
Succinate Anaplerosis Has an Onco-Driving Potential in Prostate Cancer Cells

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
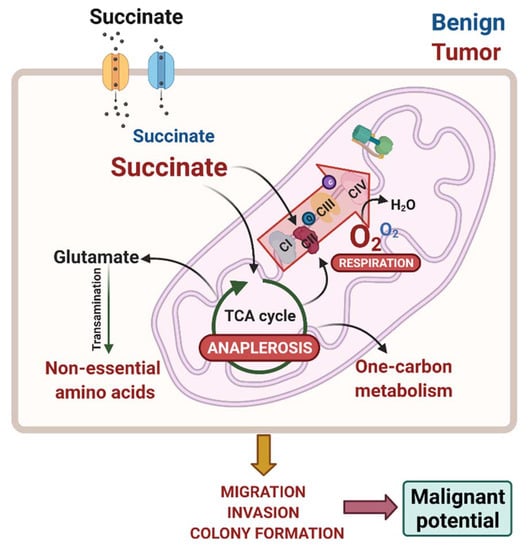
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culturing Conditions
2.2. Protein Extraction and Immunoblotting
2.3. Metabolite Extraction and Liquid Chromatography-Mass Spectrometry (LC-MS) for Metabolomics Analysis
2.4. Enzyme Activities
2.5. Dehydrogenase Activity
2.6. High-Resolution Respirometry
2.7. ATP Content
2.8. Cell Proliferation
2.9. Transwell Migration and Invasion
2.10. Anchorage-Independent Growth in Soft Agar
2.11. RNA Extraction, cDNA Synthesis and Quantitative Real Time PCR (qRT-PCR)
2.12. In silico Analysis of Prostate Cancer Gene Expression Data
2.13. Statistical Analysis
3. Results
3.1. Prostate Cells Express Plasma Membrane Dicarboxylic Transporters, Take up and Accumulate Succinate
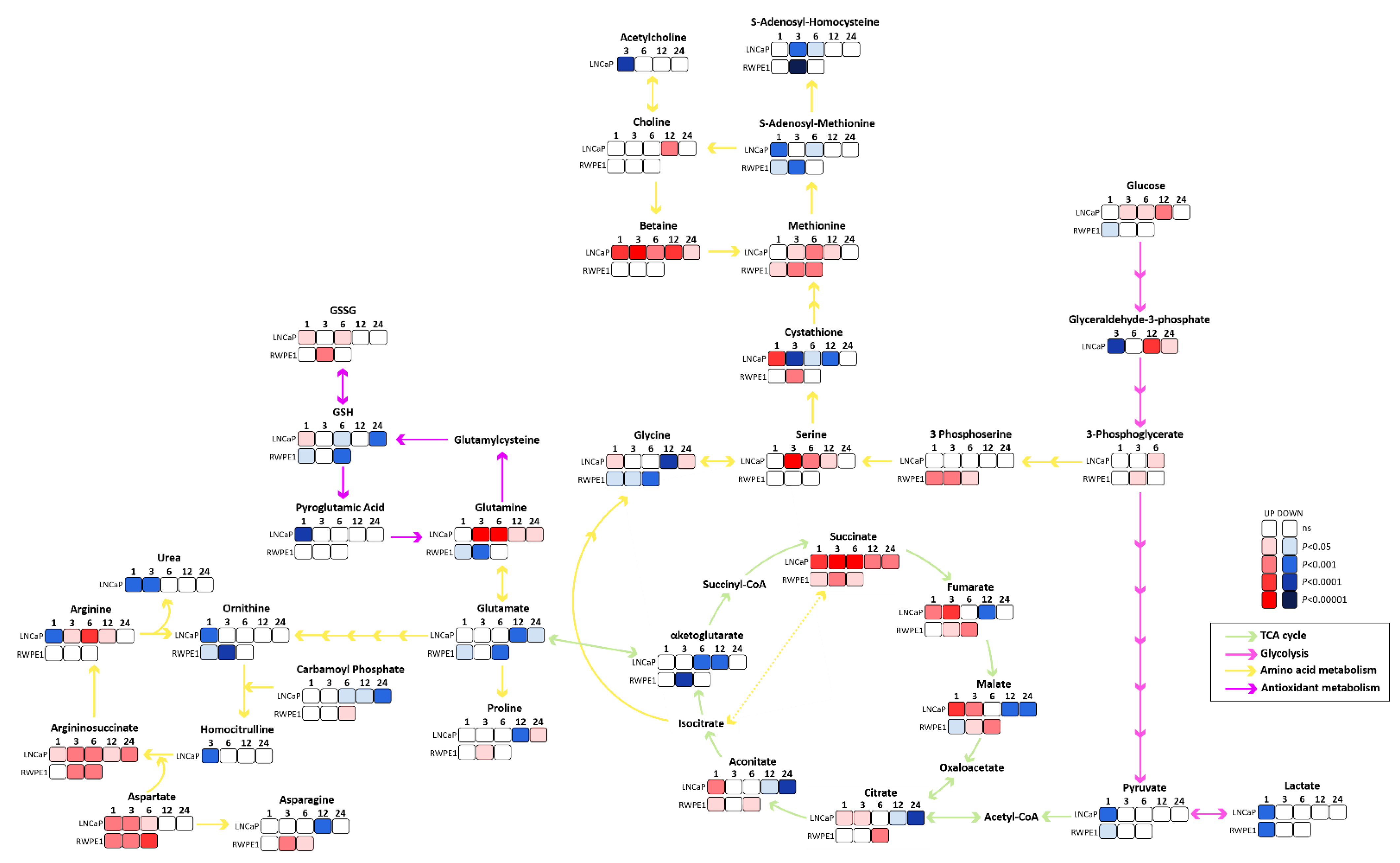
3.2. Succinate Uptake Induces Anaplerosis and Amino Acid Biosynthesis
3.3. Succinate Supports Mitochondrial Activity and Respiration
3.4. Malignant Prostate Cancer Cells Display Higher Succinate Affinity, and Succinate Incubation Induces Expression and Activity of Mitochondrial Complexes and Respiration
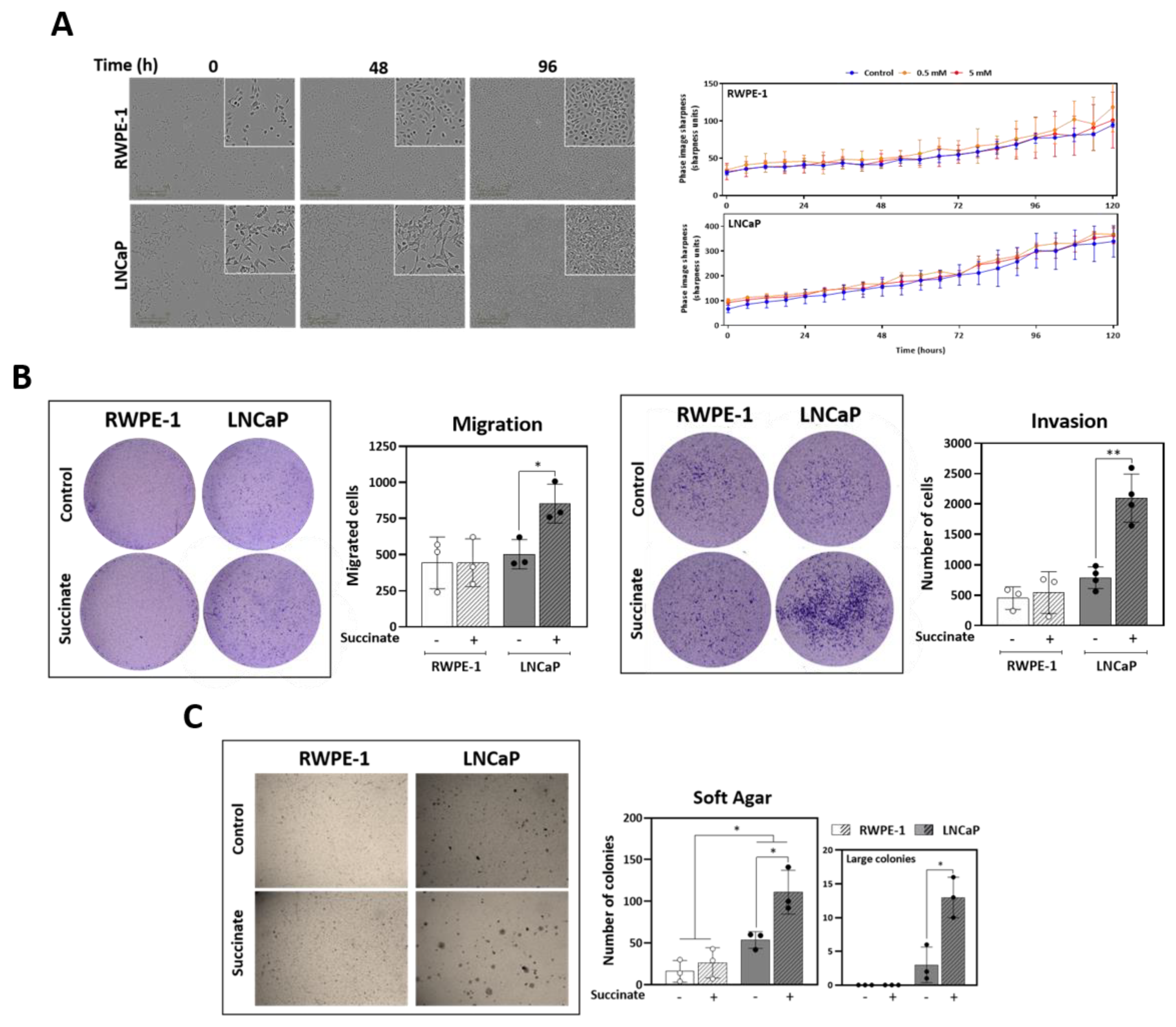
3.5. External Succinate Stimulates Migration, Invasion, and Colony Formation, but Not Proliferation, Particularly in Malignant Prostate Cells
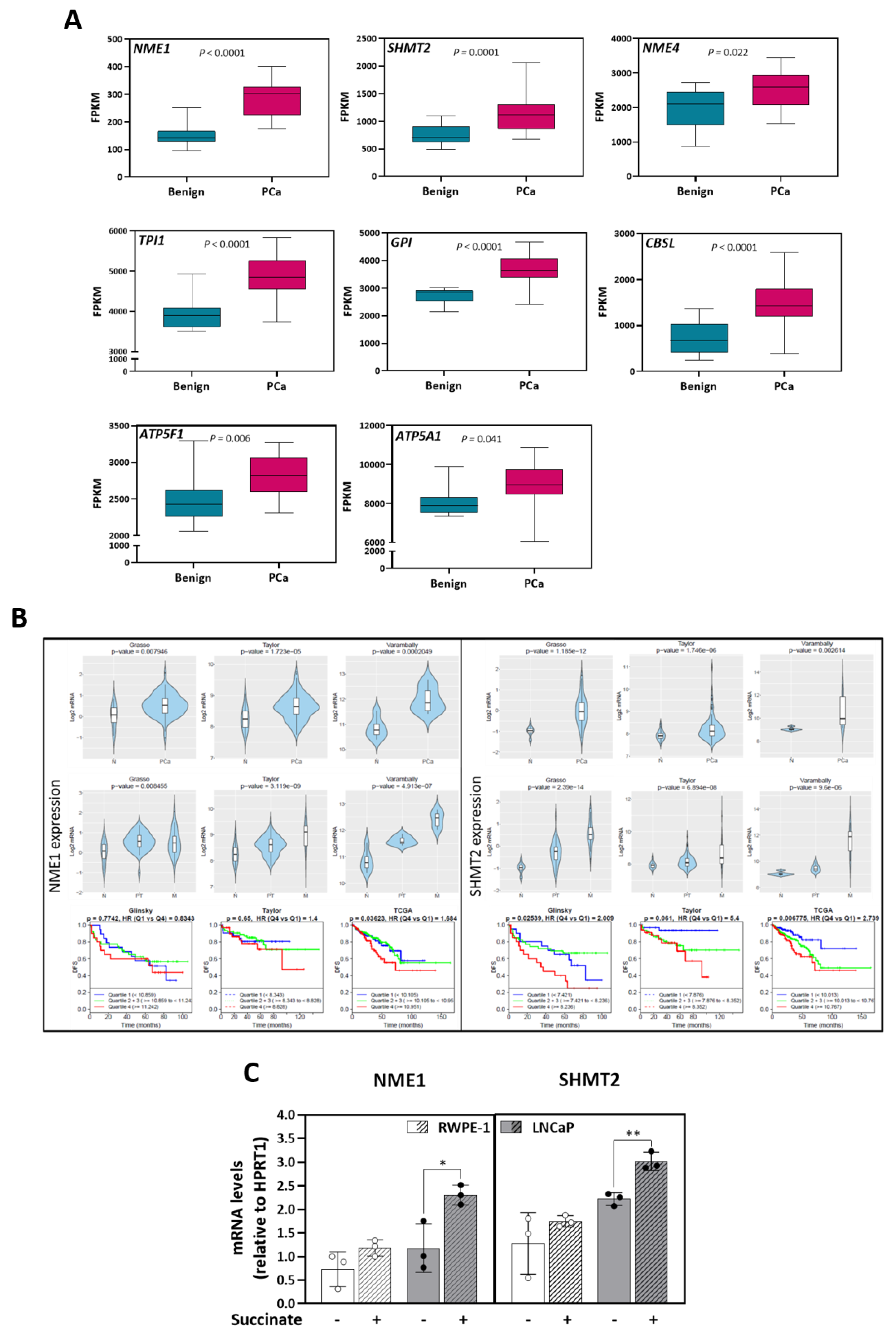
3.6. Anaplerotic and Bioenergetic Effects of Succinate Are Linked to Prostate Cancer Poor Prognosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Heiden, M.G.V.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Badrinath, N.; Yoo, S.Y. Mitochondria in cancer: In the aspects of tumorigenesis and targeted therapy. Carcinogenesis 2018, 39, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. Über den Stoffwechsel der Carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bardella, C.; Pollard, P.J.; Tomlinson, I. SDH mutations in cancer. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2011, 1807, 1432–1443. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Pollard, P.J.; Gottlieb, E. Inborn and acquired metabolic defects in cancer. J. Mol. Med. 2011, 89, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Saxena, N.; Maio, N.; Crooks, D.R.; Ricketts, C.J.; Yang, Y.; Wei, M.-H.; Fan, T.W.-M.; Lane, A.N.; Sourbier, C.; Singh, A.; et al. SDHB-Deficient Cancers: The Role of Mutations That Impair Iron Sulfur Cluster Delivery. J. Natl. Cancer Inst. 2015, 108. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Pollard, P.J. Succinate: A New Epigenetic Hacker. Cancer Cell 2013, 23, 709–711. [Google Scholar] [CrossRef] [Green Version]
- Hobert, J.A.; Mester, J.L.; Moline, J.; Eng, C. Elevated plasma succinate in PTEN, SDHB, and SDHD mutation-positive individuals. Genet. Med. 2012, 14, 616–619. [Google Scholar] [CrossRef] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Vicario, N.; Costa, A.S.; Kwok, C.K.; Leonardi, T.; Booty, L.M.; Bicci, I.; Balzarotti, B.; Volpe, G.; et al. Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell 2018, 22, 355–368.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.L.; Costa, A.S.H.; Gruszczyk, A.V.; Beach, T.E.; Allen, F.M.; Prag, H.A.; Hinchy, E.C.; Mahbubani, K.; Hamed, M.; Tronci, L.; et al. Succinate accumulation drives ischaemia-reperfusion injury during organ transplantation. Nat. Metab. 2019, 1, 966–974. [Google Scholar] [CrossRef] [Green Version]
- Jang, C.; Hui, S.; Zeng, X.; Cowan, A.J.; Wang, L.; Chen, L.; Morscher, R.J.; Reyes, J.; Frezza, C.; Hwang, H.Y.; et al. Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell Metab. 2019, 30, 594–606.e3. [Google Scholar] [CrossRef]
- Sadagopan, N.; Li, W.; Roberds, S.L.; Major, T.; Preston, G.M.; Yu, Y.; Tones, M.A. Circulating Succinate is Elevated in Rodent Models of Hypertension and Metabolic Disease. Am. J. Hypertens. 2007, 20, 1209–1215. [Google Scholar] [CrossRef]
- Alarcon, C.; Wicksteed, B.; Prentki, M.; Corkey, B.E.; Rhodes, C.J. Succinate Is a Preferential Metabolic Stimulus-Coupling Signal for Glucose-Induced Proinsulin Biosynthesis Translation. Diabetes 2002, 51, 2496–2504. [Google Scholar] [CrossRef] [Green Version]
- Fahien, L.A.; Macdonald, M.J. The Succinate Mechanism of Insulin Release. Diabetes 2002, 51, 2669–2676. [Google Scholar] [CrossRef] [Green Version]
- Serena, C.; Ceperuelo-Mallafré, V.; Keiran, N.; Queipo-Ortuño, M.I.; Bernal, R.; Gomez-Huelgas, R.; Urpi-Sarda, M.; Sabater, M.; Pérez-Brocal, V.; Andrés-Lacueva, C.; et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 2018, 12, 1642–1657. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Djukovic, D.; Deng, L.; Gu, H.; Himmati, F.; Abu Zaid, M.; Chiorean, E.G.; Raftery, D. Targeted serum metabolite profiling and sequential metabolite ratio analysis for colorectal cancer progression monitoring. Anal. Bioanal. Chem. 2015, 407, 7857–7863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.-G.; Zhao, W.; Zhang, J.; Wu, X.; Hu, J.; Yin, G.-C.; Xu, Y.-J. Metabolomics and eicosanoid analysis identified serum biomarkers for distinguishing hepatocellular carcinoma from hepatitis B virus-related cirrhosis. Oncotarget 2017, 8, 63890–63900. [Google Scholar] [CrossRef]
- Shao, Y.; Ye, G.; Ren, S.; Piao, H.-L.; Zhao, X.; Lu, X.; Wang, F.; Ma, W.; Li, J.; Yin, P.; et al. Metabolomics and transcriptomics profiles reveal the dysregulation of the tricarboxylic acid cycle and related mechanisms in prostate cancer. Int. J. Cancer 2018, 143, 396–407. [Google Scholar] [CrossRef]
- Schöpf, B.; Weissensteiner, H.; Schäfer, G.; Fazzini, F.; Charoentong, P.; Naschberger, A.; Rupp, B.; Fendt, L.; Bukur, V.; Giese, I.; et al. OXPHOS remodeling in high-grade prostate cancer involves mtDNA mutations and increased succinate oxidation. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhunussova, A.; Sen, B.; Friedman, L.; Tuleukhanov, S.; Brooks, A.D.; Sensenig, R.; Orynbayeva, Z. Tumor microenvironment promotes dicarboxylic acid carrier-mediated transport of succinate to fuel prostate cancer mitochondria. Am. J. Cancer Res. 2015, 5, 1665–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Doerrier, C.; Garcia-Souza, L.F.; Krumschnabel, G.; Wohlfarter, Y.; Mészáros, A.T.; Gnaiger, E. High-Resolution FluoRespirometry and OXPHOS Protocols for Human Cells, Permeabilized Fibers from Small Biopsies of Muscle, and Isolated Mitochondria. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; Volume 1782, pp. 31–70. [Google Scholar]
- Gnaiger, E.; Aasander Frostner, E.; Abdul, K.N.; Abdel-Rahman, E.A.; Abumrad, N.A.; Acuna-Castroviejo, D.; Adiele, R.C.; Ahn, B.; Alencar, M.B.; Ali, S.S.; et al. Mitochondrial Physiology Extended Resource of Mitochondrial Respiratory States and Rates. Bioenerg. Commun. 2020. [Google Scholar] [CrossRef]
- Gnaiger, E.; Kuznetsov, A.V.; Schneeberger, S.; Seiler, R.; Brandacher, G.; Steurer, W.; Margreiter, R. Mitochondria in the Cold. In Life in the Cold; Springer: Berlin/Heidelberg, Germany, 2000; pp. 431–442. [Google Scholar]
- Gnaiger, E. Mitochondrial pathways and respiratory control. An introduction to OXPHOS analysis. Bioenerg. Commun. 2020, 2. [Google Scholar] [CrossRef]
- Cortazar, A.R.; Torrano, V.; Martín-Martín, N.; Caro-Maldonado, A.; Camacho, L.; Hermanova, I.; Guruceaga, E.; Lorenzo-Martín, L.F.; Caloto, R.; Gomis, R.R.; et al. CANCERTOOL: A Visualization and Representation Interface to Exploit Cancer Datasets. Cancer Res. 2018, 78, 6320–6328. [Google Scholar] [CrossRef] [Green Version]
- Glinsky, G.V.; Glinskii, A.B.; Stephenson, A.J.; Hoffman, R.M.; Gerald, W.L. Gene expression profiling predicts clinical outcome of prostate cancer. J. Clin. Investig. 2004, 113, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varambally, S.; Yu, J.; Laxman, B.; Rhodes, D.R.; Mehra, R.; Tomlins, S.A.; Shah, R.B.; Chandran, U.; Monzon, F.A.; Becich, M.J.; et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005, 8, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, A.; Klocker, H.; Oberacher, H.; Gnaiger, E.; Neuwirt, H.; Sampson, N.; Eder, I.E. Succinate Accumulation Is Associated with a Shift of Mitochondrial Respiratory Control and HIF-1α Upregulation in PTEN Negative Prostate Cancer Cells. Int. J. Mol. Sci. 2018, 19, 2129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Zhuang, L.; Maddox, D.M.; Smith, S.B.; Ganapathy, V. Structure, Function, and Expression Pattern of a Novel Sodium-coupled Citrate Transporter (NaCT) Cloned from Mammalian Brain. J. Biol. Chem. 2002, 277, 39469–39476. [Google Scholar] [CrossRef] [Green Version]
- Gnaiger, E.; Lassnig, B.; Kuznetsov, A.; Rieger, G.; Margreiter, R. Mitochondrial oxygen affinity, respiratory flux control and excess capacity of cytochrome c oxidase. J. Exp. Biol. 1998, 201, 1129–1139. [Google Scholar] [PubMed]
- Schöpf, B.; Schäfer, G.; Weber, A.; Talasz, H.; Eder, I.E.; Klocker, H.; Gnaiger, E. Oxidative phosphorylation and mitochondrial function differ between human prostate tissue and cultured cells. FEBS J. 2016, 283, 2181–2196. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Culig, Z.; Hoffmann, J.; Erdel, M.; Eder, I.E.; Hobisch, A.; Hittmair, A.; Bartsch, G.; Utermann, G.; Schneider, M.R.; Parczyk, K.; et al. Switch from antagonist to agonist of the androgen receptor blocker bicalutamide is associated with prostate tumour progression in a new model system. Br. J. Cancer 1999, 81, 242–251. [Google Scholar] [CrossRef]
- Zang, L.; Frenkel, R.; Simeone, J.; Lanan, M.; Byers, M.; Lyubarskaya, Y. Metabolomics Profiling of Cell Culture Media Leading to the Identification of Riboflavin Photosensitized Degradation of Tryptophan Causing Slow Growth in Cell Culture. Anal. Chem. 2011, 83, 5422–5430. [Google Scholar] [CrossRef] [PubMed]
- Daskalaki, E.; Pillon, N.J.; Krook, A.; Wheelock, C.E.; Checa, A. The influence of culture media upon observed cell secretome metabolite profiles: The balance between cell viability and data interpretability. Anal. Chim. Acta 2018, 1037, 338–350. [Google Scholar] [CrossRef]
- De Vitto, H.; Pérez-Valencia, J.; Radosevich, J.A. Glutamine at focus: Versatile roles in cancer. Tumor Biol. 2015, 37, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yang, M.; Gaur, U.; Xu, H.; Yao, Y.; Li, D. Alpha-Ketoglutarate: Physiological Functions and Applications. Biomol. Ther. 2016, 24, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sant’Anna-Silva, A.C.B.; Santos, G.C.; Campos, S.P.C.; Gomes, A.M.O.; Pérez-Valencia, J.A.; Rumjanek, F.D. Metabolic Profile of Oral Squamous Carcinoma Cell Lines Relies on a Higher Demand of Lipid Metabolism in Metastatic Cells. Front. Oncol. 2018, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2016, 1857, 1086–1101. [Google Scholar] [CrossRef]
- Ryan, D.G.; Murphy, M.P.; Frezza, C.; Prag, H.A.; Chouchani, E.T.; O’Neill, L.A.; Mills, E.L. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 2019, 1, 16–33. [Google Scholar] [CrossRef]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Matés, J.M.; DeBerardinis, R.J. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Heiden, M.G.V. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, P. Methionine Dependence of Cancer. Biomolecules 2020, 10, 568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desvignes, T.; Pontarotti, P.; Fauvel, C.; Bobe, J. Nme protein family evolutionary history, a vertebrate perspective. BMC Evol. Biol. 2009, 9, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadrmas, E.F.; Ray, P.D.; Lambeth, D.O. Apparent ATP-linked succinate thiokinase activity and its relation to nucleoside diphosphate kinase in mitochondrial matrix preparations from rabbit. Biochim. Et Biophys. Acta (BBA) Gen. Subj. 1991, 1074, 339–346. [Google Scholar] [CrossRef]
- Kowluru, A.; Tannous, M.; Chen, H.-Q. Localization and Characterization of the Mitochondrial Isoform of the Nucleoside Diphosphate Kinase in the Pancreatic β Cell: Evidence for Its Complexation with Mitochondrial Succinyl-CoA Synthetase. Arch. Biochem. Biophys. 2002, 398, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Horak, C.E.; Lee, J.H.; Elkahloun, A.G.; Boissan, M.; Dumont, S.; Maga, T.K.; Arnaud-Dabernat, S.; Palmieri, D.; Stetler-Stevenson, W.G.; Lacombe, M.-L.; et al. Nm23-H1 Suppresses Tumor Cell Motility by Down-regulating the Lysophosphatidic Acid Receptor EDG. Cancer Res. 2007, 67, 7238–7246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Yamashita, T.; Urano, T.; Masunaga, A.; Itoyama, S.; Itoh, K.; Shiku, H.; Sugawara, I. Preferential reduction of nm23-H1 gene product in metastatic tissues from papillary and follicular carcinomas of the thyroid. Mod. Pathol. 1995, 8, 252–256. [Google Scholar]
- Liu, Y.; Jiang, C.; Tan, Y. Pathological study on the expression of cell adhesion molecules and metastasis suppressor gene in thyroid follicular carcinoma and papillary carcinoma. Zhonghua Bing Li Xue Za Zhi 2002, 31, 322–326. [Google Scholar]
- Van Noesel, M.M.; Versteeg, R. Pediatric neuroblastomas: Genetic and epigenetic ‘Danse Macabre’. Gene 2004, 325, 1–15. [Google Scholar] [CrossRef]
- Okabe-Kado, J.; Kasukabe, T.; Honma, Y. Differentiation Inhibitory Factor Nm23 as a Prognostic Factor for Acute Myeloid Leukemia. Leuk. Lymphoma 1998, 32, 19–28. [Google Scholar] [CrossRef]
- Tschiedel, S.; Gentilini, C.; Lange, T.; Wölfel, C.; Wölfel, T.; Lennerz, V.; Stevanovic, S.; Rammensee, H.-G.; Huber, C.; Cross, M.; et al. Identification of NM23-H2 as a tumour-associated antigen in chronic myeloid leukaemia. Leukemia 2008, 22, 1542–1550. [Google Scholar] [CrossRef] [Green Version]
- Andolfo, I.; De Martino, D.; Liguori, L.; Petrosino, G.; Troncone, G.; Tata, N.; Galasso, A.; Roma, C.; Chiancone, F.; Zarrilli, S.; et al. Correlation of NM23-H1 cytoplasmic expression with metastatic stage in human prostate cancer tissue. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 384, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Lucas, S.; Chen, G.; Aras, S.; Wang, J. Serine catabolism is essential to maintain mitochondrial respiration in mammalian cells. Life Sci. Alliance 2018, 1, e201800036. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.C.; Maddocks, O.D.K. One-carbon metabolism in cancer. Br. J. Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef] [Green Version]
- Labuschagne, C.F.; Broek, N.J.V.D.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but Not Glycine, Supports One-Carbon Metabolism and Proliferation of Cancer Cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Ducker, G.S.; Chen, L.; Morscher, R.J.; Ghergurovich, J.M.; Esposito, M.; Teng, X.; Kang, Y.; Rabinowitz, J.D. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016, 23, 1140–1153. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.-W.; Pawel, B.R.; Zhang, J.; Finley, L.W.; Lu, C.; Lindsten, T.; Cross, J.R.; et al. Serine Catabolism Regulates Mitochondrial Redox Control during Hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Heiden, M.G.V.; Metallo, C.M. Tracing Compartmentalized NADPH Metabolism in the Cytosol and Mitochondria of Mammalian Cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrocco, I.; Altieri, F.; Rubini, E.; Paglia, G.; Chichiarelli, S.; Giamogante, F.; Macone, A.; Perugia, G.; Magliocca, F.M.; Gurtner, A.; et al. Shmt2: A Stat3 Signaling New Player in Prostate Cancer Energy Metabolism. Cells 2019, 8, 1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Associated with Increased Metabolites in Malignant LNCaP Cells | ||||
|---|---|---|---|---|
| Genes | Mean_Benign | Mean_PCa | p value_PCa_VS_Benign | p adj_PCa_VS_Benign |
| NME1 | 149.36 | 284.76 | 8.050 × 10−15 | 2.969 × 10−12 |
| GPI | 2753.64 | 3682.55 | 1.523 × 10−09 | 7.252 × 10−08 |
| TPI1 | 3976.64 | 4868.08 | 9.558 × 10−08 | 2.137 × 10−06 |
| CBSL | 717.07 | 1481.50 | 7.901 × 10−06 | 7.883 × 10−05 |
| SHMT2 | 753.85 | 1120.12 | 2.331 × 10−05 | 1.953 × 10−04 |
| ATP5F1 | 2475.32 | 2818.06 | 1.664 × 10−03 | 6.747 × 10−03 |
| SHMT1 | 501.89 | 419.46 | 5.905 × 10−03 | 1.905 × 10−02 |
| NME4 | 1990.21 | 2545.71 | 7.191 × 10−03 | 2.238 × 10−02 |
| ATP5A1 | 8055.99 | 8915.23 | 1.514 × 10−02 | 4.106 × 10−02 |
| PGAM1 | 414.10 | 351.19 | 1.586 × 10−02 | 4.264 × 10−02 |
| GLUL | 9047.37 | 7134.23 | 1.859 × 10−02 | 4.844 × 10−02 |
| CPT1C | 75.95 | 57.83 | 2.470 × 10−02 | 6.129 × 10−02 |
| PSPH | 239.95 | 281.43 | 2.833 × 10−02 | 6.809 × 10−02 |
| ABAT | 2038.80 | 3005.96 | 3.597 × 10−02 | 8.252 × 10−02 |
| G6PC | 0.16 | 0.81 | 4.683 × 10−02 | 1.022 × 10−01 |
| ALDH7A1 | 2078.30 | 2557.72 | 5.704 × 10−02 | 1.197 × 10−01 |
| NME2 | 15.21 | 22.29 | 1.345 × 10−01 | 2.333 × 10−01 |
| CPT1B | 3.34 | 4.90 | 1.510 × 10−01 | 2.558 × 10−01 |
| NME3 | 472.52 | 621.70 | 1.655 × 10−01 | 2.745 × 10−01 |
| UPB1 | 6.78 | 5.83 | 3.837 × 10−01 | 5.170 × 10−01 |
| GLS | 1539.22 | 1470.24 | 4.210 × 10−01 | 5.523 × 10−01 |
| G6PC2 | 1.28 | 0.96 | 4.587 × 10−01 | 5.867 × 10−01 |
| PHGDH | 2123.91 | 2000.81 | 6.254 × 10−01 | 7.300 × 10−01 |
| PGK1 | 4894.12 | 4979.30 | 6.473 × 10−01 | 7.476 × 10−01 |
| PGAM2 | 7.62 | 7.41 | 6.823 × 10−01 | 7.758 × 10−01 |
| CPT1A | 1826.32 | 1809.73 | 9.266 × 10−01 | 9.522 × 10−01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sant’Anna-Silva, A.C.B.; Perez-Valencia, J.A.; Sciacovelli, M.; Lalou, C.; Sarlak, S.; Tronci, L.; Nikitopoulou, E.; Meszaros, A.T.; Frezza, C.; Rossignol, R.; et al. Succinate Anaplerosis Has an Onco-Driving Potential in Prostate Cancer Cells. Cancers 2021, 13, 1727. https://doi.org/10.3390/cancers13071727
Sant’Anna-Silva ACB, Perez-Valencia JA, Sciacovelli M, Lalou C, Sarlak S, Tronci L, Nikitopoulou E, Meszaros AT, Frezza C, Rossignol R, et al. Succinate Anaplerosis Has an Onco-Driving Potential in Prostate Cancer Cells. Cancers. 2021; 13(7):1727. https://doi.org/10.3390/cancers13071727
Chicago/Turabian StyleSant’Anna-Silva, Ana Carolina B., Juan A. Perez-Valencia, Marco Sciacovelli, Claude Lalou, Saharnaz Sarlak, Laura Tronci, Efterpi Nikitopoulou, Andras T. Meszaros, Christian Frezza, Rodrigue Rossignol, and et al. 2021. "Succinate Anaplerosis Has an Onco-Driving Potential in Prostate Cancer Cells" Cancers 13, no. 7: 1727. https://doi.org/10.3390/cancers13071727