreas and spleen exposed, Pan02 cancer cell suspension was slowly injected into the pancreatic head area using a 27G needle, following which the pancreas and spleen were pushed slightly back into the abdominal cavity and the abdominal muscle layer was closed with a single continuous 4-0 polysorb suture. The skin incision was finally closed with an autoclip wound closing system and buprenorphine (0.03 mg/mL) was administered to assist recovery. Animal health was monitored daily. Only single animals from control groups reached termination criteria. Euthanasia was conducted by means of cervical dislocation.
2.12. Orthotopic Injection of Pan02-luc Cells with Simultaneous Administration of ADSCs Pre-Infected with MYXV
C57Bl/6NCrl mice (n = 6/group) were orthotopically injected with the Pan02 cells (1 × 106 cells/25 µL PBS−), followed by immediate administration of ADSCs (5 × 105 cells/25 µL PBS−) pre-infected (MOI = 5) with vMyx-mLIGHT/Fluc/tdTr. As controls, non-infected ADSCs, unshielded vMyx-mLIGHT/Fluc/tdTr (5 × 105 FFU/25 µL PBS−) or PBS− alone were used. After 21 days, the mice were sacrificed, and pancreata and spleens were excised, weighed and measured for size.
2.13. Bioluminescence Imaging (BLI) of MYXV Distribution Following Administration to Mice
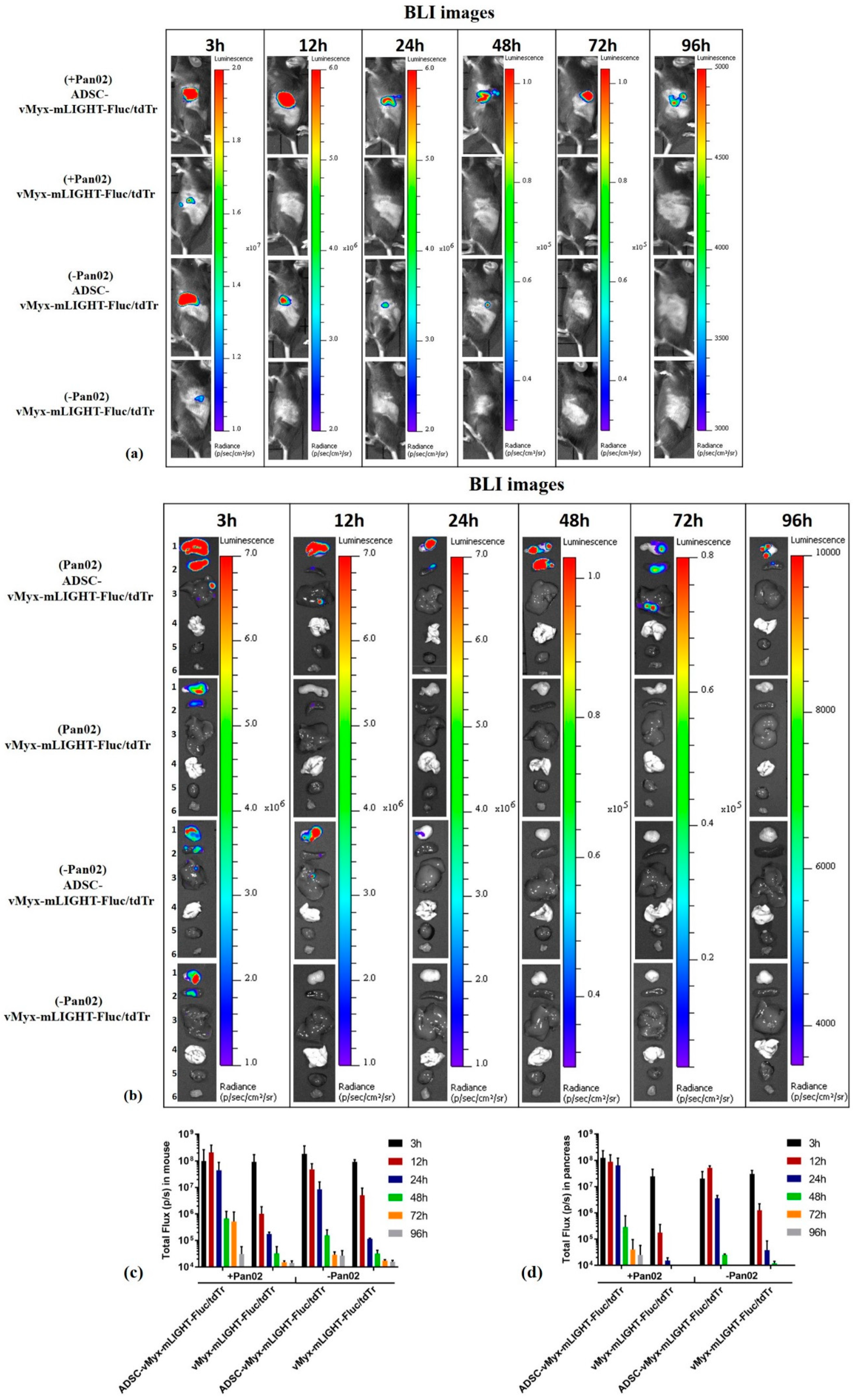
C57Bl/6NCrl mice (n = 3/group) were orthotopically implanted (day 0) with Pan02 cells (1 × 106/30 µL PBS−) (designated the +Pan02 group), or with 30 µL PBS− for unchallenged mice (referred to as the −Pan02 group). Seven days after implantation, the mice were injected intraperitoneally (IP) with either a single dose of ADSCs previously infected (MOI = 5) for 24 h with vMyx-mLIGHT-Fluc/tdTr (5 × 105 cells/100 µL PBS−), or with unshielded vMyx-mLIGHT-Fluc/tdTr (5 × 105 FFU/100 µL PBS−). Bioluminescence imaging (BLI) was performed using the Lumina IVIS Imaging System (PerkinElmer). At various time points (3–96 h) after delivery of luciferase gene-carrying MYXV, the mice were injected IP with 1.5 mg D-luciferin (Promega). BLI data were acquired and regions of interest (ROIs) determined in both intact animals and dissected organs (pancreas, spleen, liver, lungs, heart and leg muscle).
2.14. Therapy of Immunocompetent Mice Bearing Orthotopic Pancreatic Tumors
Pancreatic tumors were established in recipient C57Bl/6NCrl mice (n = 10–11/group) (day 0) by orthotopic implantation of 1 × 106 Pan02 cells/30 µL PBS−. For five-dose treatment regimens (days 4, 8, 12, 16 and 20), mice were injected IP with ADSCs infected (MOI = 5) for 24 h with vMyx-mLIGHT-Fluc/tdTr (5 × 105 cells/100 µL PBS−) or with unshielded vMyx-mLIGHT-Fluc/tdTr (5 × 105 FFU/100 µL PBS−) or 100 µL PBS− (control). After 21 days, mice (n = 3/group) were sacrificed, peripheral blood was collected, and the pancreas, spleen and liver were excised, weighed and measured for size. Peripheral blood and pancreatic tissue were used for flow cytometry studies, whereas pancreas, spleen and liver tissues were formalin-fixed and used for histological assessments. The remaining treated mice (n = 7–8/group) were monitored for survival.
2.15. Histological Assessments
Formalin-fixed and paraffin-embedded pancreas, spleen and liver sections (5 µm-thick) were H&E stained, scanned and analyzed microscopically using a digital slide scanner and CaseViewer software (3D HISTECH, Budapest, Hungary) by an experienced pathologist. Masson’s staining was performed to assess the amount and localization of connective tissue in the tumor structure. Mitotic Index [
28] was rated under 400× magnification (40 × 10) for 10 high power fields (HPF; field number 26.5).
2.16. Flow-Cytometry Analysis of CD4, CD8, CD3 Tumor-Infiltrating Lymphocytes
Pancreatic tumors were established orthotopically, and five-dose treatment conducted as described earlier. After 21 days, mice (n = 3) were sacrificed, peripheral blood samples were collected in EDTA-coated tubes, and pancreata were excised. Blood samples were treated with Red Blood Cell Lysis Buffer (BD Biosciences). Single-cell suspensions derived from pancreas tissue were obtained using a digestion mix containing 250 U/mL collagenase Type I (Gibco), 0.2 mg/mL hyaluronidase type IV-S (Sigma-Aldrich) and 0.02 mg/mL DNase I (Worthington) in DMEM + 10% fetal bovine serum (EURx). The digested samples were mashed through a sterile 70-µm nylon mesh cell strainer into ice-cold PBS− containing 1% FBS. Red blood cells in pancreatic digests were lysed (3 min) on ice using ACK Lysis Buffer (Lonza) and passed through a 40-µm nylon mesh cell strainer. To quantify percentages of CD4+, and CD8+ populations in pancreas and blood, the generated samples were treated with antibodies (20 min/RT). After fluorescent labeling, 4 × 104 cells were washed and analyzed using flow cytometry (BD FACS Canto II). The following monoclonal antibodies were used according to the manufacturer’s instructions: PerCP/Cyanine5.5 anti-mouse CD45 (clone 30-F11; BioLegend, San Diego, CA, USA), phycoerythrin (PE) anti-mouse CD3 (clone 17A2; BioLegend), fluorescein isothiocyanate (FITC) anti-mouse CD4 (clone GK1.5; BioLegend) and APC/Cyanine7 anti-mouse CD8a (clone 53-6.7; BioLegend).
2.17. Statistical Analysis
Graphs were plotted using GraphPad Prism 7 (GraphPad Software, San Diego, CA, USA) Statistical differences were determined using a one-way ANOVA test, followed by Tukey’s multiple comparisons test or two-way ANOVA with Tukey’s multiple comparisons test. Bartlett’s test was performed to ensure the suitability of the data for parametric significance tests. Kaplan–Meier survival curves were compared statistically using a log-rank test (Mantel–Cox). Data are presented as bars indicating means (±SD). The significance levels are indicated with asterisks: * p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001. p-values < 0.05 were considered statistically significant.
4. Discussion
PDAC presents a formidable challenge for oncological researchers, as the overall outcome of PDAC patients remains desperately poor. The characteristic feature of PDAC is its resistance against practically any treatment [
30,
31,
32].
Oncolytic virotherapy represents a promising approach to augmenting the PDAC therapeutic repertoire. In the current manuscript, we report the results of an experimental therapy targeting orthotopically-induced PDAC lesions in immunocompetent mice using recombinant oncolytic MYXV encoding mouse tumor necrosis factor ligand superfamily member 14 (also called LIGHT), designed to boost the anti-tumor immune response induced by the virotherapy. The vMyx-mLIGHT-Fluc/tdTr construct was pre-adsorbed ex vivo onto adipose tissue-derived stem cells (ADSCs) of mesenchymal origin and subsequently infused into the peritoneal cavity of experimental animals that had been pre-seeded orthotopically with murine PDAC into the pancreas.
We show that the cultured human ADSCs had correct morphology and multipotency. In-vitro-expanded ADSC cells were shown to differentiate into adult cells of the expected downstream lineages. The phenotype of ADSCs, as assessed by characteristic surface markers and migratory properties, was also shown to remain unchanged between MYXV-infected ADSCs and uninfected ADSCs, at least in the 1–2 day interval post-infection. We confirmed the level of permissiveness of ADSCs and three tested pancreatic cancer cell lines to MYXV. We observed significant differences between these pancreatic cancer cell lines concerning the level of infection and replication of MYXV. In general, cancer cells can be either fully permissive (i.e., produce in excess of 10 infectious progeny virus units per cell), semi-permissive (i.e., produce the progeny virus at a level below 10 infectious units/cell) or non-permissive (no detectable progeny virus) to MYXV. All three categories of cells can nevertheless express MYXV-encoded transgenes controlled by early virus promoters, but only the first two types can also express transgenes under late viral promoter control. The in vitro results indicated that MYXV productively infects the murine pancreatic cancer cell line (Pan02) with evidence of a cytopathic effect, complete viral replication and increased cell death. However, levels of productive MYXV replication in human pancreatic cancer cell lines (AsPC-1 and Panc-1) showed that they were less susceptible to infection, and we define them as semi-permissive or nonpermissive for MYXV, respectively. ADSCs remained the most viable after MYXV infection, whereas RK13 (the control rabbit cell line used to propagate the virus) was highly MYXV-susceptible.
Despite the variable levels of progeny virus generated, infection of all cultured pancreatic adenocarcinoma cells with vMyx-mLIGHT-FLuc/tdTr led to a marked-to-significant reduction in cell viability. ADSCs, in contrast, support viral infection without rapid cell death, making this viral cell carrier useful for in vivo experiments where cell viability and taxis need only be maintained for a brief period in order to ferry the virus into cancerous sites, likely hours or less. The ability of cultured ADSCs to support MYXV replication is similar to that observed with bone marrow-derived mesenchymal stem cells [
23]. This is a key feature of ADSCs, allowing the successful transfer of the virus into adjacent tumor cells via cell–cell contact. ADSCs thus seem to fulfill the criteria for an effective viral cargo carrier that is capable of transferring both the input (parental) and progeny virus to tumor cells, yielding sizeable therapeutic benefits in vivo.
We first showed that ADSCs infected with MYXV could effectively prevent the outgrowth of experimentally induced PDAC lesions when the cancer cells were co-implanted with MYXV or with ADSCs pre-infected ex vivo with MYXV. On the other hand, control tumors were rapidly induced in the absence of the virus. This pre-treatment experiment indicated that MYXV can be highly therapeutic against PDAC if the virus delivery to cancer cells in situ can be optimized. Thus, the model is a sensitive indicator of the efficiency of delivery of an OV to tumor cells at the pancreatic site of PDAC. Essentially similar outcomes seen with co-implantation of cancer cells and therapeutic constructs provide evidence of viral construct transfer from infected MSCs to cancer cells under in vivo conditions [
23].
OV-mediated systemic therapy, for example after IV infusion of the unshielded virus, faces rapid and efficient anti-viral immune host responses, as well as other clearance obstacles. During bloodstream transit, the viral cargo is largely cleared in organs like the liver and spleen, although protective carrier cells can mitigate against this [
22]. Whether IP administration of the tested therapeutic recombinant MYXV would be contingent upon, or benefit from, shielding by a protective cell carrier like ADSCs was an open question that we approached in this study. We report that IP administration of the protected viral therapeutic cargo by pre-loading onto ADSCs ex vivo yielded desirable pancreas-restricted distribution as compared to more standard IV administration. The significance of pre-loading carrier cells with oncolytic virus cannot be overestimated. Our data highlight the outcome of delivering such pre-loaded ADSCs. IVIS-generated images tracking the distribution of the viral construct (vMyx-mLIGHT-Fluc/tdTr) in PDAC-bearing mice following IP injection clearly demonstrate the delivery of the virus into the pancreas. The unshielded recombinant MYXV was rather rapidly cleared from the body after IP administration, as opposed to the virus shielded by ADSCs. The existing dogma postulates that anti-OV immune responses restrict viral replication and spread, and thus reduce direct OV-mediated killing of cancer cells. Therefore, the anti-tumor activity of the unshielded virus, although present, is likely not optimal therapeutically. This point is also illustrated by the difference in survival between the two therapeutic groups (MYXV only vs. ADSC-MYXV).
Photon flux IVIS data from intact mice demonstrate the advantage of shielding the virus using ADSCs and temporal signal changes suggest the release and transfer of the virus from carrier cells into target pancreatic cancer cells. The persistence of the signal from virus that had been pre-shielded with ADSCs implies either a different fate of the virus transferred from ADSCs or a source of the signal other than pancreas. Comparison of the signal between tumor-bearing and tumor-free control mice also confirms the protective benefits of ADSC pre-shielding and increased targeting in favor of tumor bearing mice, perhaps due to the inflammatory nature of tumor foci. Temporal differences in the signal are also suggestive of virus release and de novo infection of cancer cells, and signal differences between tumor-bearing and tumor-free animals could be ascribed to the presence or absence of tumor lesions. Total photon flux data, showing exceptionally large differences between shielded vs. unshielded virus signals from pancreata of tumor-bearing mice (several orders of magnitude), point to effective delivery and transfer of the viral construct to cancer cells within the pancreas. The potential ability of ADSCs to seek out pancreatic cancer cells and deliver oncolytic MYXV from an IP injection could be of great therapeutic value, especially when other delivery strategies are ineffective.
Ex-vivo-expanded hypoimmunogenic human adipose-derived stem cells (ADSCs) represent a unique delivery platform for OVs. They combine a natural capacity to home to inflammatory sites with a tumor tropism that is coupled with potent amplification of the OV load and transient suppression of anti-viral innate immunity, which hinders OVs from colonizing the tumor site and infecting cancer cells [
33]. Importantly, IFNγ, which is involved in the induction of an anti-viral state, also modulates immunosuppressive features of ADSCs, counteracting anti-viral immunity. Thus, the dual capacity of ADSCs to offset the innate and adaptive arms of anti-viral immunity and to allow viral load amplification potential is conducive for the success of oncolytic virotherapy. In studies involving mice and repetitive treatment in which the access route is challenging, IP delivery remains an alternative to IV when the latter strategy is clearly ineffective, as it generally is for PDAC. Targeting the pancreas with an OV using mesenchymal stem cells as carriers is not nearly as effective via IV delivery, for the same reason that targeting lung neoplasias in this manner is ineffective—the “first pass” effect [
23]. Although IP administration of pharmacological agents is minimally used in the clinic (mostly for the treatment of peritoneal cancers), in experimental animals it is a justifiable route for proof-of-concept studies where the goal is to evaluate the effect(s) of target engagement rather than the properties of a drug formulation and/or its pharmacokinetics for clinical translation [
34].
Following the postulated release of the virus from ADSCs at the targeted tumor site, some intratumoral antiviral events occur, resulting in a “cold” to “hot” transition, reversal of immunosuppression and recruitment of immune cells. Cancer cell death-associated signals further contribute to the development of a tumor-specific adaptive immune response. Overall, antiviral innate and adaptive responses targeting virus replication sites target cancer cells since OVs preferentially infect cancer cells [
35]. A decisive factor in successful immunotherapy is the presence of T cells and the reactivation of their anti-tumor properties. Even though T cells are rather low in PDAC, some data suggest that the tumor microenvironment (TME) mainly consists of various cellular components and the extracellular matrix and is highly immunosuppressive. Tumor-infiltrating lymphocytes are one of the crucial players in the TME of pancreatic cancer. On the other hand, although numbers of T cells in PDAC appear low [
36], the tumor-reactive T-cell repertoire was found to be similar to that in melanoma, in which immunotherapy does show a therapeutic impact [
37]. Since strong intra-tumoral CD8+ T cell infiltration is associated with prolonged survival, induction of anti-tumor T cell responses can indeed be a promising approach for PDAC [
38,
39,
40].
In a previous study, we showed that the therapy targeting experimentally induced lung melanoma in mice with IL-15-encoding MYXV construct (vMyx-IL15Rα-tdTr) delivered by MSCs was effective and was able to reduce the tumor burden as well as triggering the inflow of CD8+ cells [
33]. Here, we demonstrate another ADSC-shielded recombinant MYXV that expresses LIGHT protein, used to extend the survival of treated mice and to increase the influx of T lymphocytes into the tumor.
Following a five-dose therapy of orthotopic PDAC lesions with ADSC-vMyx-mLIGHT-Fluc/tdTr, we have been able to show a reduced tumor burden effect, suggesting a positive response to treatment. At the end of the therapeutic intervention (21st day), some animals were thoroughly inspected post-mortem for any signs of macroscopic pathologies and none were found, except for remaining tumor lesions. Scars surgically-induced at the onset of the experiment were healed.
An adaptive anti-tumor immune response was evidenced by the slightly decreased percentage of CD4+ helper cells among CD3+ lymphocytes, both in the blood and in the pancreas, but concurrent with a statistically significant increase in the percentage of CD8+ cells in the pancreas. Changes in the CD4+/CD8+ ratio suggest an enhanced immune response triggered by ADSC-assisted oncolytic therapy. Analysis of H&E-stained tissue specimens from all treatment groups showed that a five-dose therapy appears to be safe for the animals as no pathology was revealed in the livers. Cancer cell infiltrations in the pancreas and spleen were evidenced in the PBS− control groups, whereas high lymphocyte infiltrates were present in specimens from the group treated with the ADSC-shielded virus; and minimal lymphocyte infiltrates were present in specimens from virus-treated group. The virus-treated groups revealed fibrotic strands suggestive of the eradication of cancer cells, further supported by the decreased mitotic index. Taken together, the results of flow cytometry and microscopic analysis suggest that vMyx-mLIGHT-Fluc/tdTr delivered by ADSCs was able to modulate the immune microenviroment in PDAC tissues and contribute to the prolonged survival of mice treated with the five-dose strategy.
We show in this proof-of concept study that the recombinant MYXV used, a therapeutic agent with a dual mode of action (as a tumor oncolytic agent and an elicitor of an acquired immune response) can be an efficient component of a multi-pronged approach to PDAC therapy. ADSC-mediated IP delivery of MYXV to treat orthotopic experimental PDAC lesions in mice proved to be highly effective. Our results demonstrated increased survival of animals bearing orthotopic PDAC tumor lesions following monotherapy with an engineered recombinant MYXV delivered by ADSC; they also showed increased numbers of T cells in pancreata dissected from the treated mice. This enables a more advanced approach to virus- and ADSC-based therapies.
The levels of productive MYXV replication in two human pancreatic cancer cell lines tested (AsPC-1 and Panc-1) showed lower susceptibility to infection yet marked-to-significant reductions in cell viability. This may obviously be a disadvantage in clinical oncology. Combinatory therapeutic approaches involving more advanced armed OVs triggering enhanced adaptive immunity effects, as well as agents targeting desmoplasia, should offset this drawback. Single-agent approaches seem insufficient to improve the therapeutic outcomes of PDAC [
41].
Intelligent combinatorial therapies appear to be required to achieve significant synergism in PDAC treatment. For example, modern radiation techniques, eliciting abscopal effects via reconditioning of the tumor microenvironment, reprogramming tumor-infiltrating macrophages towards an M1-like phenotype and favoring the recruitment of adoptively transferred T cells [
42] or activating cytosolic DNA sensors, such as STING, are on the horizon [
43]. Another example are early phase clinical trials combining recombinant human hyaluronidase, gemcitabine and nab-paclitaxel, which have revealed promising results, particularly in those patients whose tumors were characterized by high levels of hyaluronan [
44]. The rationale for all these approaches is to outcompete therapy resistance; this might be challenging, however, as combined modality treatments are frequently associated with higher toxicity levels [
45]. Immunotherapy is another hope for novel strategies against pancreatic cancer, even though this deadly cancer has been so far resistant to immune checkpoint blockade and chimeric antigen receptor (CAR) T-cell therapies. Research involving the use of CAR T cells in pancreatic cancer therapy is rapidly developing, in combination with other treatments as well [
46].
Overall, the successful strategies against PDAC should thus involve combinations of modern “classical” treatments with different immunotherapeutic approaches and, hopefully, with oncovirotherapy.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





