
The Machine-Learning-Mediated Interface of Microbiome and Genetic Risk Stratification in Neuroblastoma Reveals Molecular Pathways Related to Patient Survival
 , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
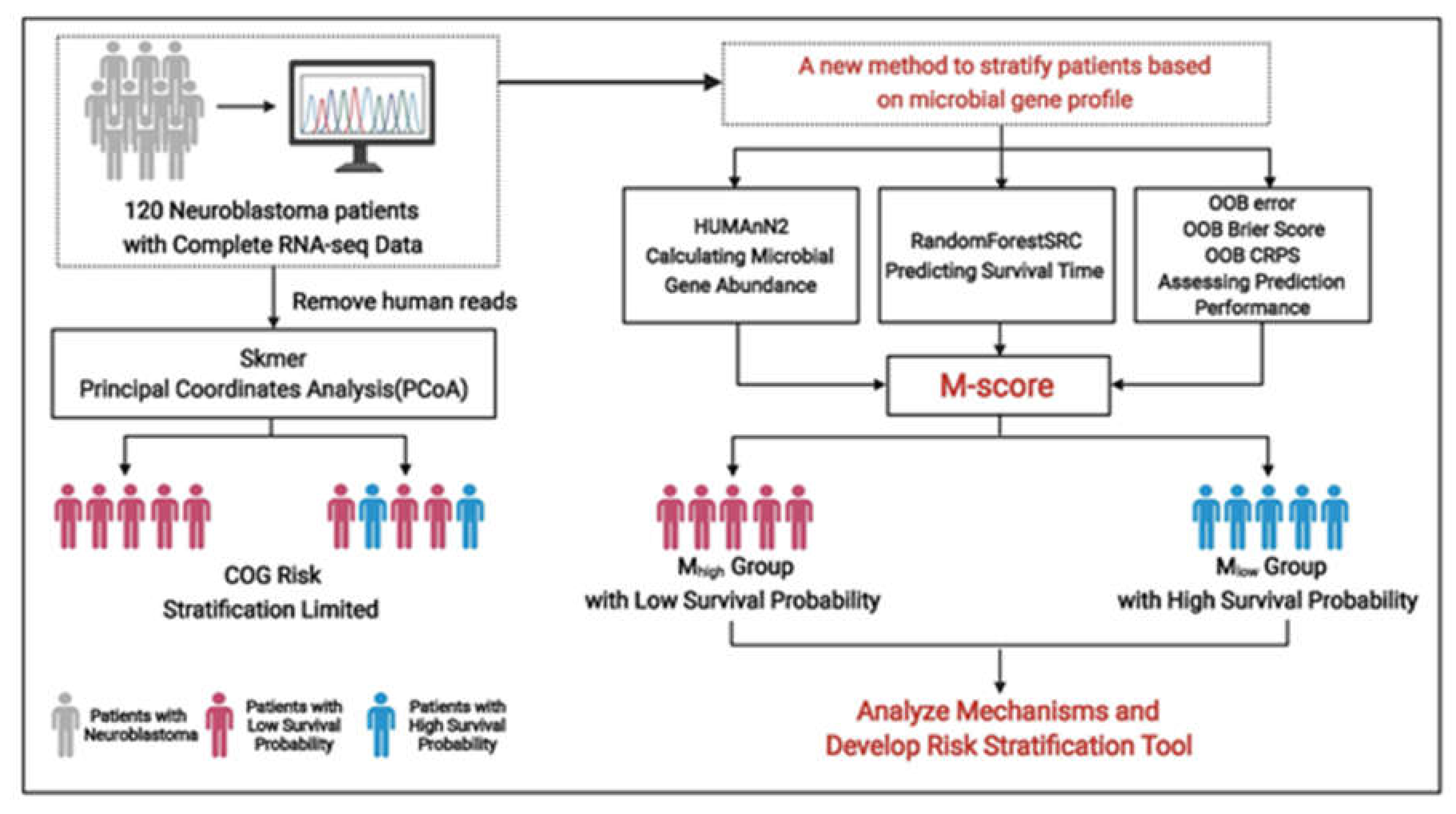
2. Methods
2.1. Datasets and Annotations
2.2. K-Mer Dissimilarity
2.3. Predicting Survival with Microbial Gene Abundance Using Random Forest Survival Analysis
2.4. Signaling Analysis in the Two Microbiome K-Mers Profile (MKP) Clusters
3. Results
3.1. The High-Risk Group of Patients Defined by the COG Criterion
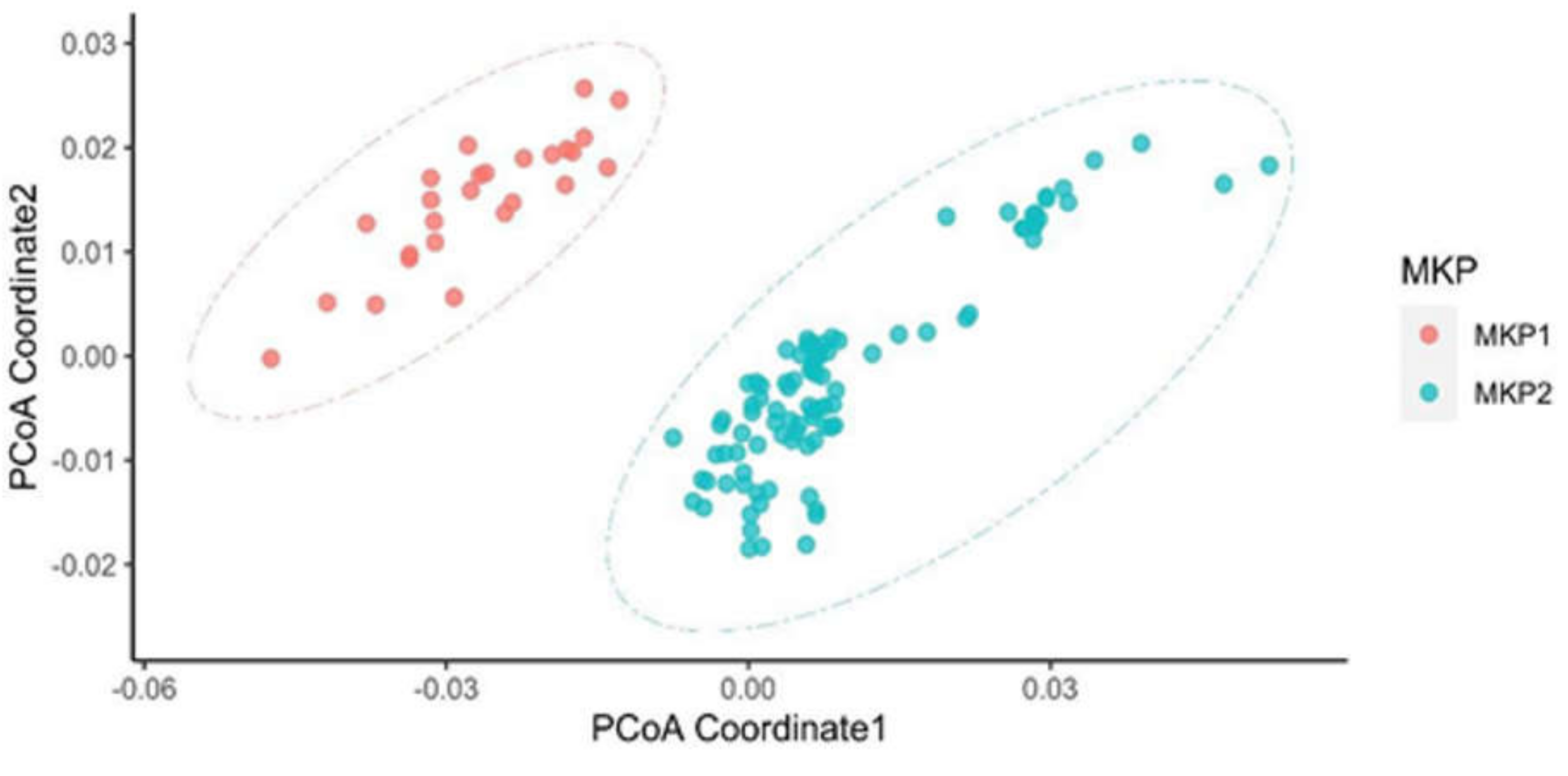
3.2. Distinct Microbiota Was Found Among Neuroblastoma Patients
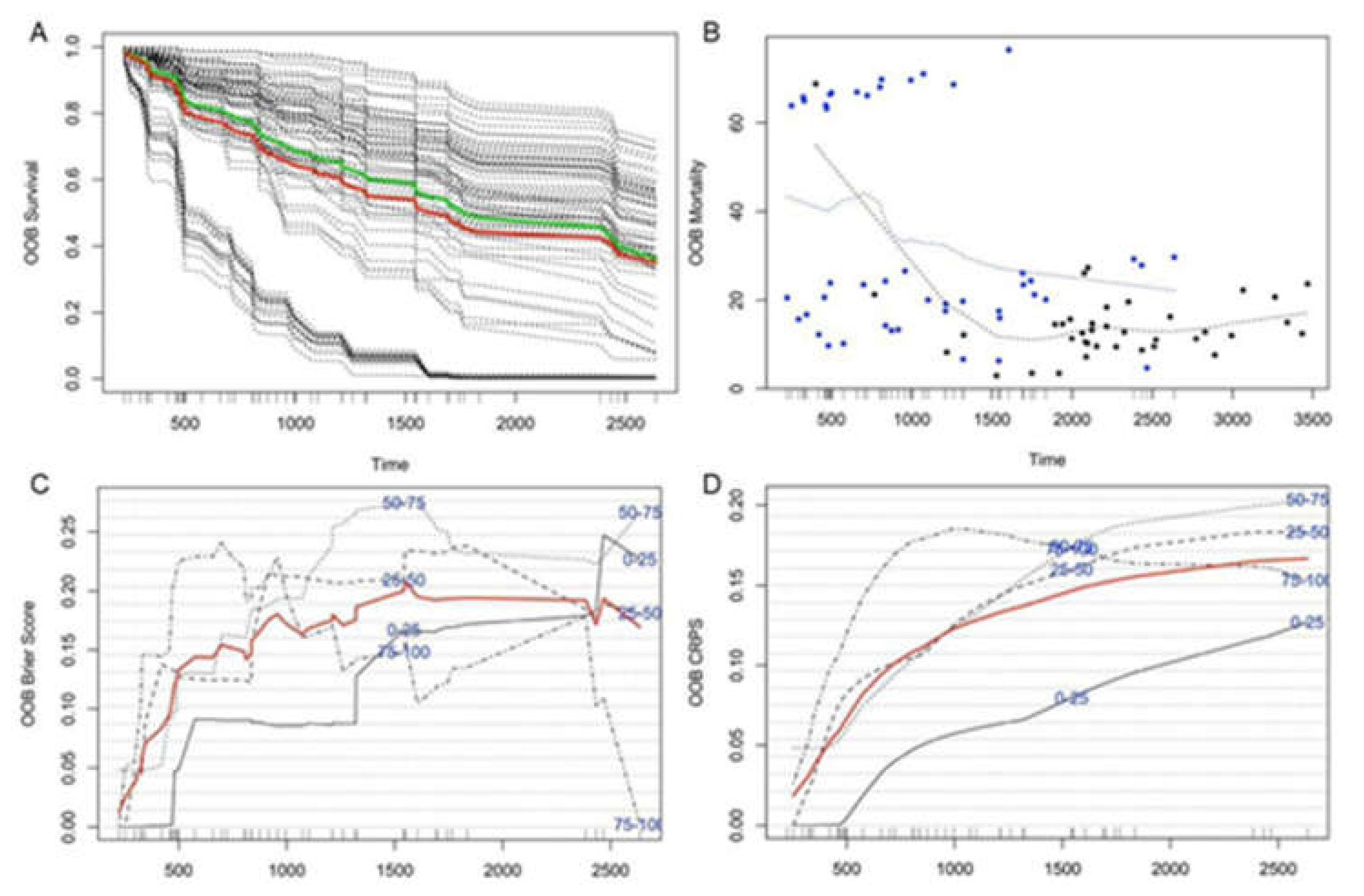
3.3. The Patient’s Survival Time Is Associated with Microbial Gene Abundance
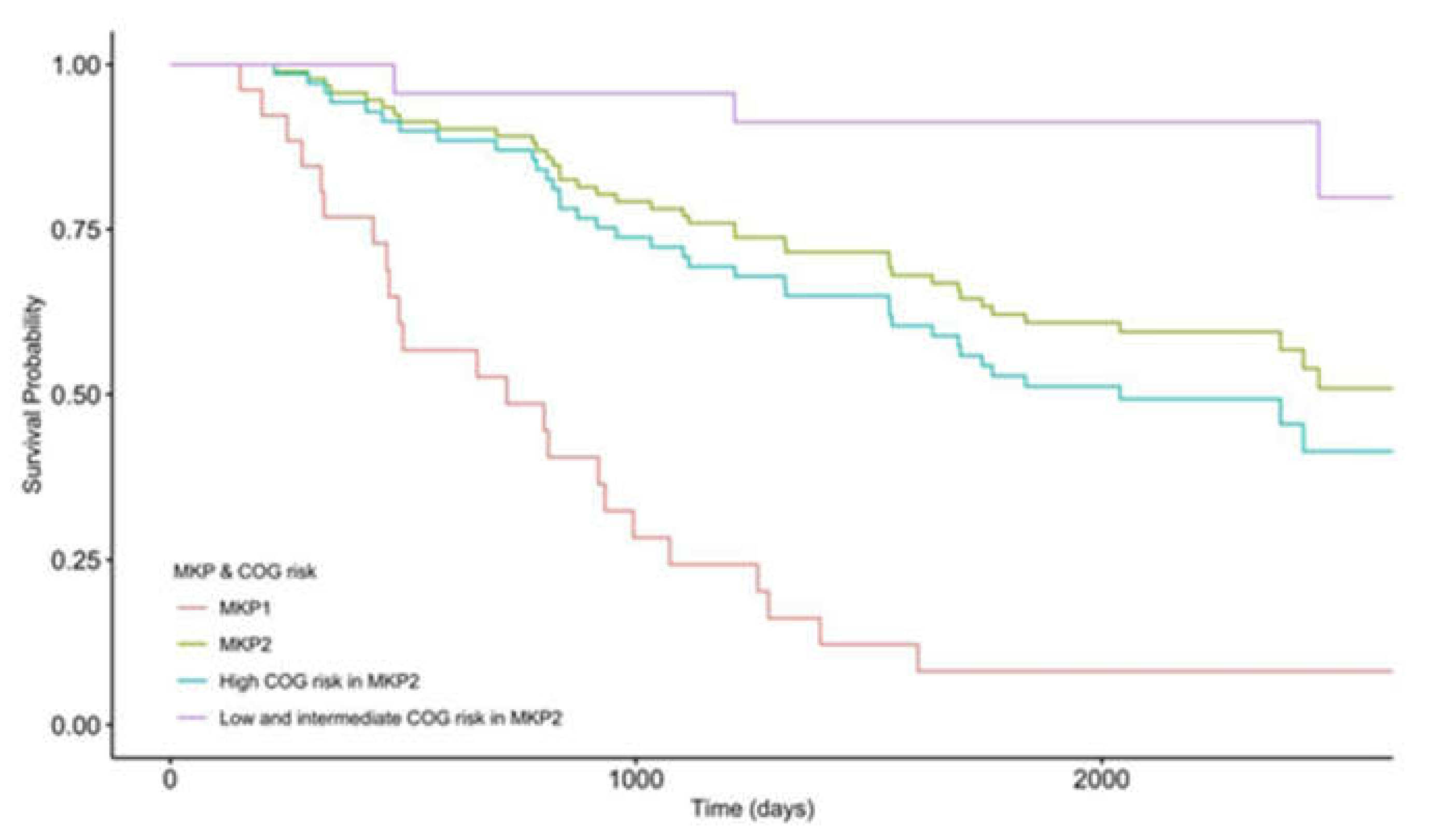
3.4. High COG Risk Patients Were Further Separated into High- and Low-Risk Groups with Differential Survival Rates
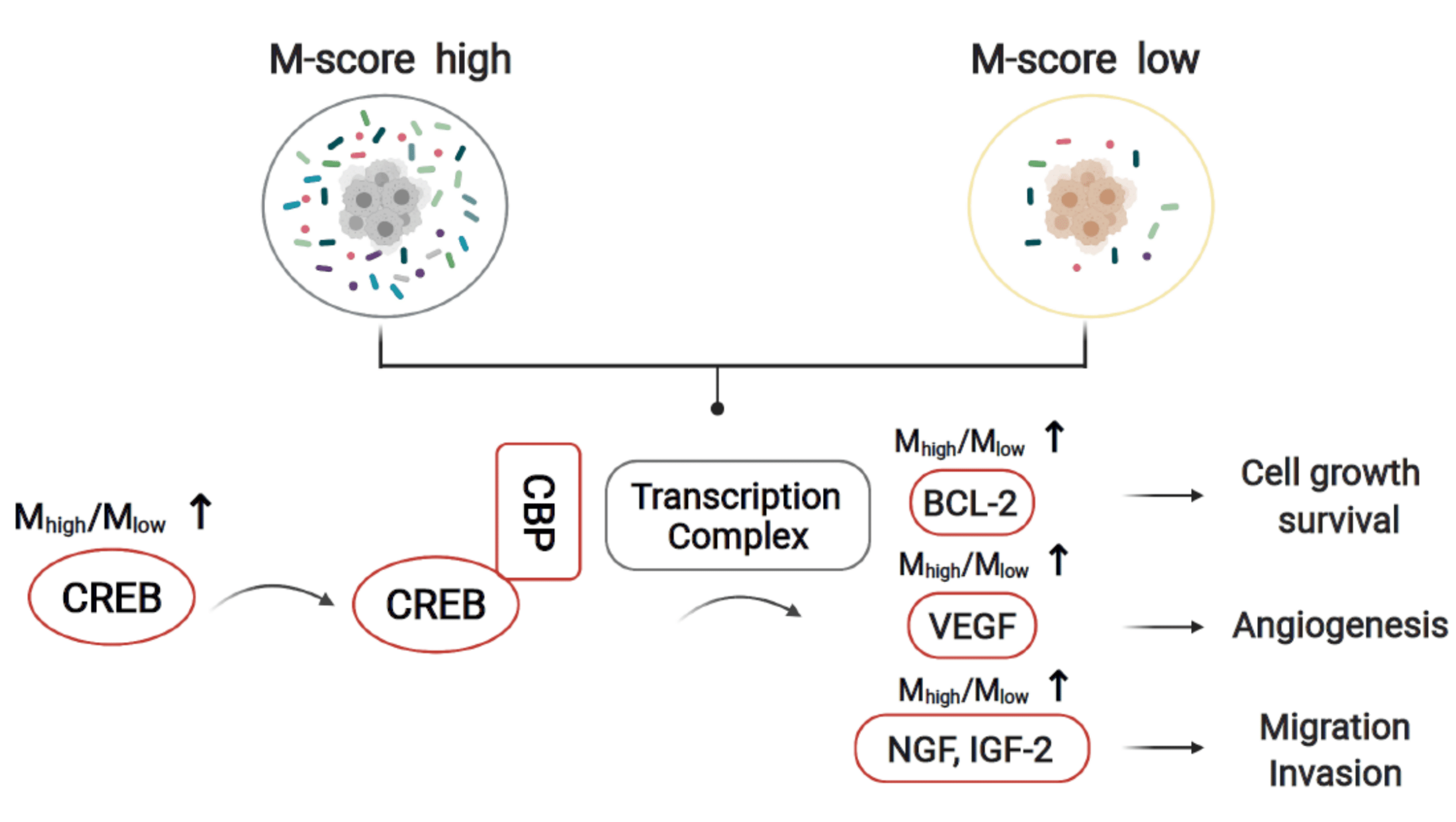
3.5. The Molecular Crosstalk between Intracellular Microbiota and Tumor Microenvironment Activates CREB and Improves Survival Probability
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| COG | Children’s Oncology Group |
| OS | Overall Survival |
| INPC | International Neuroblastoma Pathology Classification |
| NCI | National Cancer Institute |
| TARGET | Therapeutically Applicable Research To Generate Effective Treatments |
| MKP | Microbiome K-mers Profile |
| OOB | Out-Of-Bag |
| CRPS | Continuous Ranked Probability Score |
References
- Lee, L.X.; Li, S.C. Hunting down the dominating subclone of cancer stem cells as a potential new therapeutic target in multiple myeloma. An artificial intelligence perspective. World J. Stem. Cells 2020, 12, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Eggert, A.; Caron, H. Neuroblastoma: Biology, prognosis, and treatment. Pediatr. Clin. N. Am. 2008, 55, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Sokol, E.; Desai, A.V. The Evolution of Risk Classification for Neuroblastoma. Children 2019, 6, 27. [Google Scholar] [CrossRef]
- Cotterill, S.J.; Pearson, A.D.J.; Pritchard, J.; Foot, A.B.M.; Roald, B.; Kohler, J.A.; Imeson, J. Clinical prognostic factors in 1277 patients with neuroblastoma: Results of The European Neuroblastoma Study Group ‘Survey’ 1982–1992. Eur. J. Cancer 2000, 36, 901–908. [Google Scholar] [CrossRef]
- Moroz, V.; Machin, D.; Faldum, A.; Hero, B.; Iehara, T.; Mosseri, V.; Ladenstein, R.; De Bernardi, B.; Rubie, H.; Berthold, F.; et al. Changes over three decades in outcome and the prognostic influence of age-at-diagnosis in young patients with neuroblastoma: A report from the International Neuroblastoma Risk Group Project. Eur. J. Cancer 2011, 47, 561–571. [Google Scholar] [CrossRef]
- Ikeda, H.; Iehara, T.; Tsuchida, Y.; Kaneko, M.; Hata, J.; Naito, H.; Iwafuchi, M.; Ohnuma, N.; Mugishima, H.; Toyoda, Y.; et al. Experience with International Neuroblastoma Staging System and Pathology Classification. Br. J. Cancer 2002, 86, 1110–1116. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Livyatan, I.; Nejman, D.; Shental, N.; Straussman, R. Characterization of the human tumor microbiome reveals tumor-type specific intra-cellular bacteria. OncoImmunology 2020, 9, 1800957. [Google Scholar] [CrossRef] [PubMed]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Crabtree, J.; Mattick, J.S.; Anderson, K.E.; Dunning Hotopp, J.C. Distinguishing potential bacteria-tumor associations from contamination in a secondary data analysis of public cancer genome sequence data. Microbiome 2017, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-J. Segmented K-mer and its application on similarity analysis of mitochondrial genome sequences. Gene 2013, 518, 419–424. [Google Scholar] [CrossRef]
- Zhu, Z.; Ren, J.; Michail, S.; Sun, F. MicroPro: Using metagenomic unmapped reads to provide insights into human microbiota and disease associations. Genome Biol. 2019, 20, 154. [Google Scholar] [CrossRef]
- Sarmashghi, S.; Bohmann, K.; Gilbert, M.T.P.; Bafna, V.; Mirarab, S. Skmer. Assembly-free and alignment-free sample identification using genome skims. Genome Biol. 2019, 20, 34. [Google Scholar] [CrossRef]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Ishwaran, H.; Kogalur, U. randomForestSRC. Random Forests for Survival, Regression and Classification (RF-SRC). 2016. Available online: https://cran.r-project.org/web/packages/randomForestSRC/randomForestSRC.pdf (accessed on 1 May 2022).
- Segal, M.R. Regression Trees for Censored Data. Biometrics 1988, 44, 35–47. [Google Scholar] [CrossRef]
- Kaplan, E.L.; Meier, P. Nonparametric Estimation from Incomplete Observations. J. Am. Stat. Assoc. 1958, 53, 457–481. [Google Scholar] [CrossRef]
- Zhong, X.; Liu, Y.; Liu, H.; Zhang, Y.; Wang, L.; Zhang, H. Identification of Potential Prognostic Genes for Neuroblastoma. Front. Genet. 2018, 9, 589. [Google Scholar] [CrossRef] [PubMed]
- Vogtmann, E.; Goedert, J.J. Epidemiologic studies of the human microbiome and cancer. Br. J. Cancer 2016, 114, 237–242. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef]
- Shankar, D.B.; Cheng, J.C.; Kinjo, K.; Federman, N.; Moore, T.B.; Gill, A.; Rao, N.P.; Landaw, E.M.; Sakamoto, K.M. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell 2005, 7, 351–362. [Google Scholar] [CrossRef]
- Wu, D.; Zhau, H.E.; Huang, W.C.; Iqbal, S.; Habib, F.K.; Sartor, O.; Cvitanovic, L.; Marshall, F.F.; Xu, Z.; Chung, L.W.K. cAMP-responsive element-binding protein regulates vascular endothelial growth factor expression: Implication in human prostate cancer bone metastasis. Oncogene 2007, 26, 5070–5077. [Google Scholar] [CrossRef]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- Xiao, X.; Li, B.X.; Mitton, B.; Ikeda, A.; Sakamoto, K.M. Targeting CREB for cancer therapy: Friend or foe. Curr. Cancer Drug Targets 2010, 10, 384–391. [Google Scholar] [CrossRef]
- Conkright, M.D.; Montminy, M. CREB: The unindicted cancer co-conspirator. Trends Cell Biol. 2005, 15, 457–459. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Frank, D.A. CREB in the pathophysiology of cancer: Implications for targeting transcription factors for cancer therapy. Clin. Cancer Res. 2009, 15, 2583–2587. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, S.; Pigazzi, M.; Sakamoto, K.M. CREB: A Key Regulator of Normal and Neoplastic Hematopoiesis. Adv. Hematol. 2009, 2009, 634292. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | N (%) |
|---|---|
| Gender | |
| Male | 70 (58.3) |
| Female | 50 (41.7) |
| Ethnicity | |
| White | 85 (70.8) |
| Others | 35 (29.2) |
| MKI | |
| Low | 35 (29.2) |
| Intermediate | 34 (28.3) |
| High | 26 (21.7) |
| Unknown | 25 (20.8) |
| MYCN Status | |
| Amplified | 23 (19.2) |
| Not Amplified | 96 (80) |
| Unknown | 1 (0.8) |
| COG Risk | |
| Low Risk | 12 (10.0) |
| Intermediate Risk | 11 (9.2) |
| High Risk | 97 (80.8) |
| Location of tumor | |
| Abdomen | 104 (86.7) |
| Others | 16 (13.3) |
| Mean (SD) | |
| Age(in years) | 4.3 (2.5) |
| Survival Time(in days) | |
| Event | 1009.2 (617.2) |
| Censored | 2204.5 (734.5) |
| Variables | p-Value |
|---|---|
| MKP Clusters | 9.505 × 10−8 |
| Gender | 0.6899 |
| MKI | 0.0556 |
| MYCN Status | 0.2449 |
| COG Risk | 2.659 × 10−5 |
| Location | 0.9878 |
| Ethnicity | 0.5443 |
| Variables | Chi-Square (df) | p-Value |
|---|---|---|
| Gender | 0.0898(1) | 0.7645 |
| Ethnicity | 0.1997(1) | 0.655 |
| MKI | 5.0892(3) | 0.1654 |
| MYCN Status | 0.6865(1) | 0.4074 |
| COG Risk | 7.8701(2) | 0.0195 |
| Location of tumor | 0.0005(1) | 0.9827 |
| Variables | p-Value | Hazard Ratio |
|---|---|---|
| MKP1 vs. MKP2 | 9.505 × 10−8 | 5 |
| MKP1 vs. COG high risk in MKP2 | 6.42210−6 | 3.78 |
| MKP1 vs. COG low and intermediate risk in MKP2 | 4.60510−9 | 17.1 |
| MKP2 vs. COG high risk in MKP2 | 0.2119 | 0.75 |
| MKP2 vs. COG low and intermediate risk in MKP2 | 0.0041 | 4.07 |
| COG high risk in MKP2 vs. COG low/intermediate risk in MKP2 | 0.0004 | 5.56 |
| Variables | Error Rate (%) |
|---|---|
| Microbial Gene Abundance | 29.87 |
| Gender | 71.67 |
| MKI | 53.65 |
| MYCN Status | 75.21 |
| COG Risk | 68.97 |
| Location of tumor | 82.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Wang, X.; Huang, R.; Stucky, A.; Chen, X.; Sun, L.; Wen, Q.; Zeng, Y.; Fletcher, H.; Wang, C.; et al. The Machine-Learning-Mediated Interface of Microbiome and Genetic Risk Stratification in Neuroblastoma Reveals Molecular Pathways Related to Patient Survival. Cancers 2022, 14, 2874. https://doi.org/10.3390/cancers14122874
Li X, Wang X, Huang R, Stucky A, Chen X, Sun L, Wen Q, Zeng Y, Fletcher H, Wang C, et al. The Machine-Learning-Mediated Interface of Microbiome and Genetic Risk Stratification in Neuroblastoma Reveals Molecular Pathways Related to Patient Survival. Cancers. 2022; 14(12):2874. https://doi.org/10.3390/cancers14122874
Chicago/Turabian StyleLi, Xin, Xiaoqi Wang, Ruihao Huang, Andres Stucky, Xuelian Chen, Lan Sun, Qin Wen, Yunjing Zeng, Hansel Fletcher, Charles Wang, and et al. 2022. "The Machine-Learning-Mediated Interface of Microbiome and Genetic Risk Stratification in Neuroblastoma Reveals Molecular Pathways Related to Patient Survival" Cancers 14, no. 12: 2874. https://doi.org/10.3390/cancers14122874
APA StyleLi, X., Wang, X., Huang, R., Stucky, A., Chen, X., Sun, L., Wen, Q., Zeng, Y., Fletcher, H., Wang, C., Xu, Y., Cao, H., Sun, F., Li, S. C., Zhang, X., & Zhong, J. F. (2022). The Machine-Learning-Mediated Interface of Microbiome and Genetic Risk Stratification in Neuroblastoma Reveals Molecular Pathways Related to Patient Survival. Cancers, 14(12), 2874. https://doi.org/10.3390/cancers14122874