
Prediction of Adrenocortical Carcinoma Relapse and Prognosis with a Set of Novel Multigene Panels


 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. cBioPortal Database
2.2. Model Size and Variable Optimization
2.3. Assignment of Signature Scores to Individual ACCs
2.4. Cutoff Point Estimation
2.5. Time-Dependent Receiver Operating Characteristic (tROC), ROC, and PR
2.6. Examination of Gene Expression
2.7. Correlation Analyses and Heatmap
2.8. Determination of Immune Cell Infiltration
2.9. Statistical Analysis
3. Results
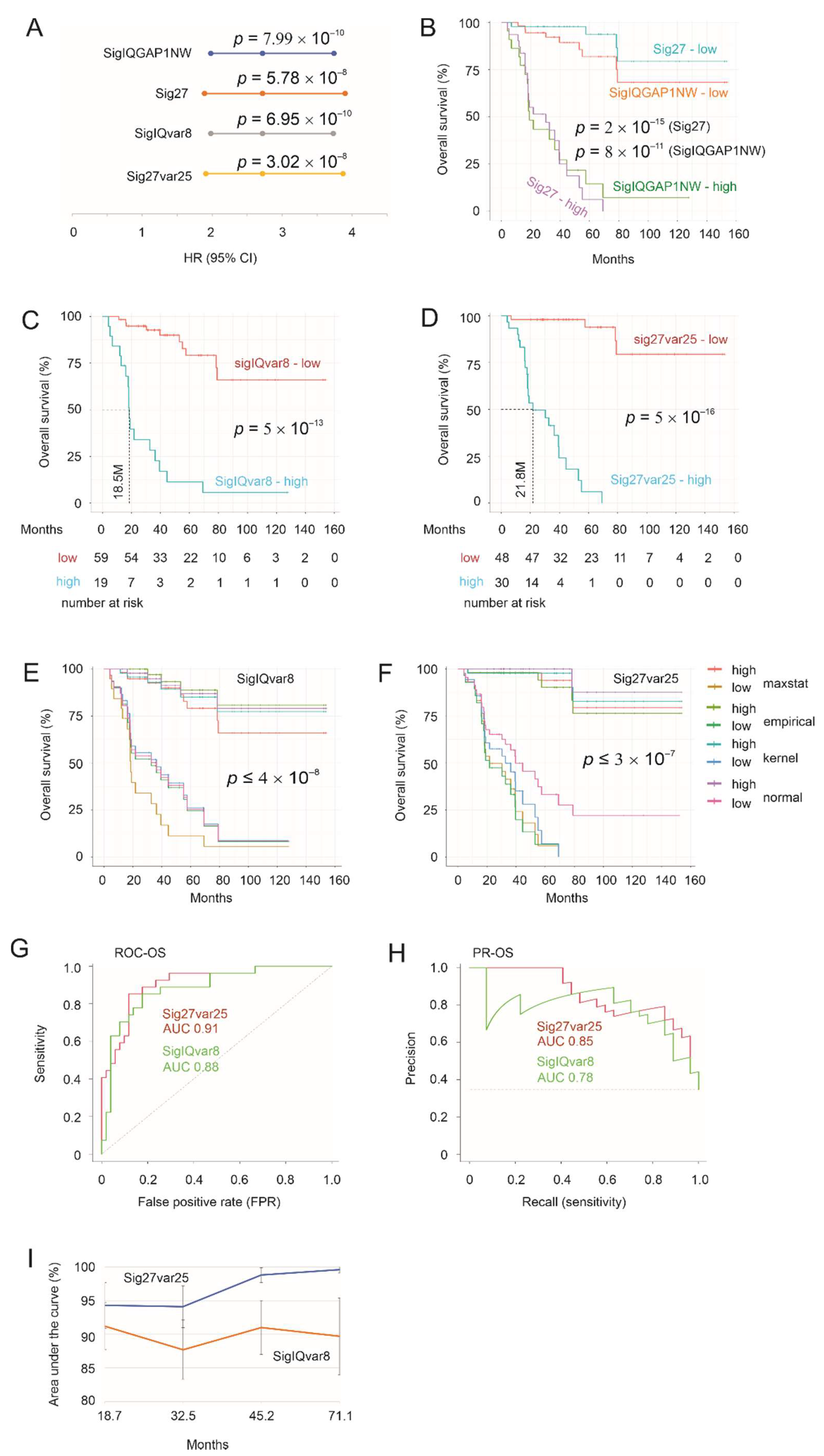
3.1. SigIQGAP1NW and Sig27gene (Sig27) as Potent Prognostic Biomarkers of ACC and Derivation of SigIQvar8 and Sig27var25
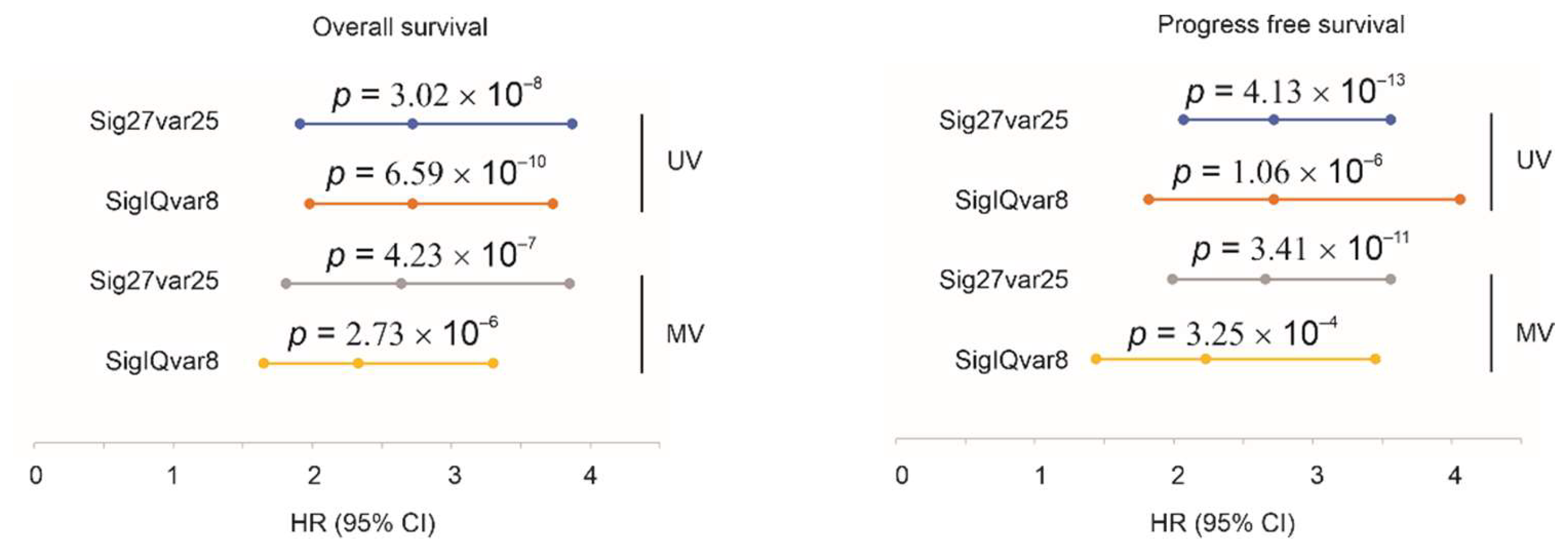
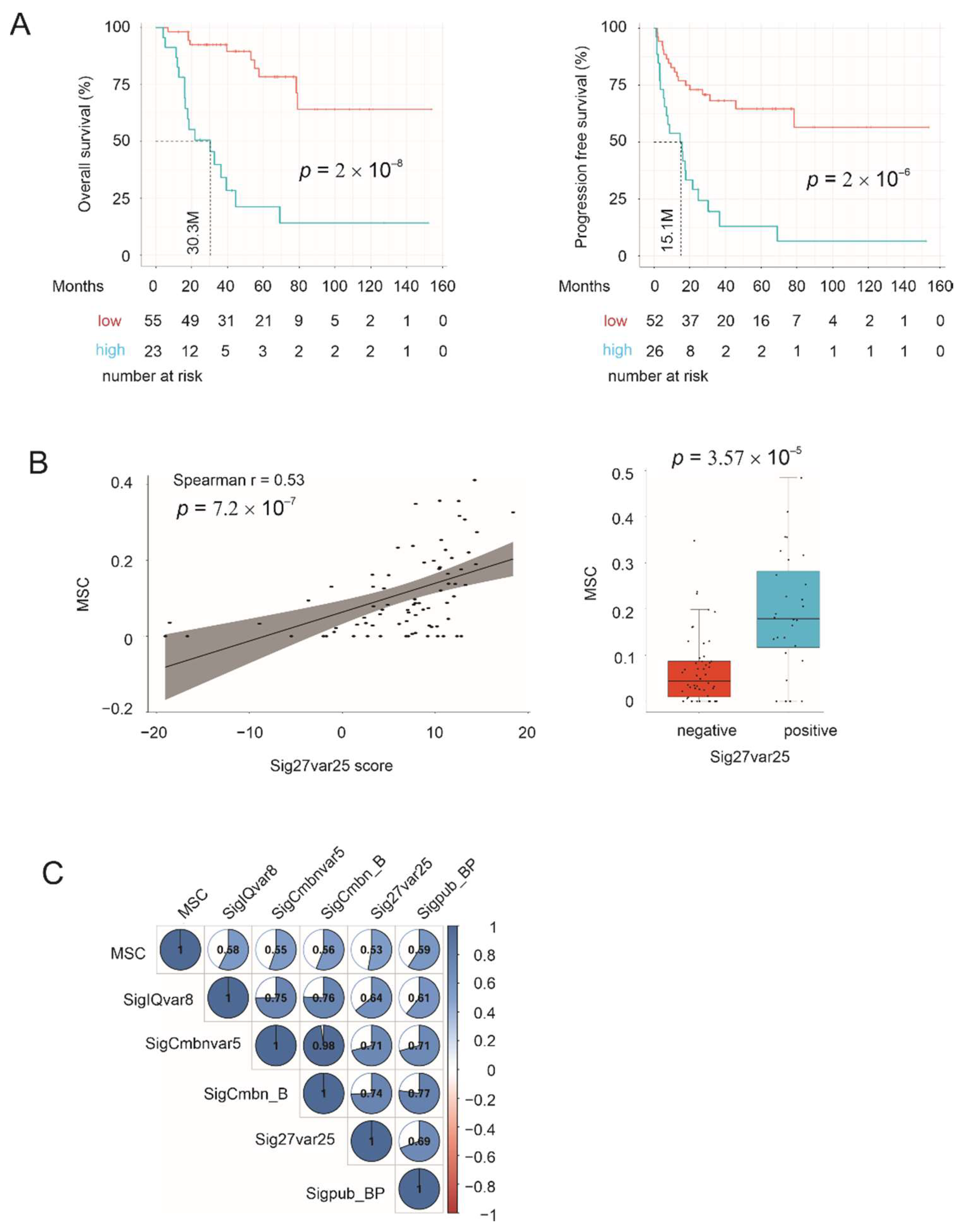
3.2. Sig27var25- and SigIQvar8-Derived Prognostic Potential for PFS
3.3. Characterization of Sig27var25 and SigIQvar8
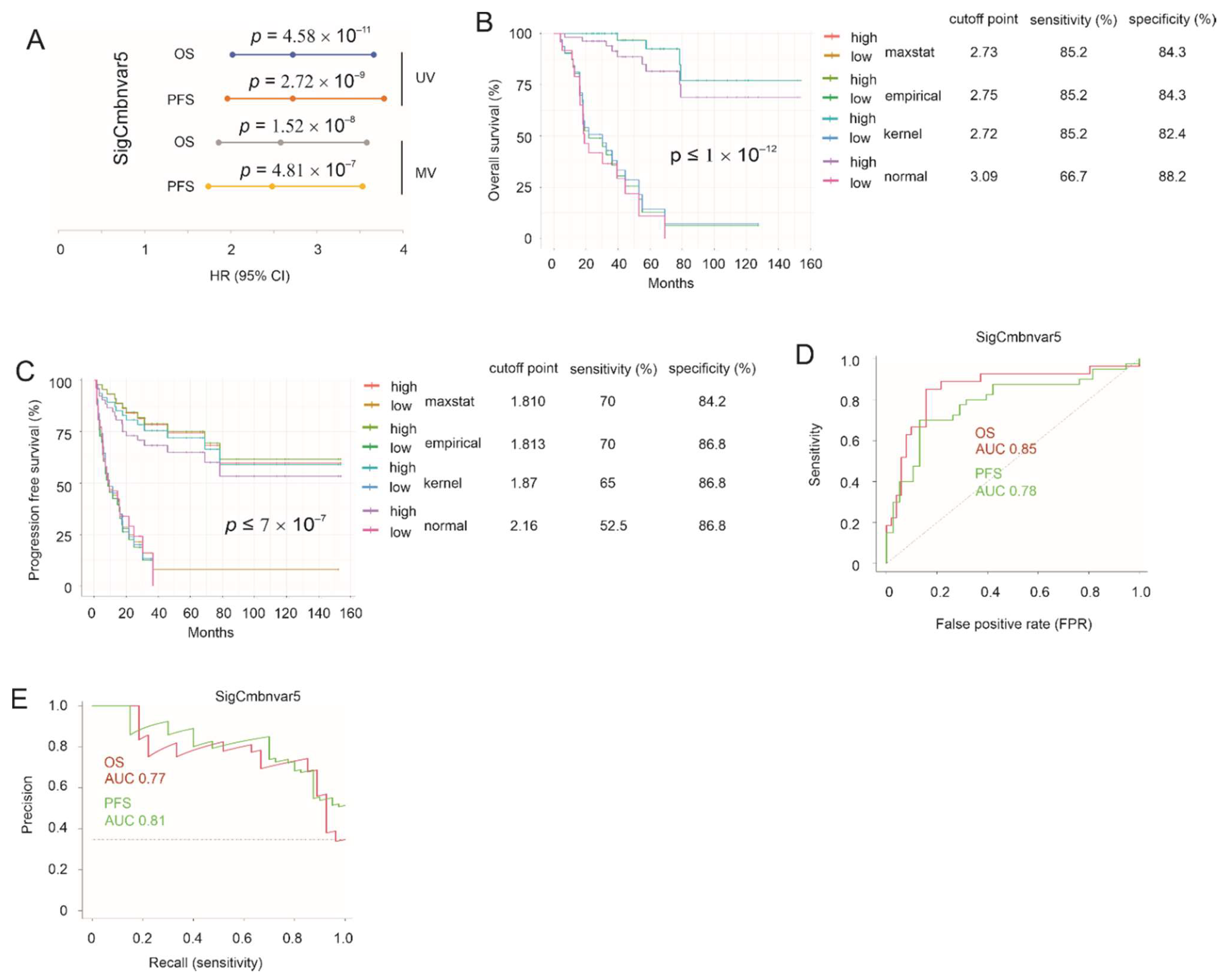
3.4. Construction of SigCmbnvar5
3.5. Superiority of Our Prognostic Multigene Signatures to Those Previously Reported
3.6. SigIQvar8, Sig27var25, SigCmbnvar5, and SigCmbn_B Being Novel to ACC
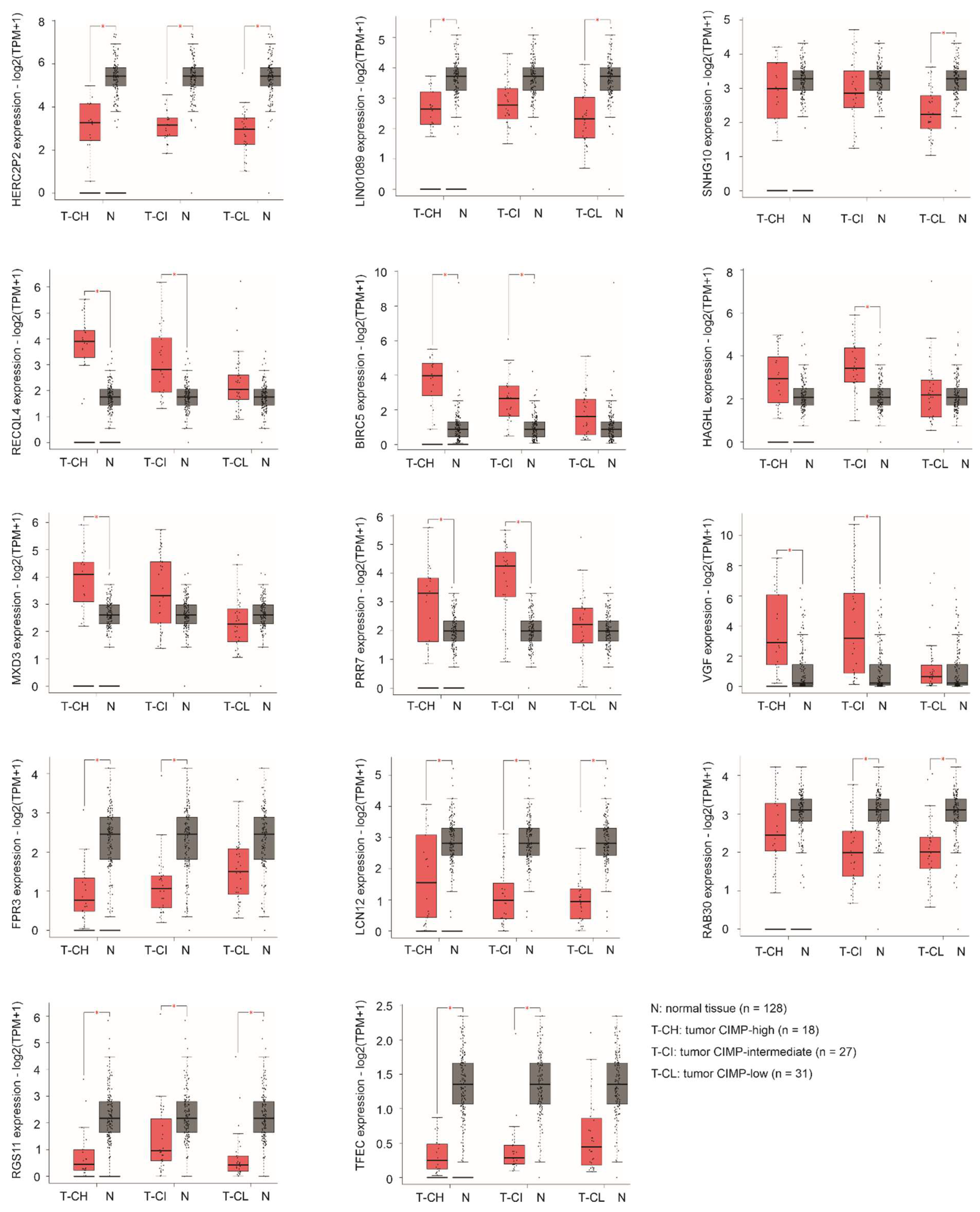
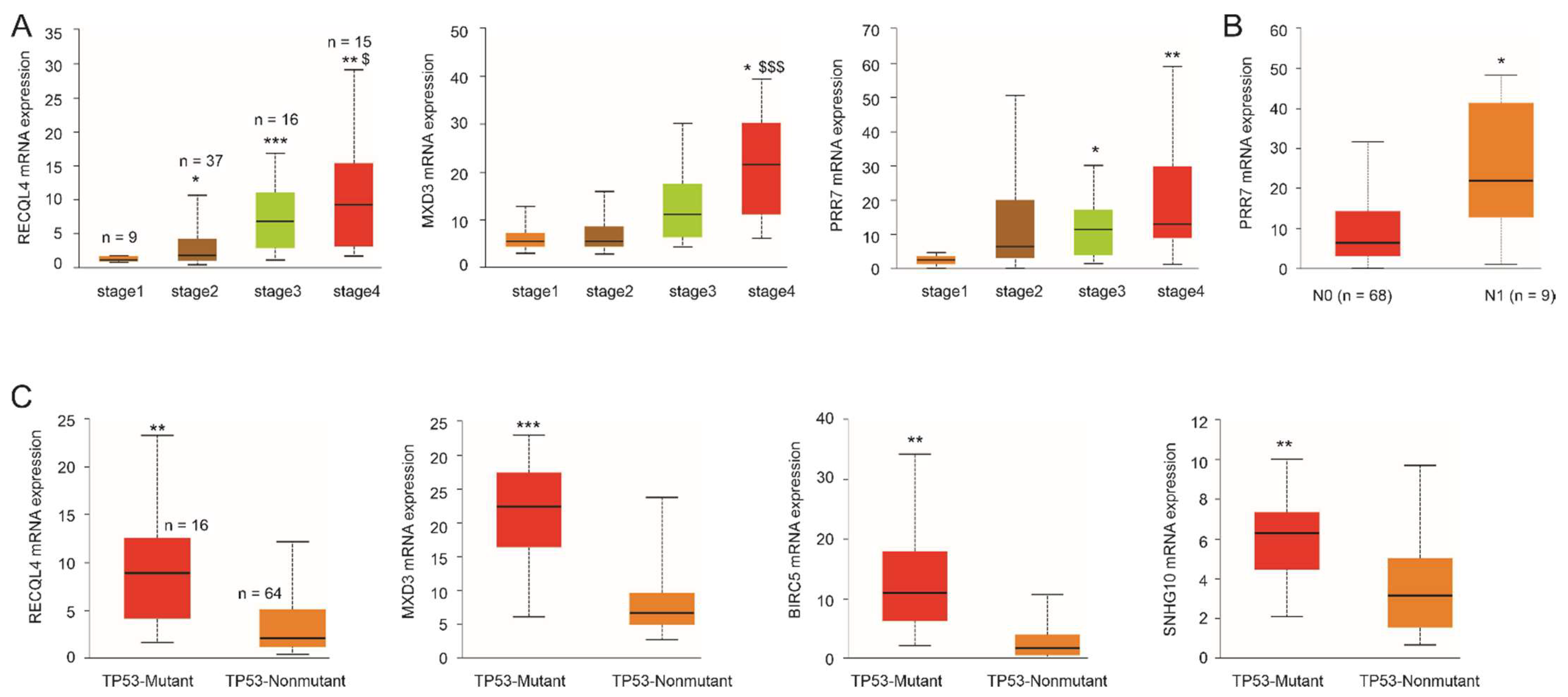
3.7. Differential Expression of Specific Component Genes during ACC Pathogenesis
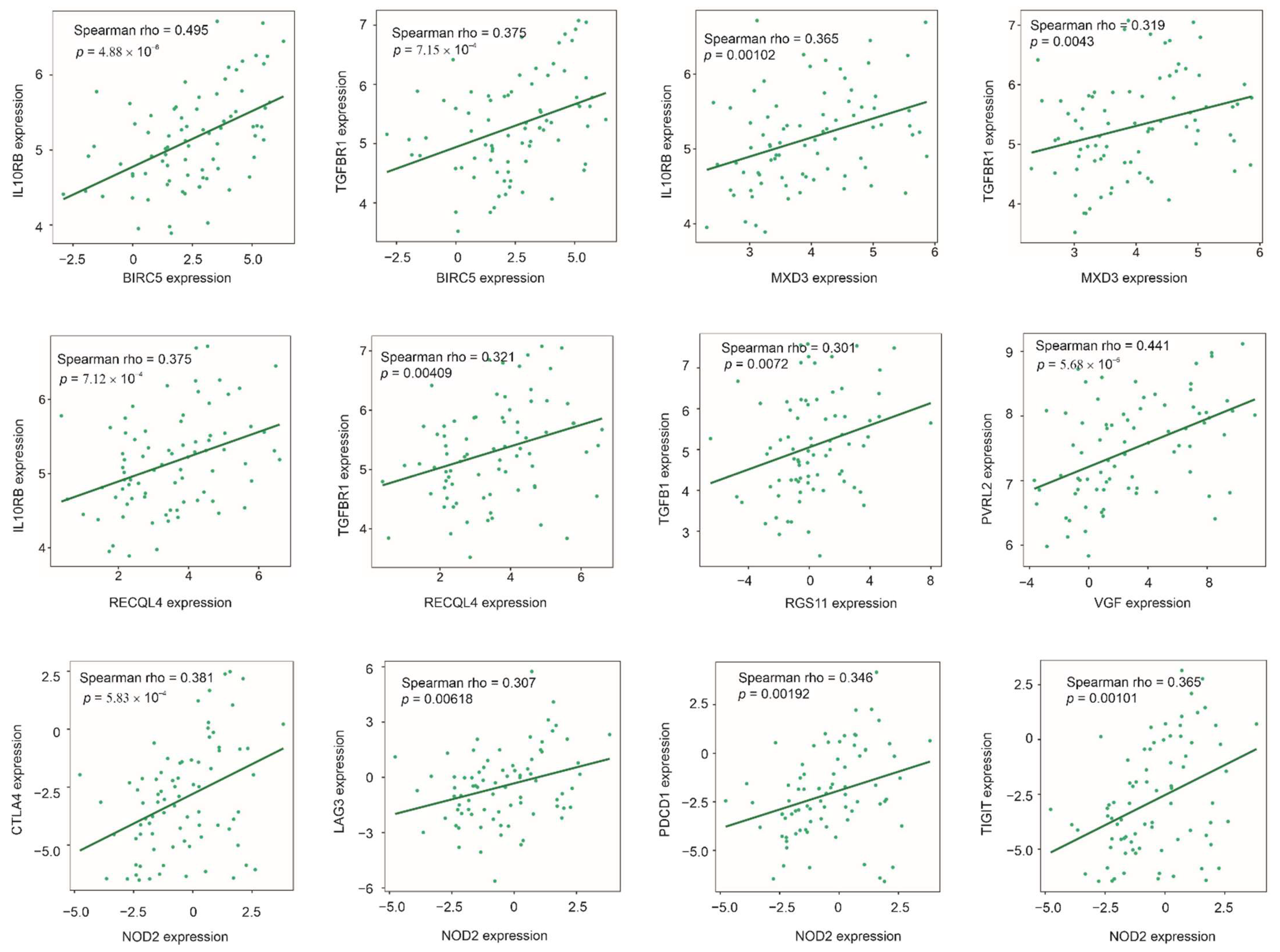
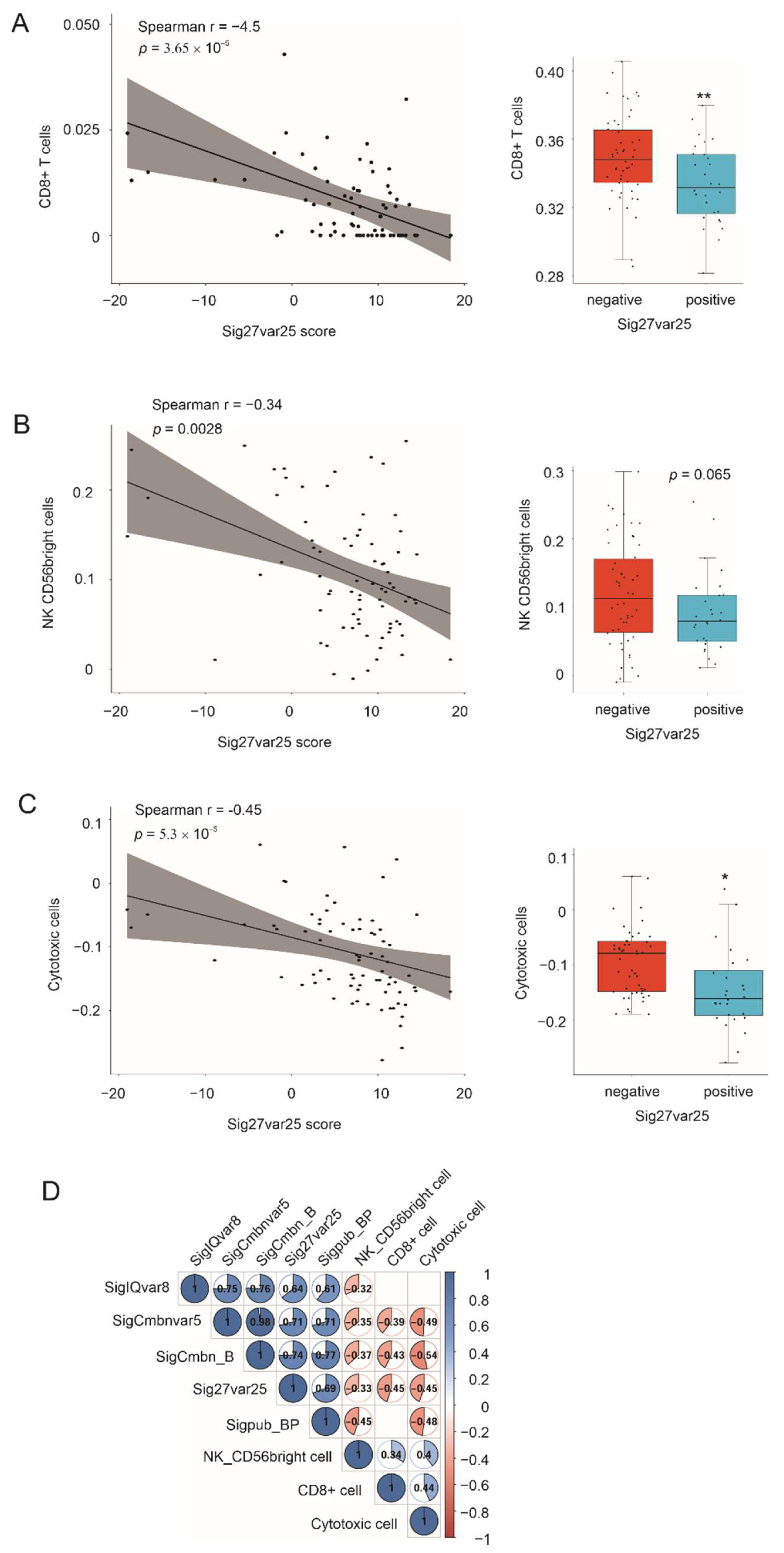
3.8. Association of Multigene Panels with Immunosuppressive ACC Microenvironment
3.9. Correlation of Multigene Signatures with Mesenchymal Stem Cells (MSCs)
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kerkhofs, T.M.; Verhoeven, R.H.; Van der Zwan, J.M.; Dieleman, J.; Kerstens, M.N.; Links, T.P.; Van de Poll-Franse, L.V.; Haak, H.R. Adrenocortical carcinoma: A population-based study on incidence and survival in the netherlands since 1993. Eur. J. Cancer 2013, 49, 2579–2586. [Google Scholar] [CrossRef] [PubMed]
- Kebebew, E.; Reiff, E.; Duh, Q.Y.; Clark, O.H.; McMillan, A. Extent of disease at presentation and outcome for adrenocortical carcinoma: Have we made progress? World J. Surg. 2006, 30, 872–878. [Google Scholar] [CrossRef] [PubMed]
- McAteer, J.P.; Huaco, J.A.; Gow, K.W. Predictors of survival in pediatric adrenocortical carcinoma: A surveillance, epidemiology, and end results (seer) program study. J. Pediatr. Surg. 2013, 48, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Sandru, F.; Petca, R.C.; Carsote, M.; Petca, A.; Dumitrascu, M.C.; Ghemigian, A. Adrenocortical carcinoma: Pediatric aspects (review). Exp. Ther. Med. 2022, 23, 287. [Google Scholar] [PubMed]
- Sharma, E.; Dahal, S.; Sharma, P.; Bhandari, A.; Gupta, V.; Amgai, B.; Dahal, S. The characteristics and trends in adrenocortical carcinoma: A united states population based study. J. Clin. Med. Res. 2018, 10, 636–640. [Google Scholar] [CrossRef] [Green Version]
- Wooten, M.D.; King, D.K. Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature. Cancer 1993, 72, 3145–3155. [Google Scholar]
- Berruti, A.; Fassnacht, M.; Haak, H.; Else, T.; Baudin, E.; Sperone, P.; Kroiss, M.; Kerkhofs, T.; Williams, A.R.; Ardito, A.; et al. Prognostic role of overt hypercortisolism in completely operated patients with adrenocortical cancer. Eur. Urol. 2014, 65, 832–838. [Google Scholar] [CrossRef]
- Ettaieb, M.; Kerkhofs, T.; van Engeland, M.; Haak, H. Past, present and future of epigenetics in adrenocortical carcinoma. Cancers 2020, 12, 1218. [Google Scholar] [CrossRef]
- Fassnacht, M.; Dekkers, O.M.; Else, T.; Baudin, E.; Berruti, A.; de Krijger, R.; Haak, H.R.; Mihai, R.; Assie, G.; Terzolo, M. European society of endocrinology clinical practice guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the european network for the study of adrenal tumors. Eur. J. Endocrinol. 2018, 179, G1–G46. [Google Scholar] [CrossRef]
- Fassnacht, M.; Johanssen, S.; Quinkler, M.; Bucsky, P.; Willenberg, H.S.; Beuschlein, F.; Terzolo, M.; Mueller, H.H.; Hahner, S.; Allolio, B.; et al. Limited prognostic value of the 2004 international union against cancer staging classification for adrenocortical carcinoma: Proposal for a revised tnm classification. Cancer 2009, 115, 243–250. [Google Scholar] [CrossRef]
- Araujo, A.N.; Bugalho, M.J. Advanced adrenocortical carcinoma: Current perspectives on medical treatment. Horm. Metab. Res. 2021, 53, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.; Nehs, M.; Kilbridge, K. Treatment of adrenocortical carcinoma. Surg. Pathol. Clin. 2019, 12, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.J.; Shapiro, S.; Sarlis, N.; Vassilopoulou-Sellin, R.; Perrier, N.D.; Evans, D.B.; Lee, J.E. Laparoscopic resection of adrenal cortical carcinoma: A cautionary note. Surgery 2005, 138, 1078–1085; discussion 1085–1076. [Google Scholar] [CrossRef] [PubMed]
- Hermsen, I.G.; Gelderblom, H.; Kievit, J.; Romijn, J.A.; Haak, H.R. Extremely long survival in six patients despite recurrent and metastatic adrenal carcinoma. Eur. J. Endocrinol. 2008, 158, 911–919. [Google Scholar] [CrossRef] [Green Version]
- Thampi, A.; Shah, E.; Elshimy, G.; Correa, R. Adrenocortical carcinoma: A literature review. Transl. Cancer Res. 2020, 9, 1253–1264. [Google Scholar] [CrossRef]
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef] [Green Version]
- Weiss, L.M.; Medeiros, L.J.; Vickery, A.L., Jr. Pathologic features of prognostic significance in adrenocortical carcinoma. Am. J. Surg. Pathol. 1989, 13, 202–206. [Google Scholar] [CrossRef]
- Barreau, O.; Assie, G.; Wilmot-Roussel, H.; Ragazzon, B.; Baudry, C.; Perlemoine, K.; Rene-Corail, F.; Bertagna, X.; Dousset, B.; Hamzaoui, N.; et al. Identification of a cpg island methylator phenotype in adrenocortical carcinomas. J. Clin. Endocrinol. Metab. 2013, 98, E174–E184. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef] [Green Version]
- de Reynies, A.; Assie, G.; Rickman, D.S.; Tissier, F.; Groussin, L.; Rene-Corail, F.; Dousset, B.; Bertagna, X.; Clauser, E.; Bertherat, J. Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J. Clin. Oncol. 2009, 27, 1108–1115. [Google Scholar] [CrossRef]
- Mohan, D.R.; Lerario, A.M.; Else, T.; Mukherjee, B.; Almeida, M.Q.; Vinco, M.; Rege, J.; Mariani, B.M.P.; Zerbini, M.C.N.; Mendonca, B.B.; et al. Targeted assessment of g0s2 methylation identifies a rapidly recurrent, routinely fatal molecular subtype of adrenocortical carcinoma. Clin. Cancer Res. 2019, 25, 3276–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragoso, M.C.; Almeida, M.Q.; Mazzuco, T.L.; Mariani, B.M.; Brito, L.P.; Goncalves, T.C.; Alencar, G.A.; Lima Lde, O.; Faria, A.M.; Bourdeau, I.; et al. Combined expression of bub1b, dlgap5, and pink1 as predictors of poor outcome in adrenocortical tumors: Validation in a brazilian cohort of adult and pediatric patients. Eur. J. Endocrinol. 2012, 166, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Kapoor, A.; Gu, Y.; Chow, M.J.; Peng, J.; Major, P.; Tang, D. Construction of a novel multigene panel potently predicting poor prognosis in patients with clear cell renal cell carcinoma. Cancers 2020, 12, 3471. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Lin, X.; Kapoor, A.; Li, T.; Major, P.; Tang, D. Effective prediction of prostate cancer recurrence through the iqgap1 network. Cancers 2021, 13, 430. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cbio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An integrated tcga pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. Gepia2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. Ualcan: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. Tisidb: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of rna-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [PubMed] [Green Version]
- Aran, D.; Hu, Z.; Butte, A.J. Xcell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [Green Version]
- Racle, J.; de Jonge, K.; Baumgaertner, P.; Speiser, D.E.; Gfeller, D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife 2017, 6, e26476. [Google Scholar]
- Sturm, G.; Finotello, F.; List, M. Immunedeconv: An R package for unified access to computational methods for estimating immune cell fractions from bulk rna-sequencing data. In Bioinformatics for Cancer Immunotherapy; Humana: New York, NY, USA, 2020; Volume 2120, pp. 223–232. [Google Scholar]
- Bie, L.; Zhao, G.; Cheng, P.; Rondeau, G.; Porwollik, S.; Ju, Y.; Xia, X.Q.; McClelland, M. The accuracy of survival time prediction for patients with glioma is improved by measuring mitotic spindle checkpoint gene expression. PLoS ONE 2011, 6, e25631. [Google Scholar] [CrossRef] [Green Version]
- Koffa, M.D.; Casanova, C.M.; Santarella, R.; Kocher, T.; Wilm, M.; Mattaj, I.W. Hurp is part of a ran-dependent complex involved in spindle formation. Curr. Biol. 2006, 16, 743–754. [Google Scholar] [CrossRef] [Green Version]
- Sillje, H.H.; Nagel, S.; Korner, R.; Nigg, E.A. Hurp is a ran-importin beta-regulated protein that stabilizes kinetochore microtubules in the vicinity of chromosomes. Curr. Biol. 2006, 16, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A cerna hypothesis: The rosetta stone of a hidden rna language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Urrutia, E.; Bustamante Montes, L.P.; Ladron de Guevara Cervantes, D.; Perez-Plasencia, C.; Campos-Parra, A.D. Crosstalk between long non-coding rnas, micro-rnas and mrnas: Deciphering molecular mechanisms of master regulators in cancer. Front. Oncol 2019, 9, 669. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, J.; Cairns, M.J. Identifying mirnas, targets and functions. Brief. Bioinform. 2014, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Qin, Y.; Zeng, B.; Feng, Y.; Li, Y.; Xiang, T.; Ren, G. Long noncoding rna linc01089 predicts clinical prognosis and inhibits cell proliferation and invasion through the wnt/beta-catenin signaling pathway in breast cancer. OncoTargets Ther. 2019, 12, 4883–4895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sas-Chen, A.; Aure, M.R.; Leibovich, L.; Carvalho, S.; Enuka, Y.; Korner, C.; Polycarpou-Schwarz, M.; Lavi, S.; Nevo, N.; Kuznetsov, Y.; et al. Limt is a novel metastasis inhibiting lncrna suppressed by egf and downregulated in aggressive breast cancer. EMBO Mol. Med. 2016, 8, 1052–1064. [Google Scholar] [CrossRef]
- Zhang, D.; Cai, X.; Cai, S.; Chen, W.; Hu, C. Long intergenic non-protein coding rna 01089 weakens tumor proliferation, migration, and invasion by sponging mir-3187-3p in non-small cell lung cancer. Cancer Manag. Res. 2020, 12, 12151–12162. [Google Scholar] [CrossRef]
- Li, X.; Lv, F.; Li, F.; Du, M.; Liang, Y.; Ju, S.; Liu, Z.; Zhou, B.; Wang, B.; Gao, Y. Linc01089 inhibits tumorigenesis and epithelial-mesenchymal transition of non-small cell lung cancer via the mir-27a/sfrp1/wnt/beta-catenin axis. Front. Oncol. 2020, 10, 532581. [Google Scholar]
- Wang, F.; Yang, Q. Long non-coding rna linc01089 enhances the development of gastric cancer by sponging mir-145-5p to mediate sox9 expression. OncoTargets Ther. 2020, 13, 9213–9224. [Google Scholar] [CrossRef]
- Sun, J.; Zheng, X.; Wang, B.; Cai, Y.; Zheng, L.; Hu, L.; Lu, X.; Xie, S.; Zhang, X.; Liu, H.; et al. Lncrna limt (linc01089) contributes to sorafenib chemoresistance via regulation of mir-665 and epithelial to mesenchymal transition in hepatocellular carcinoma cells. Acta Biochim. Biophys. Sin. 2022, 54, 261–270. [Google Scholar] [CrossRef]
- Lan, T.; Yuan, K.; Yan, X.; Xu, L.; Liao, H.; Hao, X.; Wang, J.; Liu, H.; Chen, X.; Xie, K.; et al. Lncrna snhg10 facilitates hepatocarcinogenesis and metastasis by modulating its homolog scarna13 via a positive feedback loop. Cancer Res. 2019, 79, 3220–3234. [Google Scholar] [CrossRef] [Green Version]
- Hou, H.; Lyu, Y.; Jiang, J.; Wang, M.; Zhang, R.; Liew, C.C.; Wang, B.; Cheng, C. Peripheral blood transcriptome identifies high-risk benign and malignant breast lesions. PLoS ONE 2020, 15, e0233713. [Google Scholar] [CrossRef]
- Chu, W.K.; Hickson, I.D. Recq helicases: Multifunctional genome caretakers. Nat. Rev. Cancer 2009, 9, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Meador, J.A.; Calaf, G.M.; Proietti De-Santis, L.; Zhao, Y.; Bohr, V.A.; Balajee, A.S. Human recql4 helicase plays critical roles in prostate carcinogenesis. Cancer Res. 2010, 70, 9207–9217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, D.; Fang, H.; Niu, K.; Liu, J.; Wu, M.; Li, S.; Zhu, T.; Aleskandarany, M.A.; Arora, A.; Lobo, D.N.; et al. Human helicase recql4 drives cisplatin resistance in gastric cancer by activating an akt-yb1-mdr1 signaling pathway. Cancer Res. 2016, 76, 3057–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crona, J.; Beuschlein, F. Adrenocortical carcinoma—towards genomics guided clinical care. Nat. Rev. Endocrinol. 2019, 15, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.; Chiu, Y.F.; Kuo, M.H.; Lee, K.L.; Lee, A.C.; Yu, C.C.; Chang, J.L.; Huang, W.C.; Hsiao, S.H.; Lin, S.E.; et al. Expression of neuroendocrine factor vgf in lung cancer cells confers resistance to egfr kinase inhibitors and triggers epithelial-to-mesenchymal transition. Cancer Res. 2017, 77, 3013–3026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.F.; Huang, Y.Q.; Wu, K.M.; Jou, A.F.; Shih, N.Y.; Ho, J.A. Diagnosing the rgs11 lung cancer biomarker: The integration of competitive immunoassay and isothermal nucleic acid exponential amplification reaction. Anal. Chem. 2019, 91, 3327–3335. [Google Scholar] [CrossRef]
- Barisone, G.A.; Ngo, T.; Tran, M.; Cortes, D.; Shahi, M.H.; Nguyen, T.V.; Perez-Lanza, D.; Matayasuwan, W.; Diaz, E. Role of mxd3 in proliferation of daoy human medulloblastoma cells. PLoS ONE 2012, 7, e38508. [Google Scholar] [CrossRef] [Green Version]
- Koike, H.; Morikawa, Y.; Sekine, Y.; Matsui, H.; Shibata, Y.; Suzuki, K. Survivin is associated with cell proliferation and has a role in 1a,25-dihydroxyvitamin d3 induced cell growth inhibition in prostate cancer. J. Urol. 2011, 185, 1497–1503. [Google Scholar]
- Cao, L.; Li, C.; Shen, S.; Yan, Y.; Ji, W.; Wang, J.; Qian, H.; Jiang, X.; Li, Z.; Wu, M.; et al. Oct4 increases birc5 and ccnd1 expression and promotes cancer progression in hepatocellular carcinoma. BMC Cancer 2013, 13, 82. [Google Scholar]
- Halvorsen, A.R.; Helland, A.; Fleischer, T.; Haug, K.M.; Grenaker Alnaes, G.I.; Nebdal, D.; Syljuasen, R.G.; Touleimat, N.; Busato, F.; Tost, J.; et al. Differential DNA methylation analysis of breast cancer reveals the impact of immune signaling in radiation therapy. Int. J. Cancer 2014, 135, 2085–2095. [Google Scholar] [CrossRef] [Green Version]
- Hochwagen, A.; Tham, W.H.; Brar, G.A.; Amon, A. The fk506 binding protein fpr3 counteracts protein phosphatase 1 to maintain meiotic recombination checkpoint activity. Cell 2005, 122, 861–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, P.; Yu, B.; Pan, L.; Tian, X.; Liu, F. Identification of key genes and pathways in triple-negative breast cancer by integrated bioinformatics analysis. BioMed Res. Int. 2018, 2018, 2760918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, J.; Xing, Y.; Lv, J.; Gu, D.; Pan, J.; Zhang, Y.; Liu, J. Construction of an immune-related gene signature for prediction of prognosis in patients with cervical cancer. Int. Immunopharmacol. 2020, 88, 106882. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, A.; Penachioni, J.Y.; Lamba, S.; Bleeker, F.E.; Zanon, C.; Rodolfo, M.; Vallacchi, V.; Scarpa, A.; Felicioni, L.; Buck, M.; et al. Molecular profiling of the "plexinome" in melanoma and pancreatic cancer. Hum. Mutat. 2009, 30, 1167–1174. [Google Scholar]
- Wen, H.; Lei, Y.; Eun, S.Y.; Ting, J.P. Plexin-a4-semaphorin 3a signaling is required for toll-like receptor- and sepsis-induced cytokine storm. J. Exp. Med. 2010, 207, 2943–2957. [Google Scholar] [CrossRef]
- Januchowski, R.; Sterzynska, K.; Zawierucha, P.; Rucinski, M.; Swierczewska, M.; Partyka, M.; Bednarek-Rajewska, K.; Brazert, M.; Nowicki, M.; Zabel, M.; et al. Microarray-based detection and expression analysis of new genes associated with drug resistance in ovarian cancer cell lines. Oncotarget 2017, 8, 49944–49958. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.L.; Zhan, D.M.; Chong, Y.K.; Ding, L.; Yang, Y.G. Mir-652-3p promotes bladder cancer migration and invasion by targeting kcnn3. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8806–8812. [Google Scholar]
- Castano-Rodriguez, N.; Kaakoush, N.O.; Mitchell, H.M. Pattern-recognition receptors and gastric cancer. Front. Immunol. 2014, 5, 336. [Google Scholar]
- Jiang, Y.; Zheng, X.; Jiao, D.; Chen, P.; Xu, Y.; Wei, H.; Qian, Y. Peptidase inhibitor 15 as a novel blood diagnostic marker for cholangiocarcinoma. EBioMedicine 2019, 40, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Nagelkerke, A.; Mujcic, H.; Bussink, J.; Wouters, B.G.; van Laarhoven, H.W.; Sweep, F.C.; Span, P.N. Hypoxic regulation and prognostic value of lamp3 expression in breast cancer. Cancer 2011, 117, 3670–3681. [Google Scholar] [CrossRef]
- Buckwalter, J.M.; Chan, W.; Shuman, L.; Wildermuth, T.; Ellis-Mohl, J.; Walter, V.; Warrick, J.I.; Wu, X.R.; Kaag, M.; Raman, J.D.; et al. Characterization of histone deacetylase expression within in vitro and in vivo bladder cancer model systems. Int. J. Mol. Sci. 2019, 20, 2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacey, S.N.; Helgason, H.; Gudjonsson, S.A.; Thorleifsson, G.; Zink, F.; Sigurdsson, A.; Kehr, B.; Gudmundsson, J.; Sulem, P.; Sigurgeirsson, B.; et al. New basal cell carcinoma susceptibility loci. Nat. Commun. 2015, 6, 6825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between tfeb transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Sbiera, S.; Kroiss, M.; Thamm, T.; Beyer, M.; Majidi, F.; Kuehner, D.; Wobser, M.; Becker, J.C.; Adam, P.; Ronchi, C.; et al. Survivin in adrenocortical tumors—pathophysiological implications and therapeutic potential. Horm. Metab. Res. 2013, 45, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, J.D.; Novokmet, A.; Eichler-Jonsson, C.; Ribeiro, R.C.; Rodriguez-Galindo, C.; Zambetti, G.P.; Malkin, D. Prevalence and functional consequence of tp53 mutations in pediatric adrenocortical carcinoma: A children’s oncology group study. J. Clin. Oncol. 2015, 33, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Kamilaris, C.D.C.; Hannah-Shmouni, F.; Stratakis, C.A. Adrenocortical tumorigenesis: Lessons from genetics. Best Pr. Res. Clin. Endocrinol. Metab. 2020, 34, 101428. [Google Scholar] [CrossRef]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.A.; Eksioglu, E.A.; Sullivan, A.; et al. Tp53 mutations in myelodysplastic syndromes and secondary aml confer an immunosuppressive phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Maddalena, M.; Mallel, G.; Nataraj, N.B.; Shreberk-Shaked, M.; Hassin, O.; Mukherjee, S.; Arandkar, S.; Rotkopf, R.; Kapsack, A.; Lambiase, G.; et al. Tp53 missense mutations in pdac are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proc. Natl. Acad. Sci. USA 2021, 118, e2025631118. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. Tgfbeta attenuates tumour response to pd-l1 blockade by contributing to exclusion of t cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Canellas, A.; Hernando-Momblona, X.; et al. Tgfbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Hanna, B.S.; Llao-Cid, L.; Iskar, M.; Roessner, P.M.; Klett, L.C.; Wong, J.K.L.; Paul, Y.; Ioannou, N.; Ozturk, S.; Mack, N.; et al. Interleukin-10 receptor signaling promotes the maintenance of a pd-1(int) tcf-1(+) cd8(+) t cell population that sustains anti-tumor immunity. Immunity 2021, 54, 2825–2841.e2810. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Li, B.; Lu, Z.; Liang, H.; Yang, H.; Chen, Y.; Zhu, S.; Zeng, M.; Wei, Y.; Liu, T.; et al. Ccdc137 is a prognostic biomarker and correlates with immunosuppressive tumor microenvironment based on pan-cancer analysis. Front. Mol. Biosci. 2021, 8, 674863. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, tim-3, and tigit: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Chauvin, J.M.; Zarour, H.M. Tigit in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef] [PubMed]
- Ridge, S.M.; Sullivan, F.J.; Glynn, S.A. Mesenchymal stem cells: Key players in cancer progression. Mol. Cancer 2017, 16, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assie, G.; Letouze, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; Rene-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef]
- Lippert, J.; Appenzeller, S.; Liang, R.; Sbiera, S.; Kircher, S.; Altieri, B.; Nanda, I.; Weigand, I.; Gehrig, A.; Steinhauer, S.; et al. Targeted molecular analysis in adrenocortical carcinomas: A strategy toward improved personalized prognostication. J. Clin. Endocrinol. Metab. 2018, 103, 4511–4523. [Google Scholar] [CrossRef] [PubMed]
- Kanczkowski, W.; Sue, M.; Bornstein, S.R. The adrenal gland microenvironment in health, disease and during regeneration. Hormones 2017, 16, 251–265. [Google Scholar] [CrossRef]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F., Jr.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef]
- Wagner, J.; Portwine, C.; Rabin, K.; Leclerc, J.M.; Narod, S.A.; Malkin, D. High frequency of germline p53 mutations in childhood adrenocortical cancer. J. Natl. Cancer Inst. 1994, 86, 1707–1710. [Google Scholar] [CrossRef]
- Varley, J.M.; McGown, G.; Thorncroft, M.; James, L.A.; Margison, G.P.; Forster, G.; Evans, D.G.; Harris, M.; Kelsey, A.M.; Birch, J.M. Are there low-penetrance tp53 alleles? Evidence from childhood adrenocortical tumors. Am. J. Hum. Genet. 1999, 65, 995–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, C.; Armaiz-Pena, G.; Dahia, P.L.M.; Lu, Y.; Toledo, R.A.; Varghese, J.; Habra, M.A. Endocrine and neuroendocrine tumors special issue-checkpoint inhibitors for adrenocortical carcinoma and metastatic pheochromocytoma and paraganglioma: Do they work? Cancers 2022, 14, 467. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Prados, A.; Armaka, M.; Kollias, G. The mesenchymal context in inflammation, immunity and cancer. Nat. Immunol. 2020, 21, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Rueda, M.; Alkhateeb, A. Classification of breast cancer nottingham prognostic index using high-dimensional embedding and residual neural network. Cancers 2022, 14, 934. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Univariate Cox Analysis | Multivariate Cox Analysis 1 | |||||
|---|---|---|---|---|---|---|
| Gene 2 | HR | 95% CI | p-Value | HR | 95% CI | p-Value |
| Poor OS | ||||||
| LINC01089 | 1.002 | 1–1.003 | 0.0479 * | 1.002 | 0.9997–1.003 | 0.101 |
| RGS11 | 1 | 1–1.001 | 0.0291 * | 1 | 1–1.001 | 0.0697 |
| MXD3 | 1.003 | 1.002–1.004 | 8.53 × 10−7 *** | 1.003 | 1.0016–1.004 | 2.75 × 10−5 *** |
| BIRC5 | 1.003 | 1.002–1.004 | 1.45 × 10−9 *** | 1.002 | 1.0014–1.003 | 8.79 × 10−7 *** |
| RAB30 | 1.008 | 1.003–1.013 | 0.00283 ** | 1.006 | 1.001–1.012 | 0.0122* |
| worse PFS | ||||||
| LCN12 | 1.007 | 1.001–1.013 | 0.0187 * | 1.011 | 1.0004–1.018 | 0.00172 ** |
| VGF | 1 | 1–1 | 0.00124 ** | 1 | 1–1 | 0.0463 * |
| RGS11 | 1.001 | 1–1.001 | 0.00271 ** | 1.001 | 1.0001–1.001 | 0.0207 * |
| MXD3 | 1.002 | 1.001–1.003 | 0.000676 *** | 1.0017 | 1.0005–1.003 | 0.00495 ** |
| BIRC5 | 1.002 | 1.001–1.002 | 6.13 × 10−5 *** | 1.001 | 1.0002–1.002 | 0.0196 * |
| RAB30 | 1.02 | 1.005–1.014 | 1.92 × 10−5 *** | 1.0099 | 1.0053–1.014 | 1.94 × 10−5 *** |
| NOD2 | 1.007 | 1.001–1.014 | 0.0237 * | 1.0064 | 1.0003–1.012 | 0.0394 * |
| ZFHX4 | 1 | 1–1.001 | 0.035 * | 1.001 | 1.0001–1.001 | 0.0235 * |
| Univariate Cox Analysis | Multivariate Cox Analysis 1 | |||||
|---|---|---|---|---|---|---|
| Gene 2 | HR | 95% CI | p-Value | HR | 95% CI | p-Value |
| poor OS | ||||||
| LINC01089 | 1.002 | 1–1.003 | 0.0479 * | 1.002 | 0.9997–1.003 | 0.101 |
| SNHG10 | 1.011 | 1.006–1.017 | 1.72 × 10−5 *** | 1.01 | 1.004–1.016 | 0.000758 *** |
| RECQL4 | 1.002 | 1.001–1.002 | 9.26 × 10−8 *** | 1.001 | 1.0004–1.002 | 0.00158 ** |
| worse PFS | ||||||
| LOC100128288 | 0.9897 | 0.98–0.995 | 0.0392 * | 0.993 | 0.981–1.005 | 0.2556 |
| SNHG10 | 1.006 | 1.002–1.01 | 0.00175 ** | 1.01 | 1.004–1.016 | 0.000758 *** |
| RECQL4 | 1.001 | 1.001–1.002 | 0.00015 *** | 1.001 | 1.0004–1.002 | 0.00158 ** |
| Gene | Oncogenic Role in ACC | Oncogenic Role in Others | Reference |
|---|---|---|---|
| LINC01089 | unknown | inhibition of breast cancer metastasis | [44,45] |
| LOC155060 | unknown | unknown | NA |
| LOC100128288 | unknown | unknown | NA |
| SNHG10 | unknown | promotion of resistance to tyrosine kinase inhibitors in lung cancer | [50] |
| RECQL4 | unknown | promotion of gastric cancer | [54] |
| HERC2P2 | unknown | suppression of cell proliferation | [51] |
| ATXN7L2 | unknown | unclear | NA |
| THSD7A | unknown | unclear | NA |
| Gene | Oncogenic Role in ACC | Oncogenic Role in Others | Reference |
|---|---|---|---|
| HAGHL | unknown | Unknow | NA |
| LCN12 | unknown | Unknown | NA |
| DCST2 | unknown | Unknown | NA |
| VGF | unknown | promotion of resistance to tyrosine kinase inhibitors in lung cancer | [56] |
| RGS11 | unknown | a biomarker of lung cancer | [57] |
| PRR7 | unknown | Unknown | NA |
| LINC01089 | unknown | See Table 3 | See Table 3 |
| MXD3 | unknow | promotion of medullobastoma | [58] |
| BIRC5 | Upregulation in AC and association with ACC poor prognosis [75] | promotion of cancer progression and metastasis | [59,60] |
| LTC4S | unknown | a component gene of an immune signature of breast cancer | [61] |
| FPR3 | unknown | sustain meiotic recombination checkpoint actions | [62] |
| RAB30 | unknown | association of good prognosis in triple negative breast cancer | [63] |
| RIPOR2 | unknown | association of immune cell infiltration and thus inhibition of cervical cancer | [64] |
| NOD2 | unknown | immunosuppression of tumorigenesis of gastric cancer | [69] |
| PLXNA4 | unknown | inhibition of tumor cell migration and contribution to innate immunity in working with Toll-like receptor | [65,66] |
| TFEC | unknown | regulation of lysosome biogenesis and mTOR activation | [74] |
| PI15 | unknown | biomarker of cholangiocarcinoma | [70] |
| ZFHX4 | unknown | a susceptibility locus of cutaneous basal cell carcinoma | [73] |
| LAMP3 | unknown | a hypoxia-induced gene associated with aggressive breast cancer | [71] |
| HDAC9 | unknown | increases in expression in bladder cancer | [72] |
| MCTP1 | unknown | downregulation in paclitaxel-resistant ovarian cancer cells | [67] |
| KCNN3 | unknown | suppression of bladder cancer cell migration and invasion | [68] |
| PCDHB8 | unknown | Unknown | NA |
| PCDHGB2 | unknown | Unknown | NA |
| PCDHGA5 | unknown | Unknown | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Gu, Y.; Su, Y.; Dong, Y.; Major, P.; Kapoor, A.; Tang, D. Prediction of Adrenocortical Carcinoma Relapse and Prognosis with a Set of Novel Multigene Panels. Cancers 2022, 14, 2805. https://doi.org/10.3390/cancers14112805
Lin X, Gu Y, Su Y, Dong Y, Major P, Kapoor A, Tang D. Prediction of Adrenocortical Carcinoma Relapse and Prognosis with a Set of Novel Multigene Panels. Cancers. 2022; 14(11):2805. https://doi.org/10.3390/cancers14112805
Chicago/Turabian StyleLin, Xiaozeng, Yan Gu, Yingying Su, Ying Dong, Pierre Major, Anil Kapoor, and Damu Tang. 2022. "Prediction of Adrenocortical Carcinoma Relapse and Prognosis with a Set of Novel Multigene Panels" Cancers 14, no. 11: 2805. https://doi.org/10.3390/cancers14112805
APA StyleLin, X., Gu, Y., Su, Y., Dong, Y., Major, P., Kapoor, A., & Tang, D. (2022). Prediction of Adrenocortical Carcinoma Relapse and Prognosis with a Set of Novel Multigene Panels. Cancers, 14(11), 2805. https://doi.org/10.3390/cancers14112805