
Targeting CDK9 for Anti-Cancer Therapeutics



Abstract
:Simple Summary
Abstract
1. Cyclin Dependent Kinases (CDKs)
2. CDK9
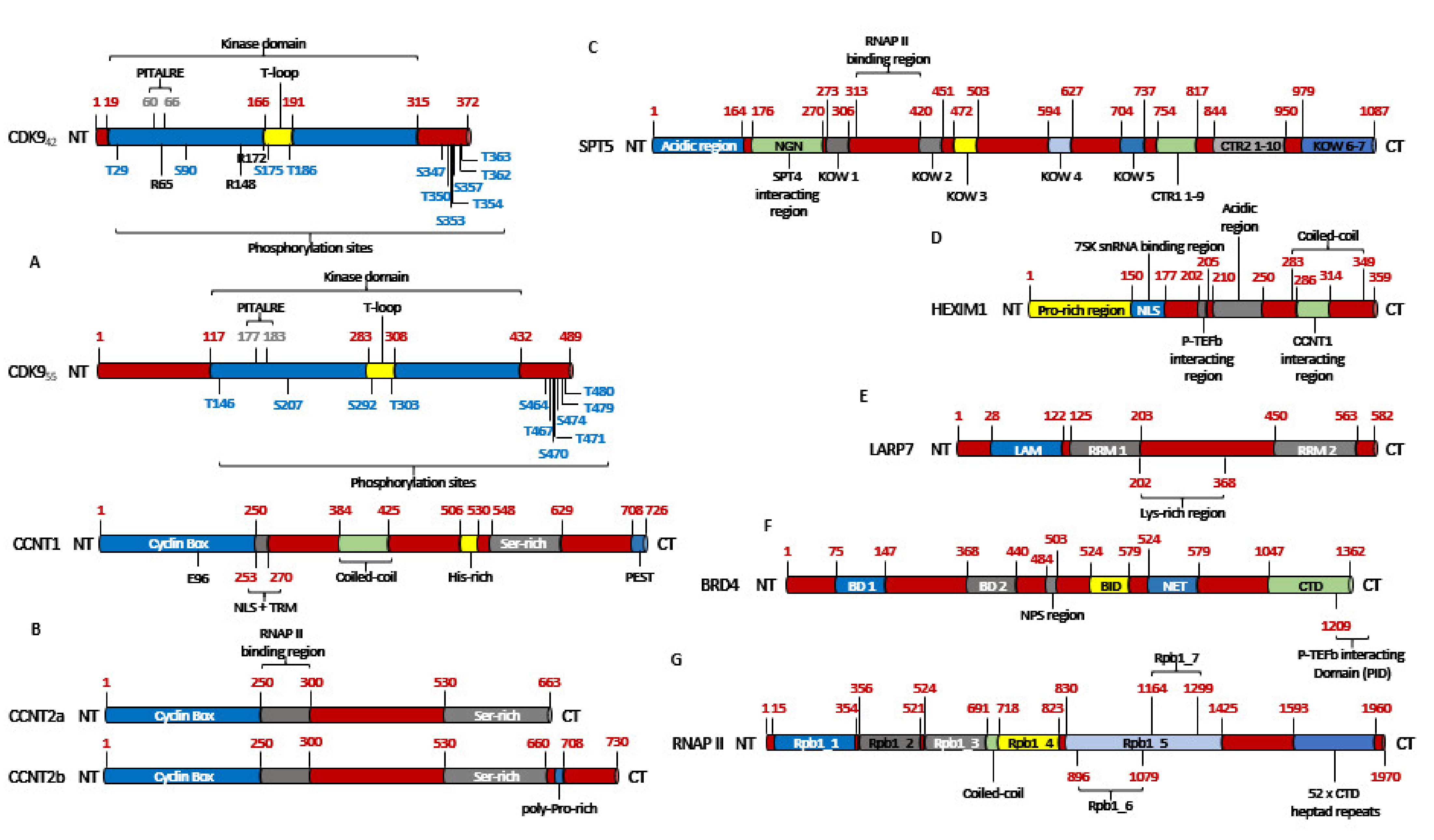
2.1. The Structure of CDK9
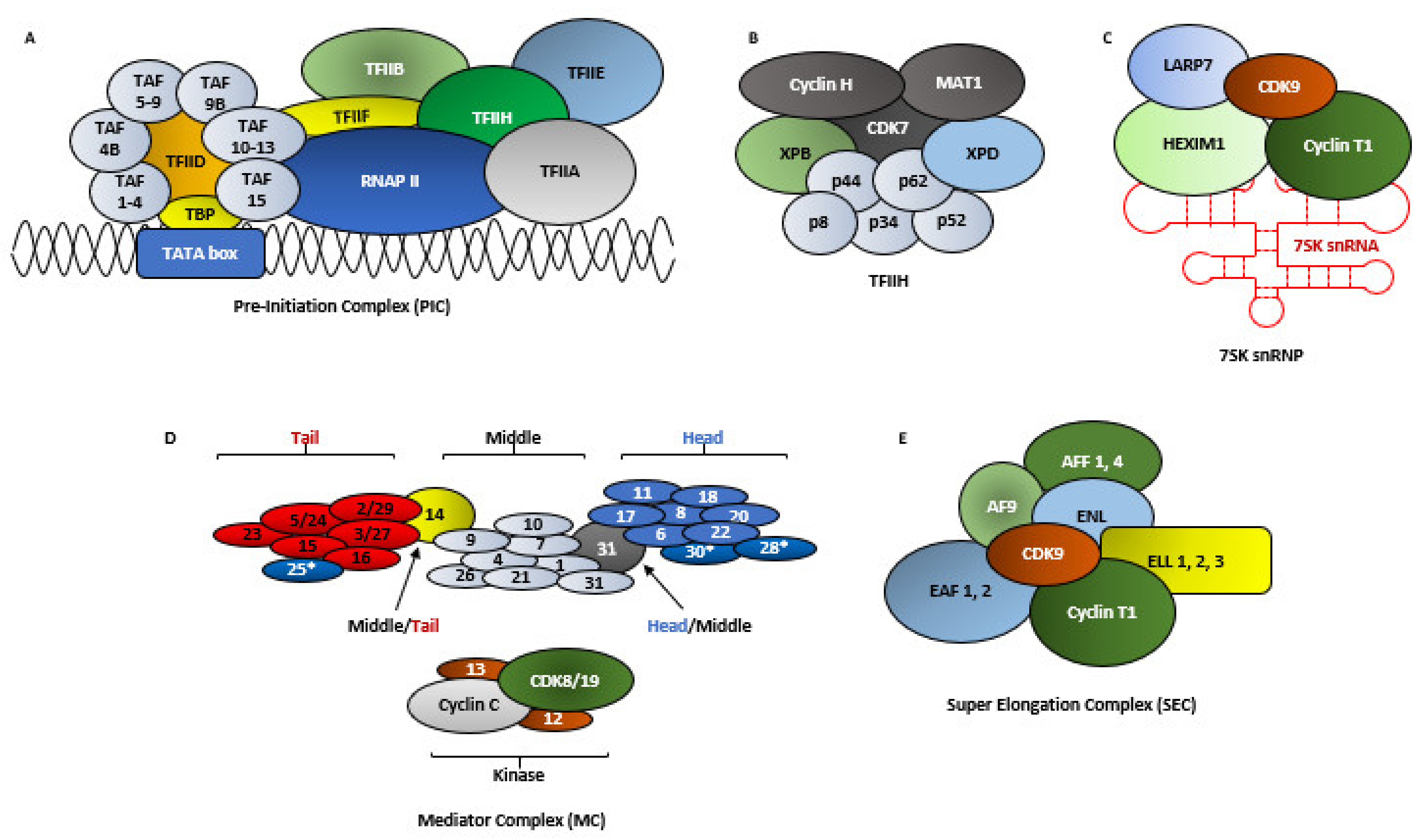
2.2. The Activation of CDK9
2.3. RNA Polymerase II-Mediated Transcription
2.4. The Molecular Functions of CDK9
2.5. The Regulation of CDK9 Activity
2.6. The Other Pool of CDK9
3. The Clinical Relevance of CDK9
3.1. Breast Cancer
3.2. Osteosarcoma
3.3. Endometrial Cancer
3.4. AML
3.5. Lung Cancer
3.6. Prostate Cancer
3.7. Melanoma
3.8. Ovarian Cancer
4. Inhibitors of CDK9
4.1. Flavivirid
4.2. AZD-4573
4.3. BAY-1143572 (Atuveciclib)
4.4. BAY-1251152
4.5. SNS-032 (BMS-387032)
4.6. AT-7519
4.7. NVP-2
4.8. JSH-150
4.9. LY-2857785
4.10. LDC000067
4.11. CDKI-73 (LS-007)

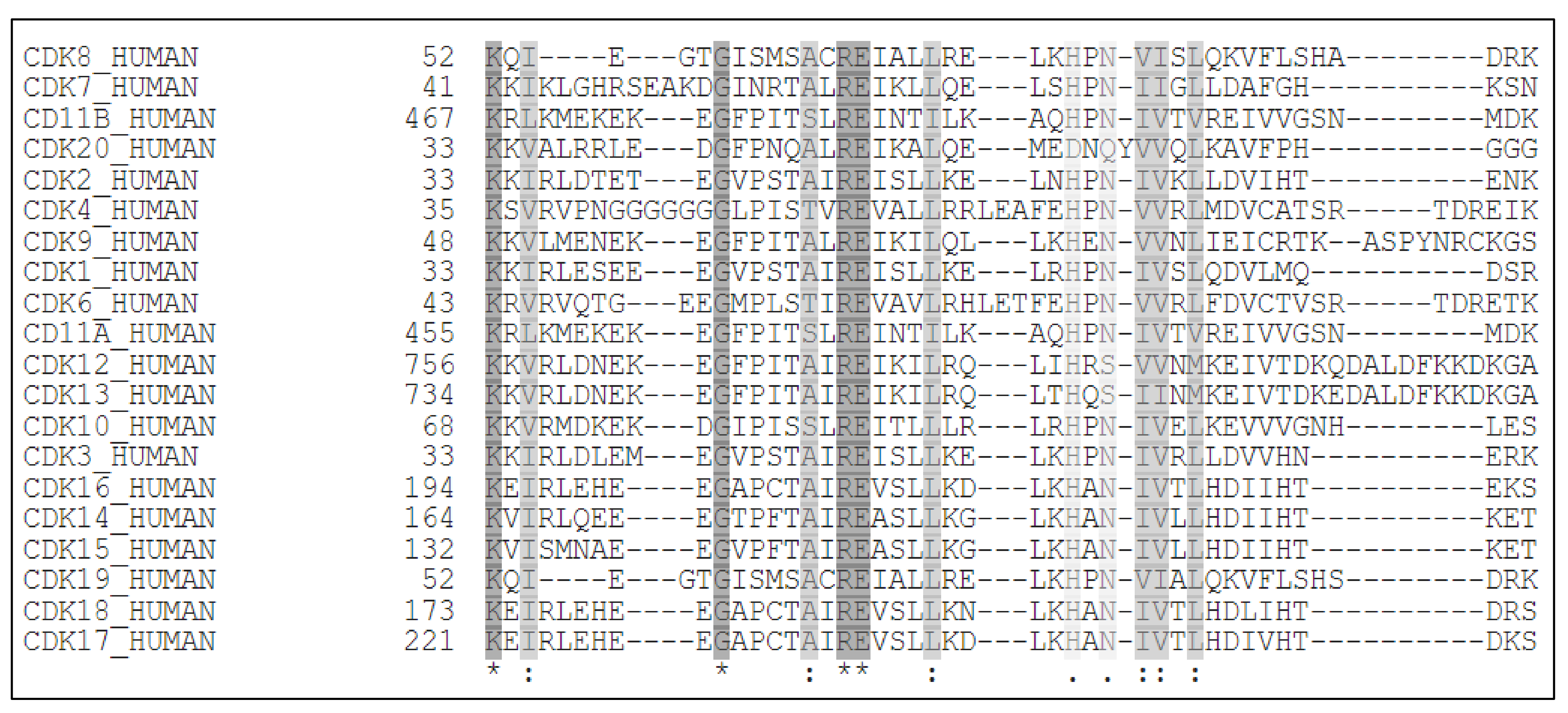{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | CDKs | Against | Clinical Trial and Status | Ref. |
|---|---|---|---|---|
Flavopiridol (Alvocidib)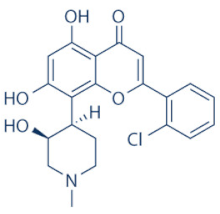 | 1, 2, 4, 6, 7 and 9 | Multiple cancer entities | Numerous; three active trials (NCT03604783; NCT03969420; NCT03593915) | |
SCH-727965 (Dinaciclib)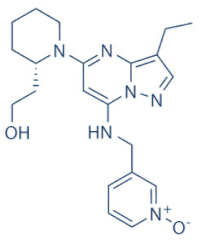 | 1, 2, 5 and 9 | Multiple cancer entities | Numerous; four active trials (NCT01676753; NCT03484520; NCT01434316; NCT00937937) | [123,124] |
LDC000067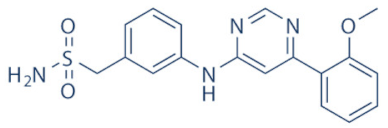 | 2 and 9 | Multiple cancer entities (pre-clinical studies) | None | [138] |
BAY-1143572 (Atuveciclib)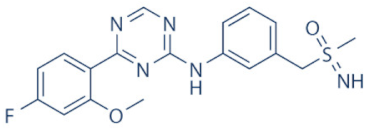 | 9 | Acute leukaemia and advanced malignancies | NCT02345382, NCT01938638 (complete/pre-maturely terminated) | [130,131] |
BAY-1251152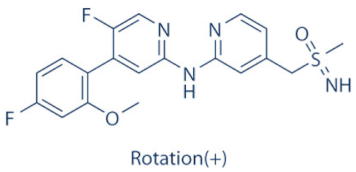 | 9 | Advanced hematological cancers and advanced malignancies | NCT02745743 (complete), NCT02635672 (active) | [132,134,135] |
SNS-032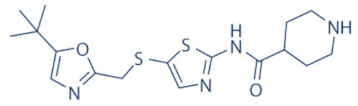 | 2, 7 and 9 | Advanced solid tumors and advanced B-lymphoid malignancies | NCT00446342, NCT00292864 (complete) | [137,139,140] |
THAL-SNS-032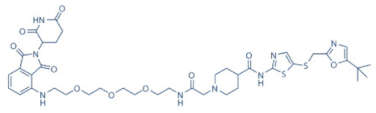 | 9 | ALL (Acute Lymphoblastic Leukemia) (pre-clinical studies) | None | [141] |
AZD-4573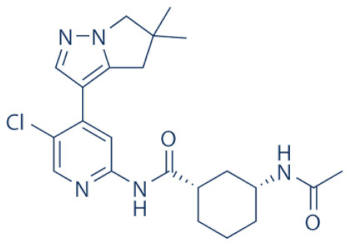 | 9 | Advanced hematological cancers and relapsed/refractory hematological cancers | NCT04630756, NCT03263637 (active) | [125,126,127,128] |
NVP-2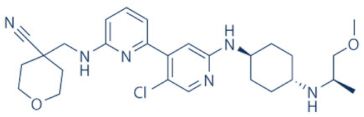 | 9 | ALL (pre-clinical studies) | None | [118,141] |
JSH-150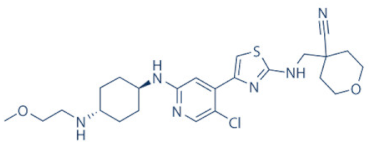 | 9 | AML and CLL (pre-clinical studies) | None | [149] |
LY-2857785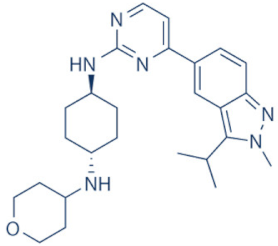 | 7 and 9 | Multiple cancer entities (pre-clinical studies) | None | [150] |
AT-7519 | 1, 2, 4, 5 and 9 | Advanced/metastatic/unresectable solid tumors, refractory NHL, MM, MCL, CLL | NCT02503709 (active), NCT00390117, NCT01183949, NCT01652144, NCT01627054 (complete) | [144,145,146,148] |
CYC-202 (Roscovitine) | 1, 2, 4, 5, 7 and 9 | TNBC, NSCLC, advanced solid tumors, Cushings disease | NCT01333423 (withdrawn), NCT00372073 (terminated), NCT00999401, NCT02160730 (terminated), NCT03774446 (recruiting) | [155,156] |
CR-8 | 1, 2, 4, 5, 7 and 9 | Neuroblastoma, (pre-clinical studies) | None | [155,156] |
CDKI-73 (LS-007)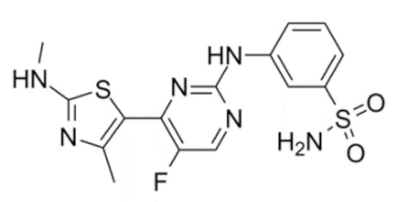 | 1, 2, 4 and 9 | CLL, AML, ovarian cancer (pre-clinical studies) | None | [152,153] |
5. Conclusions
Funding
Conflicts of Interest
References
- Cassandri, M.; Fioravanti, R.; Pomella, S.; Valente, S.; Rotili, D.; Del Baldo, G.; De Angelis, B.; Rota, R.; Mai, A. CDK9 as a Valuable Target in Cancer: From Natural Compounds Inhibitors to Current Treatment in Pediatric Soft Tissue Sarcomas. Front. Pharmacol. 2020, 11, 1230. [Google Scholar] [CrossRef] [PubMed]
- García-Reyes, B.; Kretz, A.-L.; Ruff, J.-P.; Von Karstedt, S.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; Lemke, J. The Emerging Role of Cyclin-Dependent Kinases (CDKs) in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2018, 19, 3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.J.; Endicott, J.A. Structural insights into the functional diversity of the CDK–cyclin family. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthess, Y.; Raab, M.; Sanhaji, M.; Lavrik, I.N.; Strebhardt, K. Cdk1/Cyclin B1 Controls Fas-Mediated Apoptosis by Regulating Caspase-8 Activity. Mol. Cell. Biol. 2010, 30, 5726–5740. [Google Scholar] [CrossRef] [Green Version]
- Matthess, Y.; Raab, M.; Knecht, R.; Becker, S.; Strebhardt, K. Sequential Cdk1 and Plk1 phosphorylation of caspase-8 triggers apoptotic cell death during mitosis. Mol. Oncol. 2014, 8, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, Y.A.; Taylor, M.A.; Napoleon, J.V.; Rana, S.; Contreras, J.I.; Natarajan, A. Cyclin Dependent Kinase 9 Inhibitors for Cancer Therapy. J. Med. Chem. 2016, 59, 8667–8684. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Dhavan, R.; Tsai, L.H. A decade of CDK5. Nat. Rev. Clin. Oncol. 2001, 2, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Sicinski, P. A kinase of many talents: Non-neuronal functions of CDK5 in development and disease. Open Biol. 2020, 10, 190287. [Google Scholar] [CrossRef] [Green Version]
- Diab, S.; Yu, M.; Wang, S. CDK7 Inhibitors in Cancer Therapy: The Sweet Smell of Success? J. Med. Chem. 2020, 63, 7458–7474. [Google Scholar] [CrossRef]
- Dannappel, M.V.; Sooraj, D.; Loh, J.J.; Firestein, R. Molecular and in vivo Functions of the CDK8 and CDK19 Kinase Modules. Front. Cell Dev. Biol. 2019, 6, 171. [Google Scholar] [CrossRef] [Green Version]
- Morales, F.; Giordano, A. Overview of CDK9 as a target in cancer research. Cell Cycle 2016, 15, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Guen, V.J.; Gamble, C.; Flajolet, M.; Unger, S.; Thollet, A.; Ferandin, Y.; Superti-Furga, A.; Cohen, P.A.; Meijer, L.; Colas, P. CDK10/cyclin M is a protein kinase that controls ETS2 degradation and is deficient in STAR syndrome. Proc. Natl. Acad. Sci. USA 2013, 110, 19525–19530. [Google Scholar] [CrossRef] [Green Version]
- Robert, T.; Johnson, J.L.; Guichaoua, R.; Yaron, T.M.; Bach, S.; Cantley, L.C.; Colas, P. Development of a CDK10/CycM in vitro Kinase Screening Assay and Identification of First Small-Molecule Inhibitors. Front. Chem. 2020, 8, 147. [Google Scholar] [CrossRef] [Green Version]
- Bösken, C.A.; Farnung, L.; Hintermair, C.; Schachter, M.M.; Vogel-Bachmayr, K.; Blazek, D.; Anand, K.; Fisher, R.P.; Eick, D.; Geyer, M. The structure and substrate specificity of human Cdk12/Cyclin K. Nat. Commun. 2014, 5, 3505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilarova, K.; Herudek, J.; Blazek, D. CDK12: Cellular functions and therapeutic potential of versatile player in cancer. NAR Cancer 2020, 2. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Devlin, J.R.; Hogg, S.J.; Doyle, M.A.; Harrison, P.F.; Todorovski, I.; Cluse, L.A.; Knight, D.A.; Sandow, J.J.; Gregory, G.; et al. CDK13 cooperates with CDK12 to control global RNA polymerase II processivity. Sci. Adv. 2020, 6, eaaz5041. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Song, W.; Jiang, M.; Zeng, L.; Zhu, X.; Chen, J. Phosphorylation of cyclin Y by CDK14 induces its ubiquitination and degradation. FEBS Lett. 2014, 588, 1989–1996. [Google Scholar] [CrossRef] [Green Version]
- Park, M.H.; Kim, S.Y.; Kim, Y.J.; Chung, Y.-H. ALS2CR7 (CDK15) attenuates TRAIL induced apoptosis by inducing phosphorylation of survivin Thr34. Biochem. Biophys. Res. Commun. 2014, 450, 129–134. [Google Scholar] [CrossRef]
- Mikolcevic, P.; Sigl, R.; Rauch, V.; Hess, M.W.; Pfaller, K.; Barisic, M.; Pelliniemi, L.J.; Boesl, M.; Geley, S. Cyclin-Dependent Kinase 16/PCTAIRE Kinase 1 Is Activated by Cyclin Y and Is Essential for Spermatogenesis. Mol. Cell. Biol. 2011, 32, 868–879. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, H.; Min, S.; Shen, Y.; Li, W.; Chen, Y.; Wang, X. CDK16 overexpressed in non-small cell lung cancer and regulates cancer cell growth and apoptosis via a p27-dependent mechanism. Biomed. Pharmacother. 2018, 103, 399–405. [Google Scholar] [CrossRef]
- Dixon-Clarke, S.E.; Shehata, S.N.; Krojer, T.; Sharpe, T.D.; Von Delft, F.; Sakamoto, K.; Bullock, A.N. Structure and inhibitor specificity of the PCTAIRE-family kinase CDK16. Biochem. J. 2017, 474, 699–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axtman, A.; Drewry, D.; Wells, C. CDK16: The pick of the understudied PCTAIRE kinases. Nat. Rev. Drug Discov. 2019, 18, 489. [Google Scholar] [CrossRef] [Green Version]
- Braams, E.; D’Angiolella, V. Keeping CDK18 in balance to prevent DNA replication stress in breast cancer. Oncotarget 2018, 9, 37610–37611. [Google Scholar] [CrossRef]
- Matsuda, S.; Kawamoto, K.; Miyamoto, K.; Tsuji, A.; Yuasa, K. PCTK3/CDK18 regulates cell migration and adhesion by negatively modulating FAK activity. Sci. Rep. 2017, 7, 45545. [Google Scholar] [CrossRef] [Green Version]
- Barone, G.; Staples, C.J.; Ganesh, A.; Patterson, K.W.; Bryne, D.P.; Myers, K.N.; Patil, A.A.; Eyers, C.E.; Maslen, S.; Skehel, J.M.; et al. Human CDK18 promotes replication stress signaling and genome stability. Nucleic Acids Res. 2016, 44, 8772–8785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, L.; Shin, G.Y.; Qiu, H. The Role of Cell Cycle Regulators in Cell Survival—Dual Functions of Cyclin-Dependent Kinase 20 and p21Cip1/Waf1. Int. J. Mol. Sci. 2020, 21, 8504. [Google Scholar] [CrossRef]
- Webster, K.A.; Henke, K.; Ingalls, D.M.; Nahrin, A.; Harris, M.P.; Siegfried, K.R. Cyclin-dependent kinase 21 is a novel regulator of proliferation and meiosis in the male germline of zebrafish. Reproduction 2019, 157, 383–398. [Google Scholar] [CrossRef] [Green Version]
- Grana, X.; De Luca, A.; Sang, N.; Fu, Y.; Claudio, P.P.; Rosenblatt, J.; Morgan, D.O.; Giordano, A. PITALRE, a nuclear CDC2-related protein kinase that phosphorylates the retinoblastoma protein in vitro. Proc. Natl. Acad. Sci. USA 1994, 91, 3834–3838. [Google Scholar] [CrossRef] [Green Version]
- Paparidis, N.F.D.S.; Durvale, M.C.; Canduri, F. The emerging picture of CDK9/P-TEFb: More than 20 years of advances since PITALRE. Mol. BioSyst. 2017, 13, 246–276. [Google Scholar] [CrossRef]
- Shore, S.M.; Byers, S.A.; Maury, W.; Price, D.H. Identification of a novel isoform of Cdk9. Gene 2003, 307, 175–182. [Google Scholar] [CrossRef]
- Baumli, S.; Lolli, G.; Lowe, E.D.; Troiani, S.; Rusconi, L.; Bullock, A.N.; Debreczeni, J.É.; Knapp, S.; Johnson, L.N. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008, 27, 1907–1918. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Yu, W.; Rice, A.P. Limited redundancy in genes regulated by Cyclin T2 and Cyclin T1. BMC Res. Notes 2011, 4, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, M.C.; Wong, C.; Elledge, S.J. Human Cyclin K, a Novel RNA Polymerase II-Associated Cyclin Possessing Both Carboxy-Terminal Domain Kinase and Cdk-Activating Kinase Activity. Mol. Cell. Biol. 1998, 18, 4291–4300. [Google Scholar] [CrossRef] [Green Version]
- Blazek, D. The cyclin K/Cdk12 complex: An emerging new player in the maintenance of genome stability. Cell Cycle 2012, 11, 1049–1050. [Google Scholar] [CrossRef] [Green Version]
- Kohoutek, J.; Blazek, D. Cyclin K goes with Cdk12 and Cdk13. Cell Div. 2012, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- LaRochelle, S.; Amat, R.; Glover-Cutter, K.; Sansó, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.L.; Fisher, R.P. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Mbonye, U.; Wang, B.; Gokulrangan, G.; Shi, W.; Yang, S.; Karn, J. Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. J. Biol. Chem. 2018, 293, 10009–10025. [Google Scholar] [CrossRef] [Green Version]
- Mbonye, U.R.; Gokulrangan, G.; Datt, M.; Dobrowolski, C.; Cooper, M.; Chance, M.R.; Karn, J. Phosphorylation of CDK9 at Ser175 Enhances HIV Transcription and Is a Marker of Activated P-TEFb in CD4+ T Lymphocytes. PLOS Pathog. 2013, 9, e1003338. [Google Scholar] [CrossRef] [Green Version]
- Nekhai, S.; Petukhov, M.; Breuer, D. Regulation of CDK9 Activity by Phosphorylation and Dephosphorylation. BioMed Res. Int. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Rice, A.P. Cdk9 T-loop phosphorylation is regulated by the calcium signaling pathway. J. Cell. Physiol. 2011, 227, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.B.; Sharp, P.A. Positive Transcription Elongation Factor b Phosphorylates hSPT5 and RNA Polymerase II Carboxyl-terminal Domain Independently of Cyclin-dependent Kinase-activating Kinase. J. Biol. Chem. 2001, 276, 12317–12323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grünberg, S.; Hahn, S. Structural insights into transcription initiation by RNA polymerase II. Trends Biochem. Sci. 2013, 38, 603–611. [Google Scholar] [CrossRef] [Green Version]
- Sainsbury, S.; Bernecky, C.; Cramer, P. Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.X.; Smith, E.R.; Shilatifard, A. Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2018, 19, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Greber, B.J.; Toso, D.B.; Fang, J.; Nogales, E. Author response: The complete structure of the human TFIIH core complex. Elife 2019, 8, e44771. [Google Scholar] [CrossRef] [PubMed]
- Kohoutek, J. P-TEFb- the final frontier. Cell Div. 2009, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Compe, E.; Egly, J.M. TFIIH: When transcription met DNA repair. Nat. Rev. Mol. Cell Biol. 2012, 13, 343–354. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the Elongation Phase of Transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, T.; Price, D.H. RNA Polymerase II Elongation Control. Annu. Rev. Biochem. 2012, 81, 119–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brannan, K.; Kim, H.; Erickson, B.; Glover-Cutter, K.; Kim, S.; Fong, N.; Kiemele, L.; Hansen, K.; Davis, R.; Lykke-Andersen, J.; et al. mRNA Decapping Factors and the Exonuclease Xrn2 Function in Widespread Premature Termination of RNA Polymerase II Transcription. Mol. Cell 2012, 46, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Henriques, T.; Gilchrist, D.A.; Nechaev, S.; Bern, M.; Muse, G.W.; Burkholder, A.; Fargo, D.C.; Adelman, K. Stable Pausing by RNA Polymerase II Provides an Opportunity to Target and Integrate Regulatory Signals. Mol. Cell 2013, 52, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garriga, J.; Graña, X. CDK9 inhibition strategy defines distinct sets of target genes. BMC Res. Notes 2014, 7, 301. [Google Scholar] [CrossRef] [Green Version]
- Baumli, S.; Furst, R. Perspective of Cyclin-dependent kinase 9 (CDK9) as a Drug Target. Curr. Pharm. Des. 2012, 18, 2883–2890. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, M.B.; Baltimore, D.; Frankel, A.D. The role of Tat in the human immunodeficiency virus life cycle indicates a primary effect on transcriptional elongation. Proc. Natl. Acad. Sci. 1991, 88, 4045–4049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, Y.-L.; Cao, H.; Jacque, J.-M.; Stevenson, M.; Rana, T.M. Inhibition of Human Immunodeficiency Virus Type 1 Replication by RNA Interference Directed against Human Transcription Elongation Factor P-TEFb (CDK9/CyclinT1). J. Virol. 2004, 78, 2517–2529. [Google Scholar] [CrossRef] [Green Version]
- Garber, M.E.; Mayall, T.P.; Suess, E.M.; Meisenhelder, J.; Thompson, N.E.; Jones, K.A. CDK9 Autophosphorylation Regulates High-Affinity Binding of the Human Immunodeficiency Virus Type 1 Tat–P-TEFb Complex to TAR RNA. Mol. Cell. Biol. 2000, 20, 6958–6969. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.; Garber, M.E.; Fang, S.-M.; Fischer, W.H.; Jones, K.A. A Novel CDK9-Associated C-Type Cyclin Interacts Directly with HIV-1 Tat and Mediates Its High-Affinity, Loop-Specific Binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Zhu, Y.; Milton, J.T.; Price, D.H. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998, 12, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Yamaguchi, Y.; Inukai, N.; Okamoto, S.; Mura, T.; Handa, H. P-TEFb-Mediated Phosphorylation of hSpt5 C-Terminal Repeats Is Critical for Processive Transcription Elongation. Mol. Cell 2006, 21, 227–237. [Google Scholar] [CrossRef]
- Bowman, E.A.; Kelly, W.G. RNA polymerase II transcription elongation and Pol II CTD Ser2 phosphorylation: A tail of two kinases. Nucleus 2014, 5, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.B.; Louder, R.K.; Greber, B.J.; Grünberg, S.; Luo, J.; Fang, J.; Liu, Y.; Ranish, J.; Hahn, S.; Nogales, E. Structure of human TFIID and mechanism of TBP loading onto promoter DNA. Science 2018, 362, eaau8872. [Google Scholar] [CrossRef]
- Soutourina, J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2018, 19, 262–274. [Google Scholar] [CrossRef]
- He, N.; Chan, C.K.; Sobhian, B.; Chou, S.; Xue, Y.; Liu, M.; Alber, T.; Benkirane, M.; Zhou, Q. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc. Natl. Acad. Sci. USA 2011, 108, E636–E645. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Lin, C.; Shilatifard, A. The super elongation complex (SEC) family in transcriptional control. Nat. Rev. Mol. Cell Biol. 2012, 13, 543–547. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Kiss, T.; Michels, A.A.; Bensaude, O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nat. Cell Biol. 2001, 414, 322–325. [Google Scholar] [CrossRef]
- Yang, Z.; Zhu, Q.; Luo, K.; Zhou, Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nat. Cell Biol. 2001, 414, 317–322. [Google Scholar] [CrossRef]
- Quaresma, A.J.C.; Bugai, A.; Barboric, M. Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res. 2016, 44, 7527–7539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, A.A.; Fraldi, A.; Li, Q.; Adamson, T.E.; Bonnet, F.; Nguyen, V.T.; Sedore, S.C.; Price, J.P.; Price, D.H.; Lania, L.; et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 2004, 23, 2608–2619. [Google Scholar] [CrossRef] [Green Version]
- Krueger, B.J.; Jeronimo, C.; Roy, B.B.; Bouchard, A.; Barrandon, C.; Byers, S.A.; Searcey, C.E.; Cooper, J.J.; Bensaude, O.; Cohen, É.A.; et al. LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res. 2008, 36, 2219–2229. [Google Scholar] [CrossRef] [Green Version]
- Markert, A.; Grimm, M.; Martinez, J.; Wiesner, J.; Meyerhans, A.; Meyuhas, O.; Sickmann, A.; Fischer, U. The La-related protein LARP7 is a component of the 7SK ribonucleoprotein and affects transcription of cellular and viral polymerase II genes. EMBO Rep. 2008, 9, 569–575. [Google Scholar] [CrossRef] [Green Version]
- Jeronimo, C.; Forget, D.; Bouchard, A.; Li, Q.; Chua, G.; Poitras, C.; Thérien, C.; Bergeron, D.; Bourassa, S.; Greenblatt, J.; et al. Systematic Analysis of the Protein Interaction Network for the Human Transcription Machinery Reveals the Identity of the 7SK Capping Enzyme. Mol. Cell 2007, 27, 262–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Yang, Z.; Chen, R.; Zhou, Q. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res. 2009, 38, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Yang, Z.; Zhou, Q. Phosphorylated Positive Transcription Elongation Factor b (P-TEFb) Is Tagged for Inhibition through Association with 7SK snRNA. J. Biol. Chem. 2004, 279, 4153–4160. [Google Scholar] [CrossRef] [Green Version]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 1–13. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [Green Version]
- Vollmuth, F.; Blankenfeldt, W.; Geyer, M. Structures of the Dual Bromodomains of the P-TEFb-activating Protein Brd4 at Atomic Resolution. J. Biol. Chem. 2009, 284, 36547–36556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Huang, K.; Jung, K.-J.; Cho, W.-K.; Klase, Z.; Kashanchi, F.; Pise-Masison, C.A.; Brady, J.N. Bromodomain Protein Brd4 Regulates Human Immunodeficiency Virus Transcription through Phosphorylation of CDK9 at Threonine 29. J. Virol. 2008, 83, 1036–1044. [Google Scholar] [CrossRef] [Green Version]
- Schröder, S.; Cho, S.; Zeng, L.; Zhang, Q.; Kaehlcke, K.; Mak, L.; Lau, J.; Bisgrove, D.; Schnölzer, M.; Verdin, E.; et al. Two-pronged Binding with Bromodomain-containing Protein 4 Liberates Positive Transcription Elongation Factor b from Inactive Ribonucleoprotein Complexes*. J. Biol. Chem. 2012, 287, 1090–1099. [Google Scholar] [CrossRef] [Green Version]
- Itzen, F.; Greifenberg, A.K.; Bösken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yik, J.H.N.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Michels, A.A.; Bensaude, O. Hexim1, an RNA-controlled protein hub. Transcription 2018, 9, 262–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Keeffe, B.; Fong, Y.; Chen, D.; Zhou, S.; Zhou, Q. Requirement for a Kinase-specific Chaperone Pathway in the Production of a Cdk9/Cyclin T1 Heterodimer Responsible for P-TEFb-mediated Tat Stimulation of HIV-1 Transcription. J. Biol. Chem. 2000, 275, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Kostova, I.; Mandal, R.; Becker, S.; Strebhardt, K. The role of caspase-8 in the tumor microenvironment of ovarian cancer. Cancer Metastasis Rev. 2021, 40, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.; Barrón, J.C.; Kostova, I.; Becker, S.; Strebhardt, K. Caspase-8: The double-edged sword. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188357. [Google Scholar] [CrossRef]
- Napolitano, G.; Licciardo, P.; Carbone, R.; Majello, B.; Lania, L. CDK9 has the intrinsic property to shuttle between nucleus and cytoplasm, and enhanced expression of CyclinT1 promotes its nuclear localization. J. Cell. Physiol. 2002, 192, 209–215. [Google Scholar] [CrossRef]
- Brasier, A.R. Perspective: Expanding role of cyclin dependent kinases in cytokine inducible gene expression. Cell Cycle 2008, 7, 2661–2666. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Seebacher, N.A.; Hornicek, F.J.; Duan, Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in osteosarcoma. EBioMedicine 2019, 39, 182–193. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Biarnes, M.C.; Jordan, V.C. Cyclin dependent kinase-9 mediated transcriptional de-regulation of cMYC as a critical determinant of endocrine-therapy resistance in breast cancers. Breast Cancer Res. Treat. 2014, 143, 113–124. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Fang, X.; Xia, X.; Hou, T.; Zhang, T. Targeting CDK9: A novel biomarker in the treatment of endometrial cancer. Oncol. Rep. 2020, 44, 1929–1938. [Google Scholar] [CrossRef]
- Boffo, S.; Damato, A.; Alfano, L.; Giordano, A.; Alfano, L. CDK9 inhibitors in acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- McNeil, C.M.; Sergio, C.M.; Anderson, L.R.; Inman, C.K.; Eggleton, S.A.; Murphy, N.C.; Millar, E.K.; Crea, P.; Kench, J.G.; Alles, M.C.; et al. c-Myc overexpression and endocrine resistance in breast cancer. J. Steroid Biochem. Mol. Biol. 2006, 102, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.-J. Is CDK9 a promising target for both primary and metastatic osteosarcoma? EBioMedicine 2019, 40, 27–28. [Google Scholar] [CrossRef] [Green Version]
- Franco, L.C.; Morales, F.; Boffo, S.; Giordano, A. CDK9: A key player in cancer and other diseases. J. Cell. Biochem. 2018, 119, 1273–1284. [Google Scholar] [CrossRef] [PubMed]
- Lyle, L.; Daver, N. Current and emerging therapies for patients with acute myeloid leukemia: A focus on MCL-1 and the CDK9 pathway. Am. J. Manag. Care 2018, 24, S356–S365. [Google Scholar]
- Lee, D.J.; Zeidner, J.F. Cyclin-dependent kinase (CDK) 9 and 4/6 inhibitors in acute myeloid leukemia (AML): A promising therapeutic approach. Expert Opin. Investig. Drugs 2019, 28, 989–1001. [Google Scholar] [CrossRef]
- Bogenberger, J.; Whatcott, C.; Hansen, N.; Delman, D.; Shi, C.-X.; Kim, W.; Haws, H.; Soh, K.; Lee, Y.S.; Peterson, P.; et al. Combined venetoclax and alvocidib in acute myeloid leukemia. Oncotarget 2017, 8, 107206–107222. [Google Scholar] [CrossRef] [Green Version]
- Devaraj, S.G.T.; Fiskus, W.; Shah, B.A.; Qi, J.; Sun, B.; Iyer, S.P.; Sharma, S.; Bradner, J.E.; Bhalla, K.N. HEXIM1 induction is mechanistically involved in mediating anti-AML activity of BET protein bromodomain antagonist. Leukemia 2016, 30, 504–508. [Google Scholar] [CrossRef] [Green Version]
- Lew, Q.J.; Chu, K.L.; Chia, Y.L.; Cheong, N.; Chao, S.-H. HEXIM1, a New Player in the p53 Pathway. Cancers 2013, 5, 838–856. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Shan, B.; Zhuo, Y.; Chin, D.; Morris, C.A.; Morris, G.F.; Lasky, J.A. Cyclin-dependent Kinase 9 Is Required for Tumor Necrosis Factor-α-stimulated Matrix Metalloproteinase-9 Expression in Human Lung Adenocarcinoma Cells. J. Biol. Chem. 2005, 280, 1103–1111. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yu, C.; Wang, C.; Ma, Y.; Wang, T.; Li, Y.; Huang, Z.; Zhou, M.; Sun, P.; Zheng, J.; et al. Novel cyclin-dependent kinase 9 (CDK9) inhibitor with suppression of cancer stemness activity against non-small-cell lung cancer. Eur. J. Med. Chem. 2019, 181, 111535. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Men’s Health 2019, 37, 288–295. [Google Scholar] [CrossRef]
- Rahaman, M.H.; Kumarasiri, M.; Mekonnen, L.B.; Yu, M.; Diab, S.; Albrecht, H.; Milne, R.W.; Wang, S. Targeting CDK9: A promising therapeutic opportunity in prostate cancer. Endocr. Relat. Cancer 2016, 23, T211–T226. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Gulla, S.; Cai, C.; Balk, S.P. Androgen Receptor Serine 81 Phosphorylation Mediates Chromatin Binding and Transcriptional Activation. J. Biol. Chem. 2012, 287, 8571–8583. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Xu, Y.; Yuan, X.; Bubley, G.J.; Balk, S.P. Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc. Natl. Acad. Sci. USA 2006, 103, 15969–15974. [Google Scholar] [CrossRef] [Green Version]
- Hsu, F.-N.; Chen, M.-C.; Chiang, M.-C.; Lin, E.; Lee, Y.-T.; Huang, P.-H.; Lee, G.-S.; Lin, H. Regulation of Androgen Receptor and Prostate Cancer Growth by Cyclin-dependent Kinase 5. J. Biol. Chem. 2011, 286, 33141–33149. [Google Scholar] [CrossRef] [Green Version]
- Gordon, V.; Bhadel, S.; Wunderlich, W.; Zhang, J.; Ficarro, S.B.; Mollah, S.A.; Shabanowitz, J.; Hunt, D.F.; Xenarios, I.; Hahn, W.C.; et al. CDK9 Regulates AR Promoter Selectivity and Cell Growth through Serine 81 Phosphorylation. Mol. Endocrinol. 2010, 24, 2267–2280. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, C.; Wang, X.; Becker, D. Expression analysis and molecular targeting of cyclin-dependent kinases in advanced melanoma. Cell Cycle 2011, 10, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, S.; Ye, Q.; Pan, J. Transcriptional inhibition by CDK7/9 inhibitor SNS-032 abrogates oncogene addiction and reduces liver metastasis in uveal melanoma. Mol. Cancer 2019, 18, 140. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Hou, J.; Shi, S.; Du, J.; Liu, Y.; Huang, P.; Li, Q.; Liu, L.; Hu, H.; Ji, Y.; et al. CSN6 promotes melanoma proliferation and metastasis by controlling the UBR5-mediated ubiquitination and degradation of CDK9. Cell Death Dis. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Fang, L.; Lu, W.; Choi, H.H.; Yeung, S.-C.J.; Tung, J.-Y.; Hsiao, C.-D.; Fuentes-Mattei, E.; Menter, D.; Chen, C.; Wang, L.; et al. ERK2-Dependent Phosphorylation of CSN6 Is Critical in Colorectal Cancer Development. Cancer Cell 2015, 28, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Fang, L.; Phan, L.M.; Qdaisat, A.; Yeung, S.-C.J.; Lee, M.-H. COP9 signalosome subunit 6 (CSN6) regulates E6AP/UBE3A in cervical cancer. Oncotarget 2015, 6, 28026–28041. [Google Scholar] [CrossRef]
- Wang, J.; Dean, D.C.; Hornicek, F.J.; Shi, H.; Duan, Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in ovarian cancer. FASEB J. 2019, 33, 5990–6000. [Google Scholar] [CrossRef]
- Kumara, P.M.; Soujanya, K.; Ravikanth, G.; Vasudeva, R.; Ganeshaiah, K.; Shaanker, R.U. Rohitukine, a chromone alkaloid and a precursor of flavopiridol, is produced by endophytic fungi isolated from Dysoxylum binectariferum Hook.f and Amoora rohituka (Roxb).Wight & Arn. Phytomedicine 2014, 21, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, M.D.; Bender, H.; Espinosa, J.M. Therapeutic targeting of transcriptional cyclin-dependent kinases. Transcription 2019, 10, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Myers, T.G.; O’Connor, P.M.; Friend, S.H.; Fornace, A.J.; Kohn, K.W.; Fojo, T.; Bates, S.E.; Rubinstein, L.V.; Anderson, N.L.; et al. An Information-Intensive Approach to the Molecular Pharmacology of Cancer. Science 1997, 275, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senderowicz, A.M. Flavopiridol: The First Cyclin-Dependent Kinase Inhibitor in Human Clinical Trials. Investig. New Drugs 1999, 17, 313–320. [Google Scholar] [CrossRef]
- Chao, S.-H.; Price, D.H. Flavopiridol Inactivates P-TEFb and Blocks Most RNA Polymerase II Transcription in Vivo. J. Biol. Chem. 2001, 276, 31793–31799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, J.; Quigley, D.A.; Robinson, T.M.; Feng, F.Y.; Ashworth, A. Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer Therapy. Cancer Discov. 2020, 10, 351–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a Novel and Potent Cyclin-Dependent Kinase Inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [Green Version]
- Paruch, K.; Dwyer, M.P.; Alvarez, C.; Brown, C.; Chan, T.-Y.; Doll, R.J.; Keertikar, K.; Knutson, C.; McKittrick, B.; Rivera, J.; et al. Discovery of Dinaciclib (SCH 727965): A Potent and Selective Inhibitor of Cyclin-Dependent Kinases. ACS Med. Chem. Lett. 2010, 1, 204–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlaam, B.; Casella, R.; Cidado, J.; Cook, C.; De Savi, C.; Dishington, A.; Donald, C.S.; Drew, L.; Ferguson, A.D.; Ferguson, D.; et al. Discovery of AZD4573, a Potent and Selective Inhibitor of CDK9 That Enables Short Duration of Target Engagement for the Treatment of Hematological Malignancies. J. Med. Chem. 2020, 63, 15564–15590. [Google Scholar] [CrossRef] [PubMed]
- Cidado, J.; Boiko, S.; Proia, T.; Ferguson, D.; Criscione, S.W.; Martin, M.S.; Pop-Damkov, P.; Su, N.; Franklin, V.N.R.; Chilamakuri, C.S.R.; et al. AZD4573 Is a Highly Selective CDK9 Inhibitor That Suppresses MCL-1 and Induces Apoptosis in Hematologic Cancer Cells. Clin. Cancer Res. 2019, 26, 922–934. [Google Scholar] [CrossRef] [Green Version]
- Alcon, C.; Manzano-Muñoz, A.; Montero, J. A New CDK9 Inhibitor on the Block to Treat Hematologic Malignancies. Clin. Cancer Res. 2019, 26, 761–763. [Google Scholar] [CrossRef]
- Rule, S.; Kater, A.P.; Brümmendorf, T.H.; Fegan, C.; Kaiser, M.; Radford, J.A.; Stilgenbauer, S.; Kayser, S.; Dyer, M.J.; Brossart, P.; et al. A phase 1, open-label, multicenter, non-randomized study to assess the safety, tolerability, pharmacokinetics, and preliminary antitumor activity of AZD4573, a potent and selective CDK9 inhibitor, in subjects with relapsed or refractory hematological malignancies. J. Clin. Oncol. 2018, 36, TPS7588. [Google Scholar] [CrossRef]
- Lücking, U.; Scholz, A.; Lienau, P.; Siemeister, G.; Kosemund, D.; Bohlmann, R.; Briem, H.; Terebesi, I.; Meyer, K.; Prelle, K.; et al. Identification of Atuveciclib (BAY 1143572), the First Highly Selective, Clinical PTEFb/CDK9 Inhibitor for the Treatment of Cancer. ChemMedChem 2017, 12, 1776–1793. [Google Scholar] [CrossRef]
- NCT02345382 B. An open-label Phase I dose-escalation study to characterize the safety, tolerability, pharmacokinetics, and maximum tolerated dose of BAY 1143572 given in a once-daily or an intermittent dosing schedule in subjects with advanced acute leukemia. Available online: https://clinicaltrials.bayer.com/study/?id=16520&Keyword=Atuveciclib&gender=&ageRange=&Status=&Latitude=&Longitude=&MileRadius=&page=0&SortField=Location_Distance&SortOrder=asc&conditions=&phases=&healthyVol=&studyType=&studyResult=&locationCountryInternal=&LocationName= (accessed on 14 May 2018).
- NCT01938638 B. An open-label Phase I dose-escalation study to characterize the safety tolerability, pharmacokinetics, and maximum tolerated dose of BAY1143572 given in a once-daily or an intermittent dosing schedule in subjects with advanced malignancies. Available online: https://clinicaltrials.bayer.com/study/?id=16519&Keyword=Atuveciclib&gender=&ageRange=&Status=&Latitude=&Longitude=&MileRadius=&page=0&SortField=Location_Distance&SortOrder=asc&conditions=&phases=&healthyVol=&studyType=&studyResult=&locationCountryInternal=&LocationName=. (accessed on 12 February 2021).
- Luecking, U.T.; Scholz, A.; Kosemund, D.; Bohlmann, R.; Briem, H.; Lienau, P.; Siemeister, G.; Terebesi, I.; Meyer, K.; Prelle, K.; et al. Abstract 984: Identification of potent and highly selective PTEFb inhibitor BAY 1251152 for the treatment of cancer: From p.o. to i.v. application via scaffold hops. Cancer Chem. 2017, 77, 984. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.R.; Moreno, V.; Lim, E.A.; Cordoba, R.; Cai, C.; Ince, S.J.; Lettieri, J.T.; Merz, C.; Valencia, R.; Boni, V. Phase I dose escalation study of the first-in-class selective PTEFb inhibitor BAY 1251152 in patients with advanced cancer: Novel target validation and early evidence of clinical activity. J. Clin. Oncol. 2018, 36 (Suppl. 15), 2507. [Google Scholar] [CrossRef]
- Byrne, M.; Frattini, M.G.; Ottmann, O.G.; Mantzaris, I.; Wermke, M.; Lee, D.J.; Morillo, D.; Scholz, A.; Ince, S.; Valencia, R.; et al. Phase I Study of the PTEFb Inhibitor BAY 1251152 in Patients with Acute Myelogenous Leukemia. Blood 2018, 132, 4055. [Google Scholar] [CrossRef]
- Vincera Pharma. VIP152, a highly selective PTEFb /CDK9 inhibitor with encouraging Phase 1 monotherapy activity, including complete responses in DH-DLBCL. Available online: https://apnews.com/press-release/globe-newswire/virus-outbreak-technology-business-licensing-agreements-corporate-news-bff559f275deea5fbedac769a500a11f (accessed on 8 February 2021).
- Conroy, A.; Stockett, D.E.; Walker, D.; Arkin, M.R.; Hoch, U.; Fox, J.A.; Hawtin, R.E. SNS-032 is a potent and selective CDK 2, 7 and 9 inhibitor that drives target modulation in patient samples. Cancer Chemother. Pharmacol. 2009, 64, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Albert, T.K.; Rigault, C.; Eickhoff, J.; Baumgart, K.; Antrecht, C.; Klebl, B.; Mittler, G.; Meisterernst, M. Characterization of molecular and cellular functions of the cyclin-dependent kinase CDK9 using a novel specific inhibitor. Br. J. Pharmacol. 2014, 171, 55–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, E.I.; Bible, K.; Martell, R.E.; Adelman, D.C.; Lorusso, P.M. A phase 1 study of SNS-032 (formerly BMS-387032), a potent inhibitor of cyclin-dependent kinases 2, 7 and 9 administered as a single oral dose and weekly infusion in patients with metastatic refractory solid tumors. Investig. New Drugs 2007, 26, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.-G.; Chen, R.; Plunkett, W.; Siegel, D.; Sinha, R.; Harvey, R.D.; Badros, A.Z.; Popplewell, L.; Coutre, S.; Fox, J.A.; et al. Phase I and Pharmacologic Study of SNS-032, a Potent and Selective Cdk2, 7, and 9 Inhibitor, in Patients With Advanced Chronic Lymphocytic Leukemia and Multiple Myeloma. J. Clin. Oncol. 2010, 28, 3015–3022. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar] [CrossRef]
- Wyatt, P.G.; Woodhead, A.J.; Berdini, V.; Boulstridge, J.A.; Carr, M.G.; Cross, D.M.; Davis, D.J.; Devine, L.A.; Early, T.R.; Feltell, R.E.; et al. Identification of N-(4-Piperidinyl)-4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxamide (AT7519), a Novel Cyclin Dependent Kinase Inhibitor Using Fragment-Based X-Ray Crystallography and Structure Based Drug Design. J. Med. Chem. 2008, 51, 4986–4999. [Google Scholar] [CrossRef]
- Squires, M.S.; Feltell, R.E.; Wallis, N.G.; Lewis, E.J.; Smith, D.-M.; Cross, D.M.; Lyons, J.F.; Thompson, N.T. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol. Cancer Ther. 2009, 8, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Squires, M.S.; Cooke, L.; Lock, V.; Qi, W.; Lewis, E.J.; Thompson, N.T.; Lyons, J.F.; Mahadevan, D. AT7519, a Cyclin-Dependent Kinase Inhibitor, Exerts Its Effects by Transcriptional Inhibition in Leukemia Cell Lines and Patient Samples. Mol. Cancer Ther. 2010, 9, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, D.; Plummer, R.; Squires, M.S.; Rensvold, D.; Dragovich, T.; Adams, J.; Lock, V.; Smith, D.; Von Hoff, D.D.; Calvert, H. A dose escalation, pharmacokinetic, and pharmacodynamic study of AT7519, a cyclin-dependent kinase inhibitor in patients with refractory solid tumors. J. Clin. Oncol. 2008, 26, 3533. [Google Scholar] [CrossRef]
- Chen, E.X.; Hotte, S.; Hirte, H.; Siu, L.L.; Lyons, J.; Squires, M.; Lovell, S.; Turner, S.; McIntosh, L.; Seymour, L. A Phase I study of cyclin-dependent kinase inhibitor, AT7519, in patients with advanced cancer: NCIC Clinical Trials Group IND 177. Br. J. Cancer 2014, 111, 2262–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seftel, M.D.; Kuruvilla, J.; Kouroukis, T.; Banerji, V.; Fraser, G.; Crump, M.; Kumar, R.; Chalchal, H.I.; Salim, M.; Laister, R.C.; et al. The CDK inhibitor AT7519M in patients with relapsed or refractory chronic lymphocytic leukemia (CLL) and mantle cell lymphoma. A Phase II study of the Canadian Cancer Trials Group. Leuk. Lymphoma 2017, 58, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Do, K.T.; Chen, A.P.; Meehan, R.S.; Coyne, G.H.O.; Supko, J.G.; Trepel, J.B.; Clark, J.W.; Hays, J.L.; Lyons, J.F.; Keer, H.N.; et al. Phase 1 study of onalespib, HSP90 inhibitor, and AT7519M, CDK9 inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2017, 35, TPS2617. [Google Scholar] [CrossRef]
- Wang, B.; Wu, J.; Wu, Y.; Chen, C.; Zou, F.; Wang, A.; Wu, H.; Hu, Z.; Jiang, Z.; Liu, Q.; et al. Discovery of 4-(((4-(5-chloro-2-(((1s,4s)-4-((2-methoxyethyl)amino)cyclohexyl)amino)pyridin-4-yl)thiazol-2-yl)amino)methyl)tetrahydro-2H-pyran-4-carbonitrile (JSH-150) as a novel highly selective and potent CDK9 kinase inhibitor. Eur. J. Med. Chem. 2018, 158, 896–916. [Google Scholar] [CrossRef]
- Yin, T.; Lallena, M.J.; Kreklau, E.L.; Fales, K.R.; Carballares, S.; Torrres, R.; Wishart, G.N.; Ajamie, R.T.; Cronier, D.M.; Iversen, P.W.; et al. A Novel CDK9 Inhibitor Shows Potent Antitumor Efficacy in Preclinical Hematologic Tumor Models. Mol. Cancer Ther. 2014, 13, 1442–1456. [Google Scholar] [CrossRef] [Green Version]
- Fischer Peter, M.; Wang, S.; Zaytsev, A. Pyrimidines, triazines and their use as pharmaceutical agents. WO2009118567A2, 11 March 2010. [Google Scholar]
- Walsby, E.; Pratt, G.; Shao, H.; Abbas, A.Y.; Fischer, P.M.; Bradshaw, T.D.; Brennan, P.; Fegan, C.; Wang, S.; Pepper, C. A novel Cdk9 inhibitor preferentially targets tumor cells and synergizes with fludarabine. Oncotarget 2013, 5, 375–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.; Jiang, H.; Zhai, X.-W.; Wei, F.; Wang, S.-D.; Ding, J.; Chen, Y. Antitumor action of CDK inhibitor LS-007 as a single agent and in combination with ABT-199 against human acute leukemia cells. Acta Pharmacol. Sin. 2016, 37, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Sorvina, A.; Shandala, T.; Wang, S.; Sharkey, D.J.; Parkinson-Lawrence, E.; Selemidis, S.; Brooks, D.A. CDKI-73 Is a Novel Pharmacological Inhibitor of Rab11 Cargo Delivery and Innate Immune Secretion. Cells 2020, 9, 372. [Google Scholar] [CrossRef] [Green Version]
- Bettayeb, K.; Oumata, N.; Echalier, A.; Ferandin, Y.; A Endicott, J.; Galons, H.; Meijer, L. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 2008, 27, 5797–5807. [Google Scholar] [CrossRef] [Green Version]
- Delehouze, C.; Godl, K.; Loaec, N.; Bruyere, C.; Desban, N.; Oumata, N.; Galons, H.; Roumeliotis, T.I.; Giannopoulou, E.G.; Grenet, J.; et al. CDK/CK1 inhibitors roscovitine and CR8 downregulate amplified MYCN in neuroblastoma cells. Oncogene 2013, 33, 5675–5687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Falco, G.; Bellan, C.; D’Amuri, A.; Angeloni, G.; Leucci, E.; Giordano, A.; Leoncini, L. Cdk9 regulates neural differentiation and its expression correlates with the differentiation grade of neuroblastoma and PNET tumors. Cancer Biol. Ther. 2005, 4, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Xu, S.; Fang, Y.; Chen, T.; Xie, X.; Lu, W. Cyclin-dependent kinase 9 promotes cervical cancer development via AKT2/p53 pathway. IUBMB Life 2018, 71, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Gothert, J.R.; Imsak, R.; Mollmann, M.; Kesper, S.; Gobel, M.; Duhrsen, U.; Scholz, A.; Lucking, U.; Baumann, M.; Unger, A.; et al. Potent anti-leukemic activity of a specific cyclin-dependent kinase 9 inhibitor in mouse models of chronic lymphocytic leukemia. Oncotarget 2018, 9, 26353–26369. [Google Scholar] [CrossRef] [Green Version]
- Juric, V.; Murphy, B. Cyclin-dependent kinase inhibitors in brain cancer: Current state and future directions. Cancer Drug Resist. 2020, 3, 48–62. [Google Scholar] [CrossRef] [Green Version]
- Hellvard, A.; Zeitlmann, L.; Heiser, U.; Kehlen, A.; Niestroj, A.; DeMuth, H.-U.; Koziel, J.; Delaleu, N.; Potempa, J.; Mydel, P. Inhibition of CDK9 as a therapeutic strategy for inflammatory arthritis. Sci. Rep. 2016, 6, 31441. [Google Scholar] [CrossRef]
- Matrone, G.; Wilson, K.S.; Maqsood, S.; Mullins, J.J.; Tucker, C.S.; Denvir, M.A. CDK9 and its repressor LARP7 modulate cardiomyocyte proliferation and response to injury in the zebrafish heart. J. Cell Sci. 2015, 128, 4560–4571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.W.; Tay, N.Q.; Brzostek, J.; Gascoigne, N.R.J.; Rybakin, V. A Dual Inhibitor of Cdc7/Cdk9 Potently Suppresses T Cell Activation. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Zaborowska, J.; Isa, N.F.; Murphy, S. P-TEFb goes viral. Inside Cell 2016, 1, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.; Becker, S.; Strebhardt, K. Stamping out RAF and MEK1/2 to inhibit the ERK1/2 pathway: An emerging threat to anticancer therapy. Oncogene 2016, 35, 2547–2561. [Google Scholar] [CrossRef]
- Mandal, R.; Raab, M.; Matthess, Y.; Becker, S.; Knecht, R.; Strebhardt, K. pERK 1/2 inhibit Caspase-8 induced apoptosis in cancer cells by phosphorylating it in a cell cycle specific manner. Mol. Oncol. 2014, 8, 232–249. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.-C.; Guan, Y.; Qin, W.; Zhai, X.; Yu, B.; Liu, H.-M. Targeting Brd4 for cancer therapy: Inhibitors and degraders. MedChemComm 2018, 9, 1779–1802. [Google Scholar] [CrossRef] [PubMed]
- McCalmont, H.; Li, K.L.; Jones, L.; Toubia, J.; Bray, S.C.; Casolari, D.A.; Mayoh, C.; Samaraweera, S.E.; Lewis, I.D.; Prinjha, R.K.; et al. Efficacy of combined CDK9/BET inhibition in preclinical models of MLL-rearranged acute leukemia. Blood Adv. 2020, 4, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, D.; Tontsch-Grunt, U.; Baum, A.; Popow, J.; Scharn, D.; Hofmann, M.H.; Engelhardt, H.; Kaya, O.; Beck, J.; Schweifer, N.; et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene 2018, 37, 2687–2701. [Google Scholar] [CrossRef] [PubMed]
- Moreno, N.; Holsten, T.; Mertins, J.; Zhogbi, A.; Johann, P.; Kool, M.; Meisterernst, M.; Kerl, K. Combined BRD4 and CDK9 inhibition as a new therapeutic approach in malignant rhabdoid tumors. Oncotarget 2017, 8, 84986–84995. [Google Scholar] [CrossRef] [Green Version]
- Piha-Paul, S.A.; Sachdev, J.C.; Barve, M.; Lorusso, P.; Szmulewitz, R.; Patel, S.P.; Lara, P.N., Jr.; Chen, X.; Hu, B.; Freise, K.J.; et al. First-in-Human Study of Mivebresib (ABBV-075), an Oral Pan-Inhibitor of Bromodomain and Extra Terminal Proteins, in Patients with Relapsed/Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 6309–6319. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Carvajal, R.D.; Komatsubara, K.M.; Britten, C.D.; Wesolowski, R.; Michelson, G.; Alcantar, O.; Zhang, C.; Powell, B.; Severson, P.; et al. Phase ib/2a study of PLX51107, a small molecule BET inhibitor, in subjects with advanced hematological malignancies and solid tumors. J. Clin. Oncol. 2018, 36, 2550. [Google Scholar] [CrossRef]
- Mita, M.M.; Mita, A.C. Bromodomain inhibitors a decade later: A promise unfulfilled? Br. J. Cancer 2020, 123, 1713–1714. [Google Scholar] [CrossRef]
- BRD4 inhibitors. Available online: https://clinicaltrials.gov/ct2/results?recrs=&cond=Cancer&term=BRD4&cntry=&state=&city=&dist= (accessed on 12 February 2021).
- Alfonso-Dunn, R.; Arbuckle, J.H.; Vogel, J.L.; Kristie, T.M. Inhibition of the Super Elongation Complex Suppresses Herpes Simplex Virus Immediate Early Gene Expression, Lytic Infection, and Reactivation from Latency. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Smith, E.R.; Aoi, Y.; Stoltz, K.L.; Katagi, H.; Woodfin, A.R.; Rendleman, E.J.; Marshall, S.A.; Murray, D.C.; Wang, L.; et al. Targeting Processive Transcription Elongation via SEC Disruption for MYC-Induced Cancer Therapy. Cell 2018, 175, 766–779.e17. [Google Scholar] [CrossRef] [Green Version]
- HSP90 inhibitors. Available online: https://clinicaltrials.gov/ct2/results?recrs=&cond=Cancer&term=HSP90+inhibitors&cntry=&state=&city=&dist= (accessed on 12 February 2021).
- Clinicaltrial. Available online: https://clinicaltrials.gov/ct2/results?recrs=&cond=Cancer&term=MCL-1&cntry=&state=&city=&dist= (accessed on 12 February 2021).
- Bolomsky, A.; Vogler, M.; Köse, M.C.; Heckman, C.A.; Ehx, G.; Ludwig, H.; Caers, J. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J. Hematol. Oncol. 2020, 13, 1–19. [Google Scholar] [CrossRef]
- XIAP inhibitors. Available online: https://clinicaltrials.gov/ct2/results?recrs=&cond=Cancer&term=XIAP+inhibitors&cntry=&state=&city=&dist= (accessed on 12 February 2021).
- BCL-2 Inhibitors. Available online: https://clinicaltrials.gov/ct2/results?recrs=&cond=Cancer&term=BCL2+inhibitors&cntry=&state=&city=&dist= (accessed on 12 February 2021).
- Lu, H.; Xue, Y.; Yu, G.K.; Arias, C.; Lin, J.; Fong, S.; Faure, M.; Weisburd, B.; Ji, X.; Mercier, A.; et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. Elife 2015, 4, 06535. [Google Scholar]
- Madden, S.K.; De Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Jovanović, K.K.; Roche-Lestienne, C.; Ghobrial, I.M.; Facon, T.; Quesnel, B.; Manier, S. Targeting MYC in multiple myeloma. Leukemia 2018, 32, 1295–1306. [Google Scholar] [CrossRef]
- Ohanian, M.; Rozovski, U.; Kanagal-Shamanna, R.; Abruzzo, L.V.; Loghavi, S.; Kadia, T.; Futreal, A.; Bhalla, K.; Zuo, Z.; Huh, Y.O.; et al. MYC protein expression is an important prognostic factor in acute myeloid leukemia. Leuk. Lymphoma 2019, 60, 37–48. [Google Scholar] [CrossRef]
- Polier, G.; Ding, J.; Konkimalla, B.; Eick, D.; Ribeiro, N.; Köhler, R.; Giaisi, M.; Efferth, T.; Desaubry, L.; Krammer, P.H.; et al. Wogonin and related natural flavones are inhibitors of CDK9 that induce apoptosis in cancer cells by transcriptional suppression of Mcl-1. Cell Death Dis. 2011, 2, e182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polier, G.; Giaisi, M.; Köhler, R.; Müller, W.W.; Lutz, C.; Buss, E.C.; Krammer, P.H.; Li-Weber, M. Targeting CDK9 by wogonin and related natural flavones potentiates the anti-cancer efficacy of the Bcl-2 family inhibitor ABT-263. Int. J. Cancer 2014, 136, 688–698. [Google Scholar] [CrossRef] [PubMed]




| Cyclin/Regulating Partners | Functions | Reference | |
|---|---|---|---|
| CDK1 | Cyclin B1 | Cell-cycle regulation—promotes G2/M transition, regulates G1 progress and G1/S transition | [4,5] |
| CDK2 | Cyclins A/D1/E1 | Cell-cycle regulation—G1/S transition, exit from S-phase; initiation of DNA synthesis | [6] |
| CDK4 | Cyclin D | Cell-cycle regulation—G1-phase transition; partial phosphorylation of Rb with CDK6 | [7] |
| CDK5 | p35/p39 | All aspects of neuronal physiology; immune response; angiogenesis; myogenesis; melanogenesis and regulation of insulin levels | [6,8,9] |
| CDK6 | Cyclin D | Cell-cycle regulation—G1-phase transition; partial phosphorylation of Rb with CDK4 | [7] |
| CDK7 | Cyclin H | CDK Activating Kinase (CAK)—phosphorylates cell-cycle regulating kinases; transcription regulation—S5 phosphorylation on RNAP II CTD to initiate transcription initiation | [10] |
| CDK8 | Cyclin C/Med12/Med13 | Part of Mediator Complex (MC), regulates the phosphorylation transcription factors, their activity and turn-over | [11] |
| CDK9 | Cyclins T1/T2a/T2b | Positive regulation of transcription elongation | [12] |
| CDK10 | Cyclin M | Cell-cycle regulation and tumor suppressor | [13,14] |
| CDK12 | Cyclin K | Positive regulation of transcription elongation | [15,16] |
| CDK13 | Cyclin K | Positive regulation of transcription elongation | [15,17] |
| CDK14 | Cyclin Y | Regulation of cell-cycle, proliferation, migration and invasion | [18] |
| CDK15 | Unknown | Inhibits apoptosis by phosphorylating Survivin on T34 | [19] |
| CDK16 | Cyclin Y | Promotes proliferation in medulloblastoma, prostate, breast, melanoma and cervical cancers, inhibits apoptosis by down-regulating the tumor suppressor p27 in NSCLC | [20,21,22] |
| CDK17 | Unknown | Down-regulation causes poor prognosis in glioma. Unknown functions | [23] |
| CDK18 | Cyclins A2 and E | Negative regulator of cell migration and adhesion, prevents the accumulation of DNA damage and genome instability | [24,25,26] |
| CDK19 | Cyclin C | CDK8 homolog, part of Mediator Complex (MC), promotes proliferation and mitotic gene expression in the absence of CDK8 expression, negative regulation of NOTCH signaling | [11] |
| CDK20 | Cyclin H and CK2 (generic CDK20); KCNIP2 and SNAPIN (cardiac CDK20) | Cell-cycle regulator (generic CDK20) and promotes cell survival (cardiac CDK20) | [27] |
| CDK21 | Unknown | Regulates spermatogonial proliferation and meiosis progression and germ line cell activation in testis; unknown function in cancer | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandal, R.; Becker, S.; Strebhardt, K. Targeting CDK9 for Anti-Cancer Therapeutics. Cancers 2021, 13, 2181. https://doi.org/10.3390/cancers13092181
Mandal R, Becker S, Strebhardt K. Targeting CDK9 for Anti-Cancer Therapeutics. Cancers. 2021; 13(9):2181. https://doi.org/10.3390/cancers13092181
Chicago/Turabian StyleMandal, Ranadip, Sven Becker, and Klaus Strebhardt. 2021. "Targeting CDK9 for Anti-Cancer Therapeutics" Cancers 13, no. 9: 2181. https://doi.org/10.3390/cancers13092181
APA StyleMandal, R., Becker, S., & Strebhardt, K. (2021). Targeting CDK9 for Anti-Cancer Therapeutics. Cancers, 13(9), 2181. https://doi.org/10.3390/cancers13092181