
Current and Future Immunotherapy-Based Treatments for Oesophageal Cancers

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Benefits and Efficacy of Immunotherapy in Oesophageal Cancer
2.1. Understanding the Complex Tumour Environment
2.2. Promising Start with Checkpoint Inhibitors
2.3. Checkpoint Proteins Associated with Oesophageal Cancer and Disease Progression
2.3.1. CTLA-4
2.3.2. PD-1
2.3.3. TIM 3
2.3.4. T-Cell Immunoreceptor with Ig and ITIM Domains (TIGIT)
2.3.5. LAG-3
2.4. Future for New and Effective Checkpoint Immunotherapies
2.5. Looking towards a Multi-Targeted Approach
2.6. Beyond Checkpoint Blockade Therapy
2.7. Problems with Checkpoint Blockade
2.8. Vaccine Technology in Antitumour Therapy
2.9. Adoptive Cellular Therapies
3. CAR-T Therapy
4. TIL Therapy
4.1. Novel Immune Cell Targets
4.2. Agonistic Immunostimulatory Antibody Therapy
4.3. Altering the Metabolic Tumour Microenvironment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ho, A.L.K.; Smyth, E.C. A global perspective on oesophageal cancer: Two diseases in one. Lancet Gastroenterol. Hepatol. 2020, 5, 521–522. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Uhlenhopp, D.; Then, E.; Sunkara, T.; Gaduputi, V. Epidemiology of esophageal cancer: Update in global trends, etiology and risk factors. Clin. J. Gastroenterol. 2020, 13, 1010–1021. [Google Scholar] [CrossRef]
- Thrift, A.P. Global burden and epidemiology of Barrett oesophagus and oesophageal cancer. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 432–443. [Google Scholar] [CrossRef]
- Huang, J.; Koulaouzidis, A.; Marlicz, W.; Lok, V.; Chu, C.; Ngai, C.H.; Zhang, L.; Chen, P.; Wang, S.; Yuan, J.; et al. Global Burden, Risk Factors, and Trends of Esophageal Cancer: An Analysis of Cancer Registries from 48 Countries. Cancers 2021, 13, 141. [Google Scholar] [CrossRef]
- Bray, F. Cancer Incidence in Five Continents: Volume XI; International Agency for Research on Cancer: Lyon, France, 2021.
- Xie, S.H.; Lagergren, J. Risk factors for oesophageal cancer. Best Pract. Res. Clin. Gastroenterol. 2018, 36–37, 3–8. [Google Scholar] [CrossRef]
- Medical Research Council Oesophageal Cancer Working Group. Surgical resection with or without preoperative chemotherapy in oesophageal cancer: A randomised controlled trial. Lancet 2002, 359, 1727–1733. [Google Scholar] [CrossRef]
- CRUK. Oesophageal Cancer Treatment Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/oesophageal-cancer/diagnosis-and-treatment#ref-52017 (accessed on 5 May 2020).
- CRUK. Proportion Diagnosed by Stage. Available online: https://crukcancerintelligence.shinyapps.io/EarlyDiagnosis/2019 (accessed on 5 May 2020).
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235. [Google Scholar] [CrossRef]
- Blattman, J.N.; Greenberg, P.D. Cancer immunotherapy: A treatment for the masses. Science 2004, 305, 200–205. [Google Scholar] [CrossRef]
- Franklin, C.; Livingstone, E.; Roesch, A.; Schilling, B.; Schadendorf, D. Immunotherapy in melanoma: Recent advances and future directions. Eur. J. Surg. Oncol. 2017, 43, 604–611. [Google Scholar] [CrossRef]
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10, 2040620719841581. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]
- Martinez-Perez, E.; Molina-Vila, M.A.; Marino-Buslje, C. Panels and models for accurate prediction of tumor mutation burden in tumor samples. NPJ Precis. Oncol. 2021, 5, 31. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, J.; Du, C.; Wu, Y.; Xia, D.; Lv, W.; Hu, J. The Predictive Value of Tumor Mutation Burden on Efficacy of Immune Checkpoint Inhibitors in Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2019, 9, 1161. [Google Scholar] [CrossRef] [Green Version]
- Davern, M.; Donlon, N.E.; Power, R.; Hayes, C.; King, R.; Dunne, M.R.; Reynolds, J.V. The tumour immune microenvironment in oesophageal cancer. Br. J. Cancer 2021, 125, 479–494. [Google Scholar] [CrossRef]
- Power, R.; Lowery, M.A.; Reynolds, J.V.; Dunne, M.R. The Cancer-Immune Set Point in Oesophageal Cancer. Front. Oncol. 2020, 10, 891. [Google Scholar] [CrossRef]
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel cancer immunotherapy agents with survival benefit: Recent successes and next steps. Nat. Rev. Cancer 2011, 11, 805–812. [Google Scholar] [CrossRef]
- Brunet, J.F.; Denizot, F.; Luciani, M.F.; Roux-Dosseto, M.; Suzan, M.; Mattei, M.G.; Golstein, P. A new member of the immunoglobulin superfamily--CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirschl, C.J.; Drake, C.G. Molecular pathways: Coexpression of immune checkpoint molecules: Signaling pathways and implications for cancer immunotherapy. Clin. Cancer Res. 2013, 19, 4917–4924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, R.J.; Ajani, J.A.; Kuzdzal, J.; Zander, T.; Van Cutsem, E.; Piessen, G.; Mendez, G.; Feliciano, J.; Motoyama, S.; Lièvre, A.; et al. Adjuvant Nivolumab in Resected Esophageal or Gastroesophageal Junction Cancer. N. Engl. J. Med. 2021, 384, 1191–1203. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef]
- Wagener-Ryczek, S.; Schoemmel, M.; Kraemer, M.; Bruns, C.; Schroeder, W.; Zander, T.; Gebauer, F.; Alakus, H.; Merkelbach-Bruse, S.; Buettner, R.; et al. Immune profile and immunosurveillance in treatment-naive and neoadjuvantly treated esophageal adenocarcinoma. Cancer Immunol. Immunother. 2020, 69, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Linsley, P.S.; Ledbetter, J.A. The role of the CD28 receptor during T cell responses to antigen. Annu. Rev. Immunol. 1993, 11, 191–212. [Google Scholar] [CrossRef]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. Pillars article: CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994. 1: 405–413. J. Immunol. 2011, 187, 3466–3474. [Google Scholar]
- Oyewole-Said, D.; Konduri, V.; Vazquez-Perez, J.; Weldon, S.A.; Levitt, J.M.; Decker, W.K. Beyond T-Cells: Functional Characterization of CTLA-4 Expression in Immune and Non-Immune Cell Types. Front. Immunol. 2020, 11, 608024. [Google Scholar] [CrossRef]
- Wang, W.; Chen, D.; Zhao, Y.; Zhao, T.; Wen, J.; Mao, Y.; Chen, C.; Sang, Y.; Zhang, Y.; Chen, Y. Characterization of LAG-3, CTLA-4, and CD8. Ann. Transl. Med. 2019, 7, 776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Pan, K.; Weng, D.S.; Chen, C.L.; Wang, Q.J.; Zhao, J.J.; Pan, Q.Z.; Liu, Q.; Jiang, S.S.; Li, Y.Q.; et al. Cytotoxic T lymphocyte antigen-4 expression in esophageal carcinoma: Implications for prognosis. Oncotarget 2016, 7, 26670–26679. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Däster, S.; Eppenberger-Castori, S.; Mele, V.; Schäfer, H.M.; Schmid, L.; Weixler, B.; Soysal, S.D.; Droeser, R.A.; Spagnoli, G.C.; Kettelhack, C.; et al. Low Expression of Programmed Death 1 (PD-1), PD-1 Ligand 1 (PD-L1), and Low CD8+ T Lymphocyte Infiltration Identify a Subgroup of Patients With Gastric and Esophageal Adenocarcinoma With Severe Prognosis. Front. Med. (Lausanne) 2020, 7, 144. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Saeki, H.; Nakashima, Y.; Ito, S.; Oki, E.; Morita, M.; Oda, Y.; Okano, S.; Maehara, Y. Programmed death-ligand 1 expression at tumor invasive front is associated with epithelial-mesenchymal transition and poor prognosis in esophageal squamous cell carcinoma. Cancer Sci. 2017, 108, 1119–1127. [Google Scholar] [CrossRef]
- Cui, H.; Li, Y.; Li, S.; Liu, G. Prognostic Function of Programmed Cell Death-Ligand 1 in Esophageal Squamous Cell Carcinoma Patients Without Preoperative Therapy: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 693886. [Google Scholar] [CrossRef]
- Qu, H.X.; Zhao, L.P.; Zhan, S.H.; Geng, C.X.; Xu, L.; Xin, Y.N.; Jiang, X.J. Clinicopathological and prognostic significance of programmed cell death ligand 1 (PD-L1) expression in patients with esophageal squamous cell carcinoma: A meta-analysis. J. Thorac. Dis. 2016, 8, 3197–3204. [Google Scholar] [CrossRef] [Green Version]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef]
- Anderson, A.C. Tim-3: An emerging target in the cancer immunotherapy landscape. Cancer Immunol. Res. 2014, 2, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Shan, B.; Man, H.; Liu, J.; Wang, L.; Zhu, T.; Ma, M.; Xv, Z.; Chen, X.; Yang, X.; Li, P. TIM-3 promotes the metastasis of esophageal squamous cell carcinoma by targeting epithelial-mesenchymal transition via the Akt/GSK-3β/Snail signaling pathway. Oncol. Rep. 2016, 36, 1551–1561. [Google Scholar] [CrossRef] [Green Version]
- Blake, S.J.; Dougall, W.C.; Miles, J.J.; Teng, M.W.; Smyth, M.J. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 5183–5188. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Joller, N.; Hafler, J.P.; Brynedal, B.; Kassam, N.; Spoerl, S.; Levin, S.D.; Sharpe, A.H.; Kuchroo, V.K. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 2011, 186, 1338–1342. [Google Scholar] [CrossRef] [Green Version]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [Green Version]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.M.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8, 1014. [Google Scholar] [CrossRef]
- Workman, C.J.; Cauley, L.S.; Kim, I.J.; Blackman, M.A.; Woodland, D.L.; Vignali, D.A. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J. Immunol. 2004, 172, 5450–5455. [Google Scholar] [CrossRef]
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; De Marzo, A.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Investig. 2007, 117, 3383–3392. [Google Scholar] [CrossRef] [Green Version]
- Kojima, T.; Shah, M.A.; Muro, K.; Francois, E.; Adenis, A.; Hsu, C.H.; Doi, T.; Moriwaki, T.; Kim, S.B.; Lee, S.H.; et al. Randomized Phase III KEYNOTE-181 Study of Pembrolizumab Versus Chemotherapy in Advanced Esophageal Cancer. J. Clin. Oncol. 2020, 38, 4138–4148. [Google Scholar] [CrossRef]
- Kato, K.; Cho, B.C.; Takahashi, M.; Okada, M.; Lin, C.Y.; Chin, K.; Kadowaki, S.; Ahn, M.J.; Hamamoto, Y.; Doki, Y.; et al. Nivolumab versus chemotherapy in patients with advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION-3): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 1506–1517. [Google Scholar] [CrossRef]
- Shen, L.; Kato, K.; Kim, S.-B.; Ajani, J.A.; Zhao, K.; He, Z.; Yu, X.; Shu, Y.; Luo, Q.; Wang, J.; et al. RATIONALE 302: Randomized, phase 3 study of tislelizumab versus chemotherapy as second-line treatment for advanced unresectable/metastatic esophageal squamous cell carcinoma. In Proceedings of the 2021 ASCO Annual Meeting I, Chicago, IL, USA, 4–8 June 2021; Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.15_suppl.4012 (accessed on 20 May 2020).
- Huang, J.; Xu, J.; Chen, Y.; Zhuang, W.; Zhang, Y.; Chen, Z.; Chen, J.; Zhang, H.; Niu, Z.; Fan, Q.; et al. Camrelizumab versus investigator’s choice of chemotherapy as second-line therapy for advanced or metastatic oesophageal squamous cell carcinoma (ESCORT): A multicentre, randomised, open-label, phase 3 study. Lancet Oncol. 2020, 21, 832–842. [Google Scholar] [CrossRef]
- Sun, J.M.; Shen, L.; Shah, M.A.; Enzinger, P.; Adenis, A.; Doi, T.; Kojima, T.; Metges, J.P.; Li, Z.; Kim, S.B.; et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (KEYNOTE-590): A randomised, placebo-controlled, phase 3 study. Lancet 2021, 398, 759–771. [Google Scholar] [CrossRef]
- Bang, Y.J.; Ruiz, E.Y.; Van Cutsem, E.; Lee, K.W.; Wyrwicz, L.; Schenker, M.; Alsina, M.; Ryu, M.H.; Chung, H.C.; Evesque, L.; et al. Phase III, randomised trial of avelumab versus physician’s choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: Primary analysis of JAVELIN Gastric 300. Ann. Oncol. 2018, 29, 2052–2060. [Google Scholar] [CrossRef]
- Shitara, K.; Özgüroğlu, M.; Bang, Y.J.; Di Bartolomeo, M.; Mandalà, M.; Ryu, M.H.; Fornaro, L.; Olesiński, T.; Caglevic, C.; Chung, H.C.; et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): A randomised, open-label, controlled, phase 3 trial. Lancet 2018, 392, 123–133. [Google Scholar] [CrossRef]
- Sundar, R.; Smyth, E.C.; Peng, S.; Yeong, J.P.S.; Tan, P. Predictive Biomarkers of Immune Checkpoint Inhibition in Gastroesophageal Cancers. Front. Oncol. 2020, 10, 763. [Google Scholar] [CrossRef]
- Yuki, M.; Toriyama, K.; Kadowaki, S.; Ogata, T.; Nakazawa, T.; Kato, K.; Nozawa, K.; Narita, Y.; Honda, K.; Masuishi, T.; et al. Impact of PD-L1 Combined Positive Score (CPS) on Clinical Response to Nivolumab in Patients with Advanced Esophageal Squamous Cell Carcinoma. 2021 ASCO Annual Meeting I. Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.15_suppl.e160452021 (accessed on 20 May 2022).
- Paterson, A.L.; Shannon, N.B.; Lao-Sirieix, P.; Ong, C.A.; Peters, C.J.; O’Donovan, M.; Fitzgerald, R.C. A systematic approach to therapeutic target selection in oesophago-gastric cancer. Gut 2013, 62, 1415–1424. [Google Scholar] [CrossRef]
- Findlay, J.M.; Middleton, M.R.; Tomlinson, I. A systematic review and meta-analysis of somatic and germline DNA sequence biomarkers of esophageal cancer survival, therapy response and stage. Ann. Oncol. 2015, 26, 624–644. [Google Scholar] [CrossRef]
- Zhou, X.W.; Qu, M.Y.; Tebon, P.; Jiang, X.; Wang, C.R.; Xue, Y.M.; Zhu, J.X.; Zhang, S.M.; Oklu, R.; Sengupta, S.; et al. Screening Cancer Immunotherapy: When Engineering Approaches Meet Artificial Intelligence. Adv. Sci. 2020, 7, 2001447. [Google Scholar] [CrossRef]
- Deng, X.; Nakamura, Y. Cancer Precision Medicine: From Cancer Screening to Drug Selection and Personalized Immunotherapy. Trends Pharmacol. Sci. 2017, 38, 15–24. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Xiong, M.; Liu, R.; Lei, X.; Fan, D.; Lin, F.; Hao, W.; Yuan, X.; Yang, Y.; Zhang, X.; Ye, Z.; et al. A Novel CD3/BCMA Bispecific T-cell Redirecting Antibody for the Treatment of Multiple Myeloma. J. Immunother. 2022, 45, 78–88. [Google Scholar] [CrossRef]
- Yang, Y.; Fang, W.; Huang, Y.; Li, X.; Huang, S.; Wu, J.; Li, Y.; Baoping, C.; Hu, S.; Yang, S.; et al. A phase 2, open-label, multicenter study to evaluate the efficacy, safety, and tolerability of KN046 in combination with chemotherapy in subjects with advanced non-small cell lung cancer. J. Clin. Oncol. 2021, 39, 9060. [Google Scholar] [CrossRef]
- Temel, J.S.; Gainor, J.F.; Sullivan, R.J.; Greer, J.A. Keeping Expectations in Check With Immune Checkpoint Inhibitors. J. Clin. Oncol. 2018, 36, 1654–1657. [Google Scholar] [CrossRef]
- Gu, Y.M.; Zhuo, Y.; Chen, L.Q.; Yuan, Y. The Clinical Application of Neoantigens in Esophageal Cancer. Front. Oncol. 2021, 11, 703517. [Google Scholar] [CrossRef]
- Fujita, S.; Wada, H.; Jungbluth, A.A.; Sato, S.; Nakata, T.; Noguchi, Y.; Doki, Y.; Yasui, M.; Sugita, Y.; Yasuda, T.; et al. NY-ESO-1 expression and immunogenicity in esophageal cancer. Clin. Cancer Res. 2004, 10, 6551–6558. [Google Scholar] [CrossRef] [Green Version]
- Raza, A.; Merhi, M.; Inchakalody, V.P.; Krishnankutty, R.; Relecom, A.; Uddin, S.; Dermime, S. Unleashing the immune response to NY-ESO-1 cancer testis antigen as a potential target for cancer immunotherapy. J. Transl. Med. 2020, 18, 140. [Google Scholar] [CrossRef] [Green Version]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 569. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer vaccines as promising immuno-therapeutics: Platforms and current progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, T.; Rezvani, K. Adoptive cell therapy: Living drugs against cancer. J. Exp. Med. 2020, 217, e20200377. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.E.; Badie, B. Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front. Immunol. 2017, 8, 1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, A.J.; Chen, L.C.; Chen, Y.Y. Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 2021, 20, 531–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L. Expression of cancer-testis antigens in esophageal cancer and their progress in immunotherapy. J. Cancer Res. Clin. Oncol. 2019, 145, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Cai, S.; Wang, L.; Zhang, X.; Li, W.; Cao, X. Analysis of the function of MAGE-A in esophageal carcinoma by bioinformatics. Medicine 2019, 98, e15774. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94 (Suppl. S1), S3–S9. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Riddell, S.R. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015, 36, 494–502. [Google Scholar] [CrossRef] [Green Version]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Kelderman, S.; Heemskerk, B.; Fanchi, L.; Philips, D.; Toebes, M.; Kvistborg, P.; van Buuren, M.M.; van Rooij, N.; Michels, S.; Germeroth, L.; et al. Antigen-specific TIL therapy for melanoma: A flexible platform for personalized cancer immunotherapy. Eur. J. Immunol. 2016, 46, 1351–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Berg, J.H.; Heemskerk, B.; van Rooij, N.; Gomez-Eerland, R.; Michels, S.; van Zon, M.; de Boer, R.; Bakker, N.A.M.; Jorritsma-Smit, A.; van Buuren, M.M.; et al. Tumor infiltrating lymphocytes (TIL) therapy in metastatic melanoma: Boosting of neoantigen-specific T cell reactivity and long-term follow-up. J. Immunother. Cancer 2020, 8, e000848. [Google Scholar] [CrossRef] [PubMed]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Li, J.; Xie, Y.; Wang, X.; Li, F.; Li, S.; Li, M.; Peng, H.; Yang, L.; Liu, C.; Pang, L.; et al. Prognostic impact of tumor-associated macrophage infiltration in esophageal cancer: A meta-analysis. Future Oncol. 2019, 15, 2303–2317. [Google Scholar] [CrossRef] [PubMed]
- Sumitomo, R.; Hirai, T.; Fujita, M.; Murakami, H.; Otake, Y.; Huang, C.L. M2 tumor-associated macrophages promote tumor progression in non-small-cell lung cancer. Exp. Ther. Med. 2019, 18, 4490–4498. [Google Scholar] [CrossRef] [Green Version]
- Verneau, J.; Sautés-Fridman, C.; Sun, C.M. Dendritic cells in the tumor microenvironment: Prognostic and theranostic impact. Semin. Immunol. 2020, 48, 101410. [Google Scholar] [CrossRef]
- Amigorena, S.; Savina, A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr. Opin. Immunol. 2010, 22, 109–117. [Google Scholar] [CrossRef]
- Bobryshev, Y.V.; Tran, D.; Killingsworth, M.C.; Buckland, M.; Lord, R.V. Dendritic cells in Barrett’s esophagus and esophageal adenocarcinoma. J. Gastrointest. Surg. 2009, 13, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Smits, E.L.; Stein, B.; Nijs, G.; Lion, E.; Van Tendeloo, V.F.; Willemen, Y.; Anguille, S.; Berneman, Z.N. Generation and Cryopreservation of Clinical Grade Wilms’ Tumor 1 mRNA-Loaded Dendritic Cell Vaccines for Cancer Immunotherapy. Methods Mol. Biol. 2016, 1393, 27–35. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X.; Zhou, X.; Lyerly, H.K.; Morse, M.A.; Ren, J. DC-CIK as a widely applicable cancer immunotherapy. Expert Opin. Biol. Ther. 2020, 20, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, J.; Zhang, G.; Song, L.; Su, Y.; Shi, Y.; Zhang, M.; He, J.; Song, D.; Lv, F.; et al. 5 years of clinical DC-CIK/NK cells immunotherapy for acute myeloid leukemia—A summary. Immunotherapy 2020, 12, 63–74. [Google Scholar] [CrossRef] [PubMed]
- White, M.; Hayden, A.; Derouet, M.; Thomas, G.; Primrose, J.; Underwood, T. Correlation of cancer-associated fibroblasts with tumour cell invasion and chemoresistance in oesophageal adenocarcinoma. Lancet 2014, 383, 108. [Google Scholar] [CrossRef]
- Van Kooten, C.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17. [Google Scholar] [CrossRef]
- Enell Smith, K.; Deronic, A.; Hägerbrand, K.; Norlén, P.; Ellmark, P. Rationale and clinical development of CD40 agonistic antibodies for cancer immunotherapy. Expert Opin. Biol. Ther. 2021, 21, 1635–1646. [Google Scholar] [CrossRef]
- Van Hooren, L.; Vaccaro, A.; Ramachandran, M.; Vazaios, K.; Libard, S.; van de Walle, T.; Georganaki, M.; Huang, H.; Pietilä, I.; Lau, J.; et al. Agonistic CD40 therapy induces tertiary lymphoid structures but impairs responses to checkpoint blockade in glioma. Nat. Commun. 2021, 12, 4127. [Google Scholar] [CrossRef]
- Byrne, K.T.; Betts, C.B.; Mick, R.; Sivagnanam, S.; Bajor, D.L.; Laheru, D.A.; Chiorean, E.G.; O’Hara, M.H.; Liudahl, S.M.; Newcomb, C.; et al. Neoadjuvant Selicrelumab, an Agonist CD40 Antibody, Induces Changes in the Tumor Microenvironment in Patients with Resectable Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 4574–4586. [Google Scholar] [CrossRef]
- Gray, J.C.; French, R.R.; James, S.; Al-Shamkhani, A.; Johnson, P.W.; Glennie, M.J. Optimising anti-tumour CD8 T-cell responses using combinations of immunomodulatory antibodies. Eur. J. Immunol. 2008, 38, 2499–2511. [Google Scholar] [CrossRef]
- Lee, S.J.; Myers, L.; Muralimohan, G.; Dai, J.; Qiao, Y.; Li, Z.; Mittler, R.S.; Vella, A.T. 4-1BB and OX40 dual costimulation synergistically stimulate primary specific CD8 T cells for robust effector function. J. Immunol. 2004, 173, 3002–3012. [Google Scholar] [CrossRef] [Green Version]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 2020, 9, e55185. [Google Scholar] [CrossRef]
- Watson, M.J.; Vignali, P.D.A.; Mullett, S.J.; Overacre-Delgoffe, A.E.; Peralta, R.M.; Grebinoski, S.; Menk, A.V.; Rittenhouse, N.L.; DePeaux, K.; Whetstone, R.D.; et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 2021, 591, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Pommey, S.; Smyth, M.J.; Stagg, J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res. 2013, 19, 5626–5635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
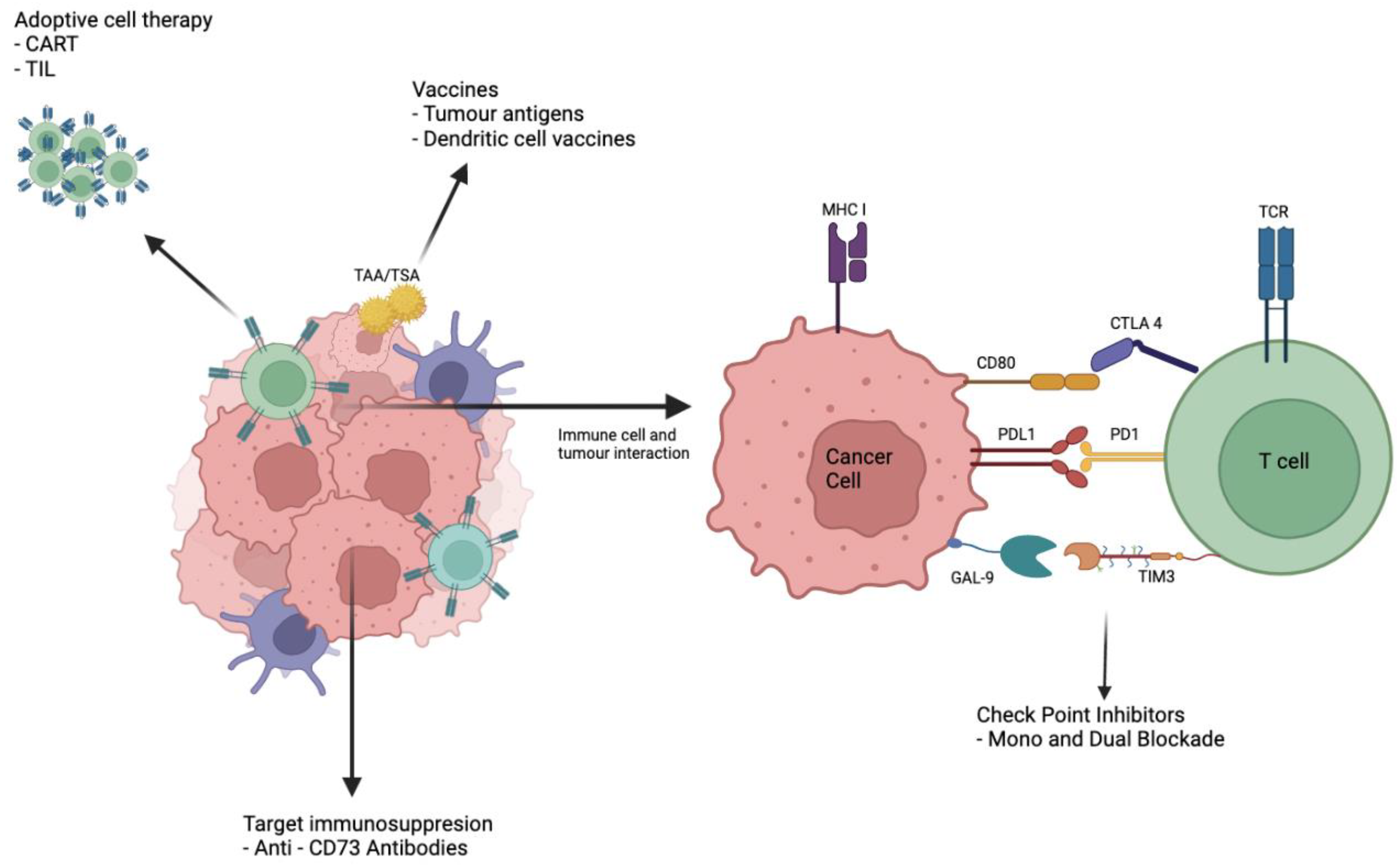

{kind=link}
| Name | Trial | Trial Details | Country Approved | Histological Type | Resectable | Unresectable /Advanced/ Metastatic | Recurrence | Summary of Results |
|---|---|---|---|---|---|---|---|---|
| Pembrolizumab | Keynote 590 | Pembrolizumab vs. placebo and chemotherapy Locally advanced, unresectable or metastatic oesophageal cancer or Siewert type 1 gastro-oesophageal junction cancer (regardless of PD-L1 status) | Japan/China | SCC, OAC | ✓ | ✓ | Prolonged overall survival (OS) in response to Pembrolizumb and chemotherapy vs. chemotherapy alone in the following groups:
| |
| US/UK/EU/Canada | SCC, OAC | ✓ | ||||||
| Keynote 181 | Pembrolizumab vs. chemotherapy Advanced/metastatic SCC or AC of the oesophagus, which progressed after one previous therapy session | Japan/China | SCC, OAC | ✓ | ✓ | Prolonged OS with pembrolizumab versus chemotherapy
| ||
| Trial not mentioned | Australia | SCC, OAC | ✓ | |||||
| Nivolumab | Attraction 3 | Nivolumab vs chemotherapy Advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy | US/EU | SCC | ✓ | ✓ | Prolonged OS in the nivolumab group compared with the chemotherapy group (median 10.9 months, 95% CI 9.2–13.3 vs. 8.4 months, 7.2–9.9; hazard ratio for death 0.77, 95% CI 0.62–0.96; p = 0.019) | |
| Checkmate 577 | Nivolumab vs. placebo Resected (R0) stage II or III oesophageal or gastroesophageal junction cancer in patients who had received neoadjuvant chemoradiotherapy and had residual pathological disease | US/UK/EU/Korea/Canada/Japan/Australia | SCC, OAC | ✓ | Prolonged disease free survival (DFS) in those that received nivolumab vs. placebo
| |||
| Checkmate 649 | Nivolumab plus chemotherapy vs. nivolumab plus ipilimumab vs. chemotherapy Previously untreated, unresectable, non-HER2-positive gastric, gastro-oesophageal junction or oesophageal adenocarcinoma, regardless of PD-ligand 1 (PD-L1) | US | OAC | ✓ | Prolonged OS in Nivolumab plus chemotherapy vs. chemotherapy alone (hazard ratio [HR] 0.71 [98.4% CI 0.59–0.86]; p < 0.0001) | |||
| Canada | OAC | ✓ | ||||||
| EU | OAC | ✓ | ||||||
| Taiwan | OAC | ✓ | ||||||
| CheckMate -648 | Nivolumab plus chemotherapy vs. nivolumab plus ipilimumab vs. chemotherapy Untreated, unresectable advanced, recurrent or metastatic oesophageal squamous-cell carcinoma | EU | SCC | ✓ | Prolonged OS with nivolumab plus chemotherapy vs. chemotherapy alone in these groups:
| |||
| Tislelizumab | RATIONALE 302 | Tislelizumab vs. Chemotherapy Advanced or metastatic OSCC with progression during or after first-line systemic treatment | EMA/China | SCC | ✓ | ✓ | Prolonged OS in tislelizumab group vs. chemotherapy in the following groups:
|
| NCT Number | Phase | Cancer Type | Location | Status | Bispecific Antibody Type | Enrolment |
|---|---|---|---|---|---|---|
| NCT03708328 | Phase 1 | SCC | Multinational | Active, not recruiting | PD-1 (CD279) and TIM-3 | 134 |
| NCT04982276 | Phase 1|Phase 2 | AC | China | Recruiting | PD-1 and CTLA-4 | 87 |
| NCT04440943 | Phase 1 | Oesophageal (histology not stated) | US | Recruiting | PD-L1 and CD27 | 40 |
| NCT03925870 | Phase 2 | SCC | China | Recruiting | PD-L1 and CTLA-4 | 30 |
| NCT04171141 | Phase 1 | AC | Multinational | Recruiting | GUCY2C and CD3 T-Cell Engaging | 130 |
| NCT04785820 | Phase 2 | SCC | Multinational | Recruiting | PD-1 (CD279) and TIM-3 | 210 |
| NCT04140500 | Phase 1 | SCC | Multinational | Recruiting | PD1 and LAG3 | 320 |
| NCT Number | Phase | Location | Status | Type | TAA Target | Enrolment |
|---|---|---|---|---|---|---|
| NCT00003125 | Phase 2 | US | Completed | Vaccine | CEA | 24 |
| NCT00948961 | Phase 1|Phase 2 | US | Completed | Vaccine | NY-ESO-1 | 70 |
| NCT01522820 * | Phase 1 | US | Completed | Vaccine | NY-ESO-1 | 18 |
| NCT01003808 | Phase 1 | Japan | Completed | Vaccine | NY-ESO-1 | 25 |
| NCT00561275 | Phase 1 | Japan | Completed | Vaccine | LY6K | 6 |
| NCT00623831 | Phase 1 | Germany | Completed | Vaccine | NY-ESO-1 | 17 |
| NCT00199849 | Phase 1 | US | Completed | Vaccine | NY-ESO-1 and LAGE-1 | 18 |
| NCT00291473 | Phase 1 | Japan | Completed | Vaccine | HER2 protein and NY-ESO-1 | 9 |
| NCT05307835 * | Phase 1 | China | Recruiting | Vaccine | Personalised to patient-specific antigen | 40 |
| NCT05192460 | Not Applicable | China | Recruiting | Vaccine | Personalised to patient-specific antigen | 36 |
| NCT03132922 | Phase 1 | USA | Active, not recruiting | Modified T-cell therapy | MAGE A4 | 52 |
| NCT04044859 | Phase 1 | Multi national | Recruiting | Modified T-cell therapy | MAGE A4 | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
To, N.; Evans, R.P.T.; Pearce, H.; Kamarajah, S.K.; Moss, P.; Griffiths, E.A. Current and Future Immunotherapy-Based Treatments for Oesophageal Cancers. Cancers 2022, 14, 3104. https://doi.org/10.3390/cancers14133104
To N, Evans RPT, Pearce H, Kamarajah SK, Moss P, Griffiths EA. Current and Future Immunotherapy-Based Treatments for Oesophageal Cancers. Cancers. 2022; 14(13):3104. https://doi.org/10.3390/cancers14133104
Chicago/Turabian StyleTo, Natalie, Richard P. T. Evans, Hayden Pearce, Sivesh K. Kamarajah, Paul Moss, and Ewen A. Griffiths. 2022. "Current and Future Immunotherapy-Based Treatments for Oesophageal Cancers" Cancers 14, no. 13: 3104. https://doi.org/10.3390/cancers14133104