

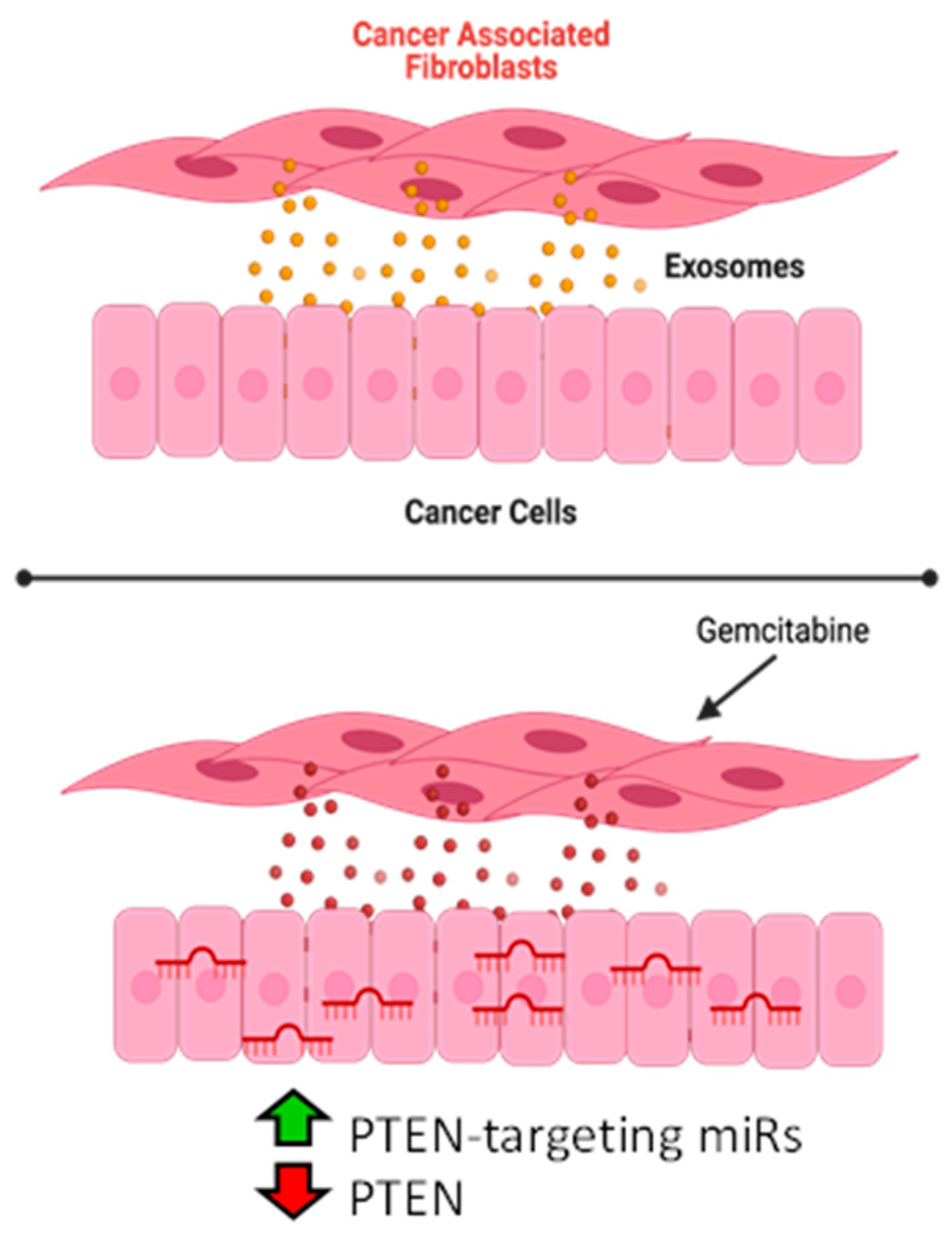
Cancer-Associated Fibroblasts Confer Gemcitabine Resistance to Pancreatic Cancer Cells through PTEN-Targeting miRNAs in Exosomes
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.1.1. Conditioned-Cell Media Transfer
2.1.2. Co-Culture Studies
2.2. Exosome Isolation, RNA Extraction, and MicroRNA-Seq
2.3. Target Gene Prediction and Pathway Analysis
2.4. RNA Isolation and RT-PCR
2.4.1. Exosome MicroRNA
2.4.2. Cellular MicroRNA and mRNA
2.5. Mimic Transfection
2.6. Mouse Studies
2.7. Western Blot
2.8. Statistical Analysis
3. Results
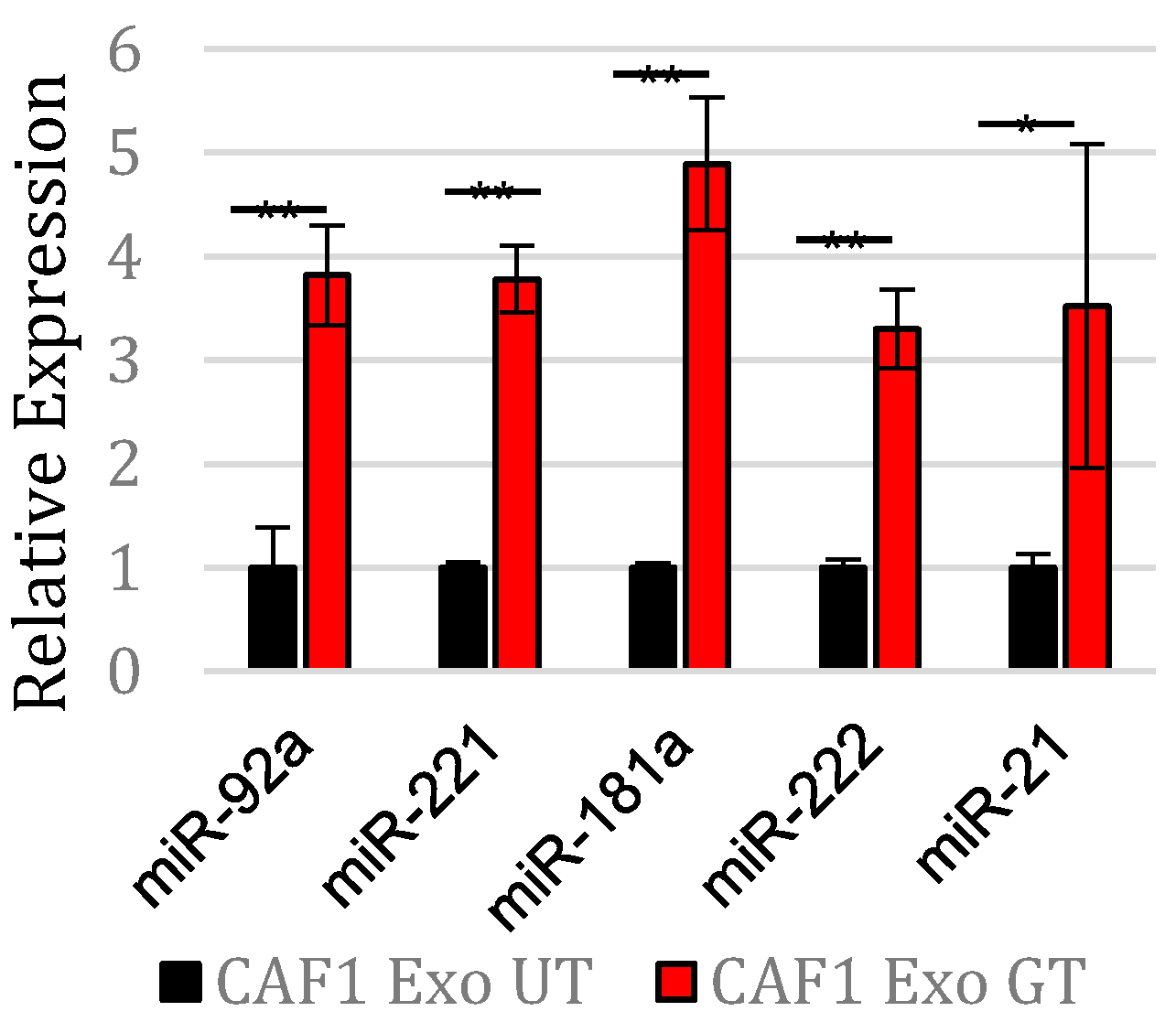
3.1. Gemcitabine Exposure Increases Expression of Five OncomiRs within Pancreatic CAF Exosomes
3.2. PTEN Is the Predicted Target of miRNAs Released by Gemcitabine-Treated CAF-Derived Exosomes
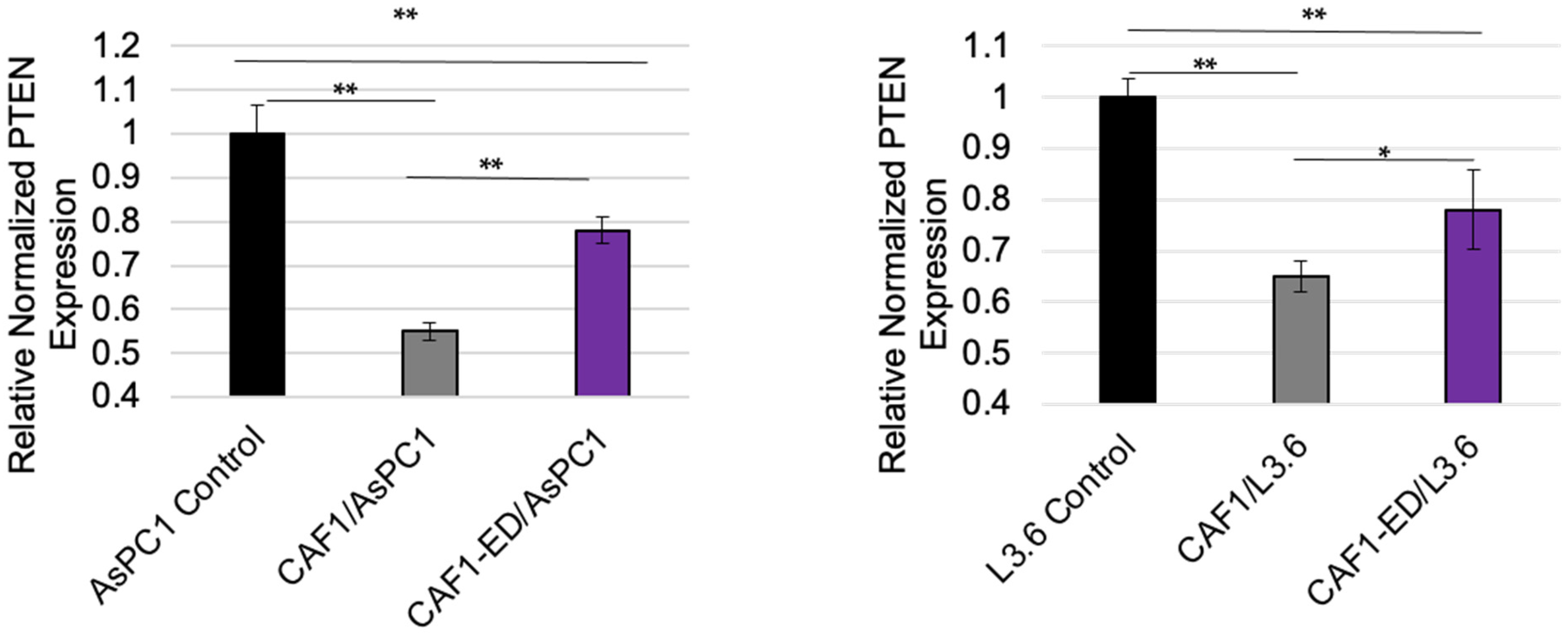
3.3. PTEN Levels Are Suppressed by Exosomes from CAFs
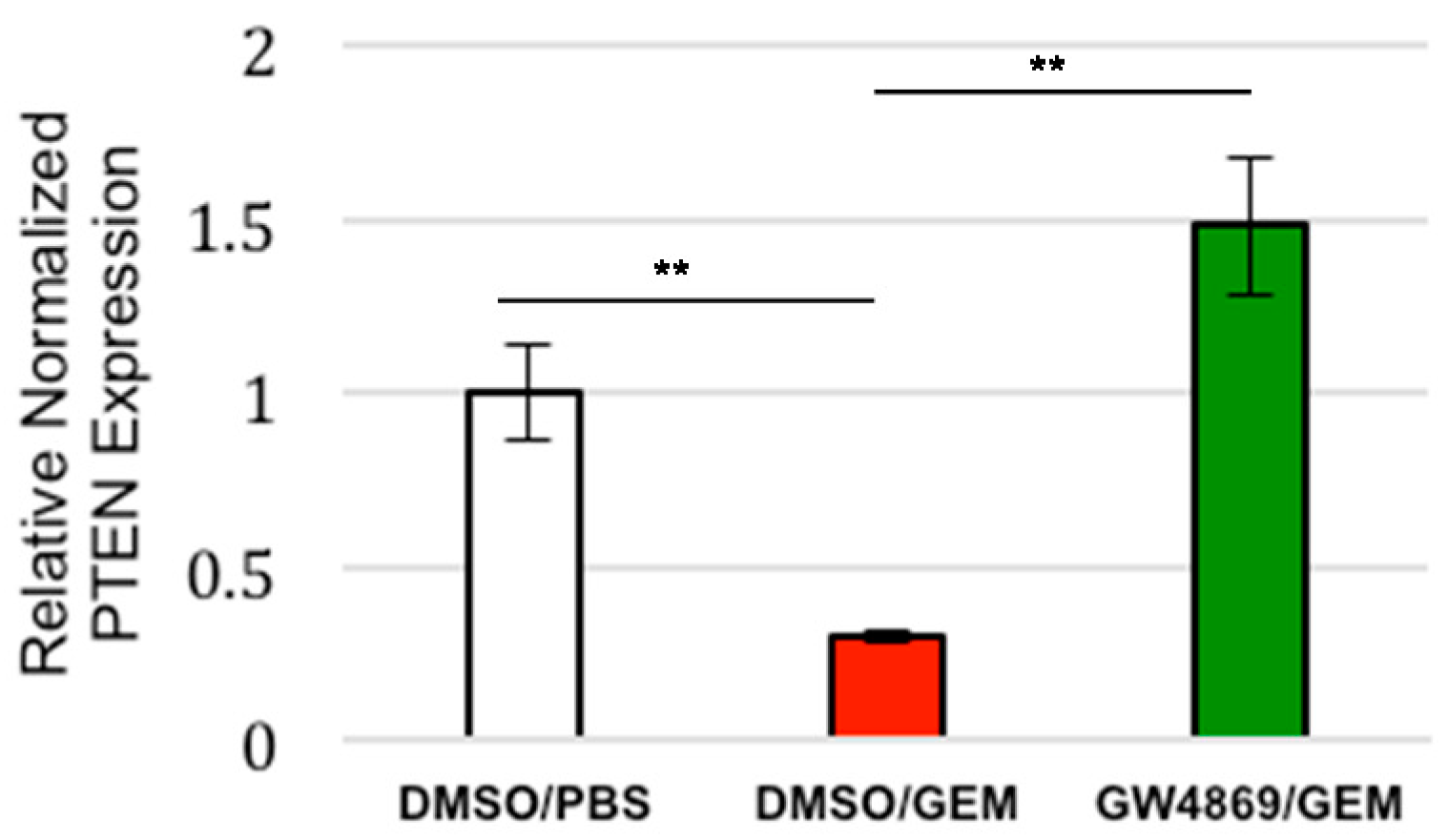
3.4. Exosome Inhibition Restores PTEN Expression to Tumors Treated with Gemcitabine In Vivo
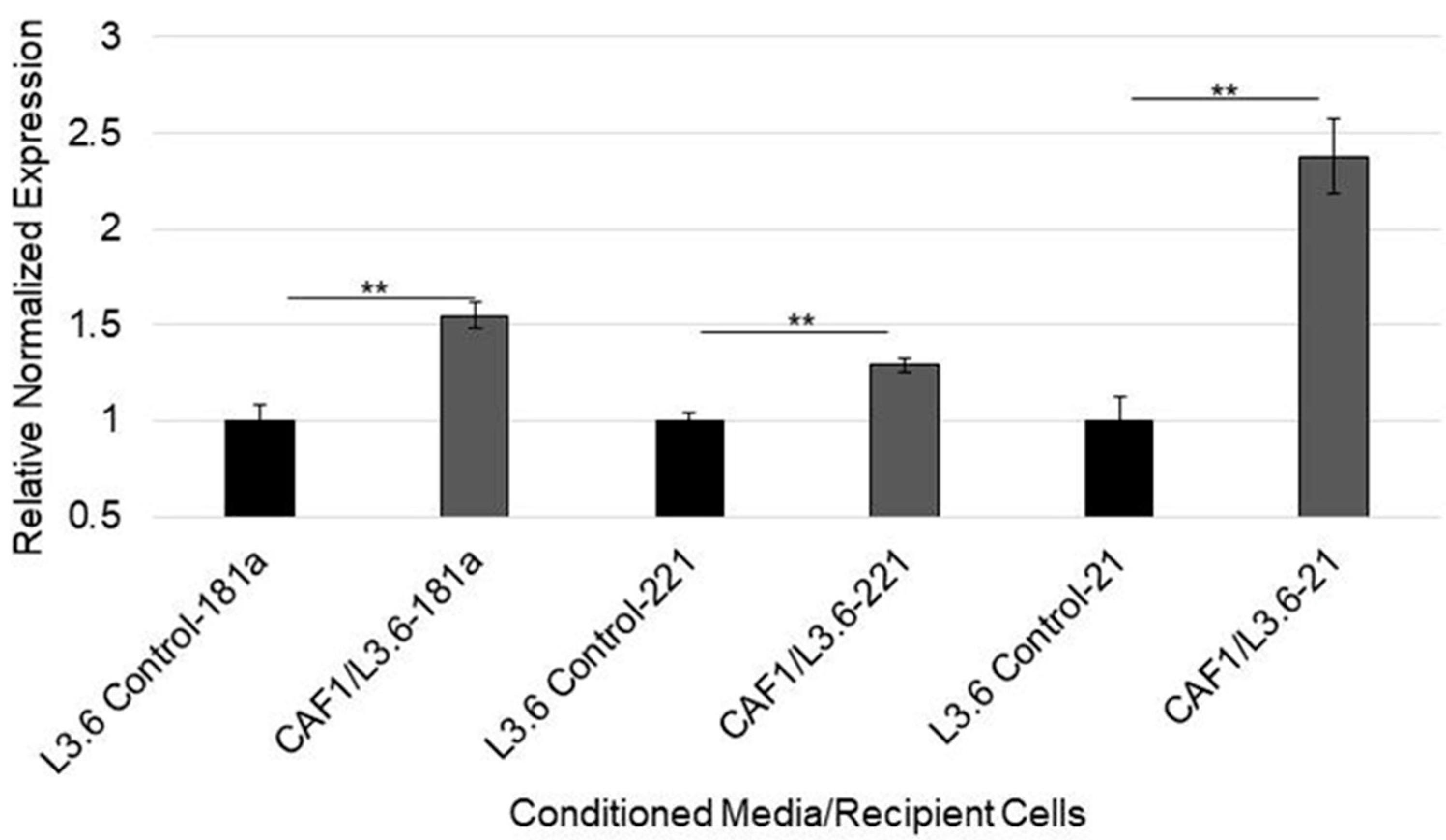
3.5. MicroRNA-92a Targets PTEN mRNA in PDAC Cells and Is Distributed through CAF Exosomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Society, A.C. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2022. [Google Scholar]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manji, G.A.; Olive, K.; Saenger, Y.M.; Oberstein, P. Current and Emerging Therapies in Metastatic Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 1670–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant Chemotherapy With Gemcitabine vs. Observation in Patients Undergoing Curative-Intent Resection of Pancreatic Cancer. JAMA J. Am. Med Assoc. 2007, 297, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Stopa, K.B.; Kusiak, A.A.; Szopa, M.D.; Ferdek, P.E.; Jakubowska, M.A. Pancreatic Cancer and Its Microenvironment—Recent Advances and Current Controversies. Int. J. Mol. Sci. 2020, 21, 3218. [Google Scholar] [CrossRef]
- Kleeff, J.; Beckhove, P.; Esposito, I.; Herzig, S.; Huber, P.E.; Löhr, J.M.; Friess, H. Pancreatic cancer microenvironment. Int. J. Cancer 2007, 121, 699–705. [Google Scholar] [CrossRef]
- Von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-Associated Stromal Fibroblasts Promote Pancreatic Tumor Progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.-C.; Wang, T.-T.; Liu, W.; Fu, B.-S.; Hua, X.; Wang, G.-Y.; Li, T.-J.; Li, X.; Wu, X.-Y.; Tai, Y.; et al. Cancer-Associated Fibroblasts from Hepatocellular Carcinoma Promote Malignant Cell Proliferation by HGF Secretion. PLoS ONE 2013, 8, e63243. [Google Scholar] [CrossRef] [Green Version]
- Ying, L.; Zhu, Z.; Xu, Z.; He, T.; Li, E.; Guo, Z.; Liu, F.; Jiang, C.; Wang, Q. Cancer Associated Fibroblast-Derived Hepatocyte Growth Factor Inhibits the Paclitaxel-Induced Apoptosis of Lung Cancer A549 Cells by Up-Regulating the PI3K/Akt and GRP78 Signaling on a Microfluidic Platform. PLoS ONE 2015, 10, e0129593. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Mark, M.T.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leca, J.; Martinez, S.; Lac, S.; Nigri, J.; Secq, V.; Rubis, M.; Bressy, C.; Sergé, A.; Lavaut, M.-N.; Dusetti, N.; et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J. Clin. Investig. 2016, 126, 4140–4156. [Google Scholar] [CrossRef] [Green Version]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.; Singh, A.P. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Mikamori, M.; Yamada, D.; Eguchi, H.; Hasegawa, S.; Kishimoto, T.; Tomimaru, Y.; Asaoka, T.; Noda, T.; Wada, H.; Kawamoto, K.; et al. MicroRNA-155 Controls Exosome Synthesis and Promotes Gemcitabine Resistance in Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2017, 7, 42339. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [Green Version]
- Okami, K.; Wu, L.; Riggins, G.; Cairns, P.; Goggins, M.; Evron, E.; Halachmi, N.; Ahrendt, S.; Reed, A.L.; Hilgers, W.; et al. Analysis of PTEN/MMAC1 alterations in aerodigestive tract tumors. Cancer Res. 1998, 58, 509–511. [Google Scholar]
- Ying, H.; Elpek, K.G.; Vinjamoori, A.; Zimmerman, S.M.; Chu, G.C.; Yan, H.; Fletcher-Sananikone, E.; Zhang, H.; Liu, Y.; Wang, W.; et al. Pten is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discov. 2011, 1, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.; Calvopina, J.H.; Kim, C.; Wang, Y.; Dawson, D.W.; Donahue, T.R.; Dry, S.; Wu, H. PTEN Loss AcceleratesKrasG12D-Induced Pancreatic Cancer Development. Cancer Res. 2010, 70, 7114–7124. [Google Scholar] [CrossRef] [Green Version]
- Wartenberg, M.; Centeno, I.; Haemmig, S.; Vassella, E.; Zlobec, I.; Galván, J.A.; Neuenschwander, M.; Schlup, C.; Gloor, B.; Lugli, A.; et al. PTEN alterations of the stromal cells characterise an aggressive subpopulation of pancreatic cancer with enhanced metastatic potential. Eur. J. Cancer 2016, 65, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cobb, G.; Anderson, T. MicroRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Sharples, R.A.; Scicluna, B.J.; Hill, A.F. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J. Extracell. Vesicles 2014, 3, 23743. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F. Oncomirs—microRNAs with a role in cancer. Nat. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Libra, M.; Hafsi, S.; Pezzino, F.M.; Candido, S.; Ligresti, G.; Spandidos, D.A.; Soua, Z.; McCubrey, J.A.; Travali, S. Gene alterations in the PI3K/PTEN/AKT pathway as a mechanism of drug-resistance (Review). Int. J. Oncol. 2011, 40, 639–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Fang, Y.-X.; Xue, J.-L.; Chen, J.-Z. Four MicroRNAs Promote Prostate Cell Proliferation with Regulation of PTEN and Its Downstream Signals In Vitro. PLoS ONE 2013, 8, e75885. [Google Scholar] [CrossRef]
- Chun-Zhi, Z.; Lei, H.; An-Ling, Z.; Yan-Chao, F.; Xiao, Y.; Guang-Xiu, W.; Zhi-Fan, J.; Pei-Yu, P.; Qing-Yu, Z.; Chun-Sheng, K. MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell proliferation and radioresistance by targeting PTEN. BMC Cancer 2010, 10, 367. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xu, D.; Wang, Q.; Zheng, D.; Jiang, X.; Xu, L. LPS Induced miR-181a Promotes Pancreatic Cancer Cell Migration via Targeting PTEN and MAP2K4. Am. J. Dig. Dis. 2014, 59, 1452–1460. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Henson, R.; Wehbe–Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 Regulates Expression of the PTEN Tumor Suppressor Gene in Human Hepatocellular Cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandol, S.; Edderkaoui, M.; Gukovsky, I.; Lugea, A.; Gukovskaya, A. Desmoplasia of Pancreatic Ductal Adenocarcinoma. Clin. Gastroenterol. Hepatol. 2009, 7, S44–S47. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Suresh, R.; Banerjee, S.; Bao, B.; Xu, Z.; Wilson, J.; Philip, P.A.; Apte, M.; Sarkar, F.H. Contribution of microRNAs in understanding the pancreatic tumor microenvironment involving cancer associated stellate and fibroblast cells. Am. J. Cancer Res. 2015, 5, 1251–1264. [Google Scholar] [PubMed]
- Foo, W.C.; Rashid, A.; Wang, H.; Katz, M.H.; Lee, J.E.; Pisters, P.W.; Wolff, R.A.; Abbruzzese, J.L.; Fleming, J.B.; Wang, H. Loss of phosphatase and tensin homolog expression is associated with recurrence and poor prognosis in patients with pancreatic ductal adenocarcinoma. Hum. Pathol. 2012, 44, 1024–1030. [Google Scholar] [CrossRef] [Green Version]
- Lefler, J.E.; Seward, C.; Ostrowski, M.C. PTEN in cancer associated fibroblasts. Adv. Cancer Res. 2022, 154, 203–226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.-C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zeng, Z.; He, Z.; Lei, S. Hypoxic pancreatic stellate cell-derived exosomal mirnas promote proliferation and invasion of pancreatic cancer through the PTEN/AKT pathway. Aging 2021, 13, 7120–7132. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, W.; Yang, Y.; Zhang, Z. miRNA-93-5p Promotes Gemcitabine Resistance in Pancreatic Cancer Cells by Targeting the PTEN-Mediated PI3K/Akt Signaling Pathway. Ann. Clin. Lab. Sci. 2021, 51, 310–320. [Google Scholar]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [Green Version]
- Roscigno, G.; Quintavalle, C.; Donnarumma, E.; Puoti, I.; Lagares, A.D.; Iaboni, M.; Fiore, D.; Russo, V.; Todaro, M.; Romano, G.; et al. MiR-221 promotes stemness of breast cancer cells by targeting DNMT3b. Oncotarget 2015, 7, 580–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Au Yeung, C.L.; Co, N.-N.; Tsuruga, T.; Yeung, T.-L.; Kwan, S.Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.-K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Zhou, W.; Rong, Y.; Kuang, T.; Xu, X.; Wu, W.; Wang, D.; Lou, W. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2019, 383, 111543. [Google Scholar] [CrossRef]
- Comandatore, A.; Immordino, B.; Balsano, R.; Capula, M.; Garajovà, I.; Ciccolini, J.; Giovannetti, E.; Morelli, L. Potential Role of Exosomes in the Chemoresistance to Gemcitabine and Nab-Paclitaxel in Pancreatic Cancer. Diagnostics 2022, 12, 286. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, X. Organotropic metastasis: Role of tumor exosomes. Cell Res. 2015, 26, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, D.A.; Patel, S.H.; Gucek, M.; Hendrix, A.; Westbroek, W.; Taraska, J.W. Exosomes Released from Breast Cancer Carcinomas Stimulate Cell Movement. PLoS ONE 2015, 10, e0117495. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-X.; Cai, Y.-Q.; Lv, M.-M.; Chen, L.; Zhong, S.; Ma, T.-F.; Zhao, J.-H.; Tang, J.-H. Exosomes from docetaxel-resistant breast cancer cells alter chemosensitivity by delivering microRNAs. Tumor Biol. 2014, 35, 9649–9659. [Google Scholar] [CrossRef] [PubMed]
- Jorfi, S.; Ansa-Addo, E.A.; Kholia, S.; Stratton, D.; Valley, S.; Lange, S.; Inal, J. Inhibition of microvesiculation sensitizes prostate cancer cells to chemotherapy and reduces docetaxel dose required to limit tumor growth in vivo. Sci. Rep. 2015, 5, 13006. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [Green Version]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lu, J.; Liu, J.; Zhang, G.; Lu, A. Advances in the discovery of exosome inhibitors in cancer. J. Enzym. Inhib. Med. Chem. 2020, 35, 1322–1330. [Google Scholar] [CrossRef]
- Hwang, J.-H.; Voortman, J.; Giovannetti, E.; Steinberg, S.M.; Leon, L.G.; Kim, Y.-T.; Funel, N.; Park, J.K.; Kim, M.A.; Kang, G.H.; et al. Identification of MicroRNA-21 as a Biomarker for Chemoresistance and Clinical Outcome Following Adjuvant Therapy in Resectable Pancreatic Cancer. PLoS ONE 2010, 5, e10630. [Google Scholar] [CrossRef]
- Lai, X.; Wang, M.; McElyea, S.D.; Sherman, S.; House, M.; Korc, M. A microRNA signature in circulating exosomes is superior to exosomal glypican-1 levels for diagnosing pancreatic cancer. Cancer Lett. 2017, 393, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Lai, L.A.; Sullivan, Y.; Wong, M.; Wang, L.; Riddell, J.; Jung, L.; Pillarisetty, V.G.; Brentnall, T.A.; Pan, S. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Xia, R.; Zhang, X.; Li, T.; Ye, Y.; Li, G.; He, R.; Li, Z.; Lin, Q.; Zheng, S.; et al. circFARP1 enables cancer-associated fibroblasts to promote gemcitabine resistance in pancreatic cancer via the LIF/STAT3 axis. Mol. Cancer 2022, 21, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, S.; Wang, H.; Cao, J.; Huang, X.; Chen, Z.; Xu, P.; Sun, G.; Xu, J.; Lv, J.; et al. Circular RNA circNRIP1 acts as a microRNA-149-5p sponge to promote gastric cancer progression via the AKT1/mTOR pathway. Mol. Cancer 2019, 18, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kγ to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018, 78, 4586–4598. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MicroRNA | Fold Change (GEM vs. NT) | log2 Fold Change (GEM vs. NT) | p-Value |
|---|---|---|---|
| hsa-miR-92a-3p ** | 4.65992105 | 2.22030551 | 0.0042333 |
| hsa-miR-221-3p ** | 4.32490131 | 2.11266721 | 0.0064948 |
| hsa-miR-181a-5p * | 3.50850376 | 1.81085591 | 0.0132411 |
| hsa-miR-222-3p * | 2.64868585 | 1.40527674 | 0.0426011 |
| hsa-miR-21-5p ** | 2.53082673 | 1.33960874 | 0.003991 |
| MicroRNA | Predicted Targeted 3′UTR Position of PTEN Transcript | miTG Score |
|---|---|---|
| hsa-miR-92a-3p | 59–84; 2842–2865; 3987–4009 | 0.971 |
| hsa-miR-181a-5p | 1261–1287; 1869–1887; 2255–2282; 2289–2316; 2780–2801; 3923–3933; 4680–4700; 5114–5137; 5249–5275; 5881–5908; 6194–6220 | 0.882 |
| hsa-miR-222-3p | 180–205; 2668–2685; 4381–4408; 5908–5928 | 0.812 |
| hsa-miR-221-3p | 180–205; 2668–2685; 4381–4408; 5908–5928 | 0.755 |
| hsa-miR-21-5p | 1588–1607; 4789–4814 | 0.406 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richards, K.E.; Xiao, W.; Hill, R.; on behalf of the USC Pancreas Research Team, on behalf of the USC Pancreas Research Team. Cancer-Associated Fibroblasts Confer Gemcitabine Resistance to Pancreatic Cancer Cells through PTEN-Targeting miRNAs in Exosomes. Cancers 2022, 14, 2812. https://doi.org/10.3390/cancers14112812
Richards KE, Xiao W, Hill R, on behalf of the USC Pancreas Research Team on behalf of the USC Pancreas Research Team. Cancer-Associated Fibroblasts Confer Gemcitabine Resistance to Pancreatic Cancer Cells through PTEN-Targeting miRNAs in Exosomes. Cancers. 2022; 14(11):2812. https://doi.org/10.3390/cancers14112812
Chicago/Turabian StyleRichards, Katherine E., Weikun Xiao, Reginald Hill, and on behalf of the USC Pancreas Research Team on behalf of the USC Pancreas Research Team. 2022. "Cancer-Associated Fibroblasts Confer Gemcitabine Resistance to Pancreatic Cancer Cells through PTEN-Targeting miRNAs in Exosomes" Cancers 14, no. 11: 2812. https://doi.org/10.3390/cancers14112812
APA StyleRichards, K. E., Xiao, W., Hill, R., & on behalf of the USC Pancreas Research Team, on behalf of the USC Pancreas Research Team. (2022). Cancer-Associated Fibroblasts Confer Gemcitabine Resistance to Pancreatic Cancer Cells through PTEN-Targeting miRNAs in Exosomes. Cancers, 14(11), 2812. https://doi.org/10.3390/cancers14112812