
Identification of Src as a Therapeutic Target in Oesophageal Adenocarcinoma through Functional Genomic and High-Throughput Drug Screening Approaches

 , ,
, ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Analysis
2.3. Combination Index Analysis
2.4. Cell Proliferation Rates
2.5. Flow Cytometry
2.6. Western Blotting
2.7. High-Throughput Compound Screening
2.8. Bioinformatic Analysis of OAC Clinical Dataset
2.9. Generation of CDDP-Resistant Cell Line
2.10. siRNA Transfection
2.11. Quantitative PCR (Q-PCR)
2.12. Statistical Analysis of In Vitro Experimental Replicates
3. Results
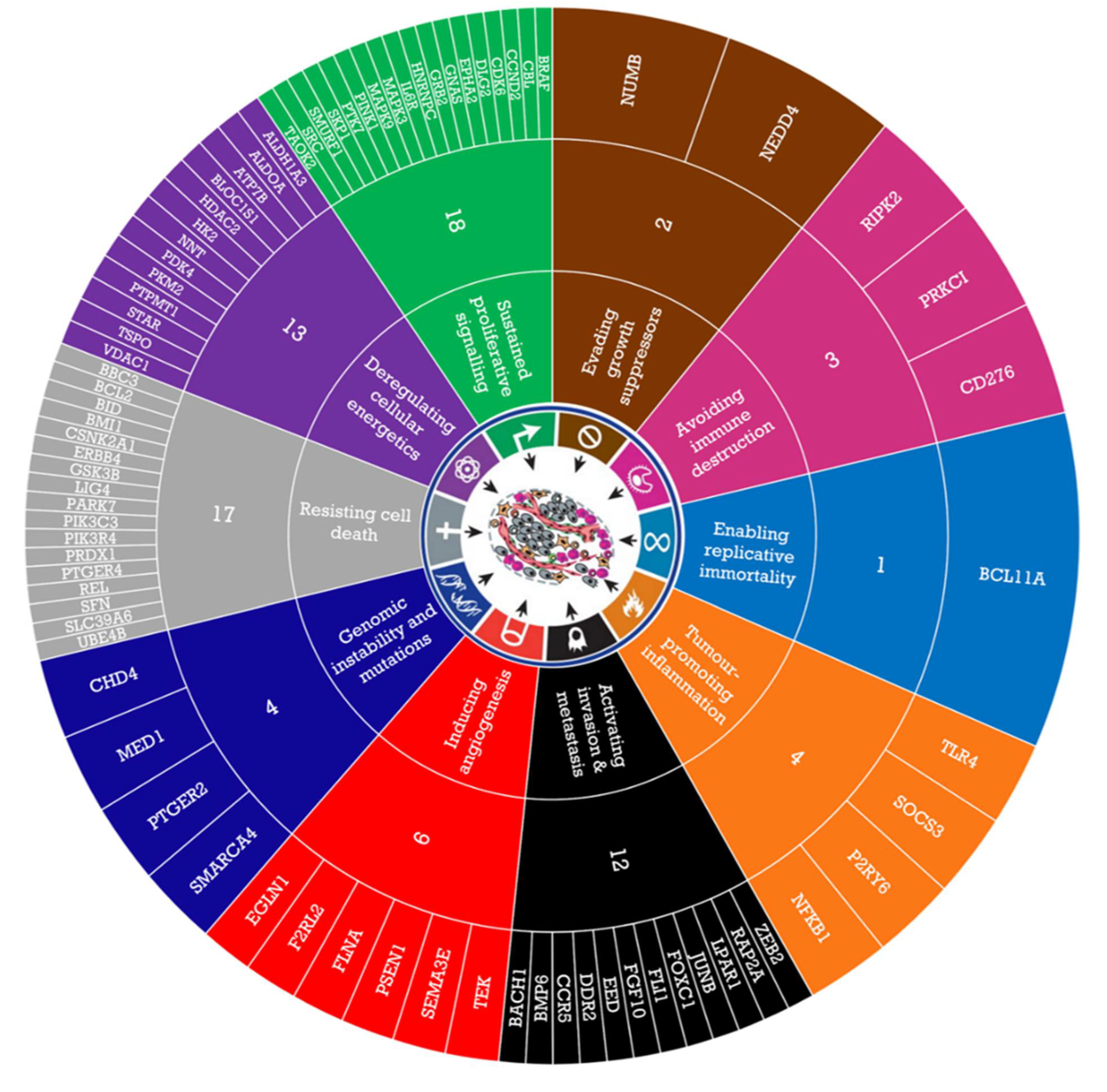
3.1. GSEA Identified Enriched Novel Pathways and Genes in Responder and Non-Responder Oac Tumours
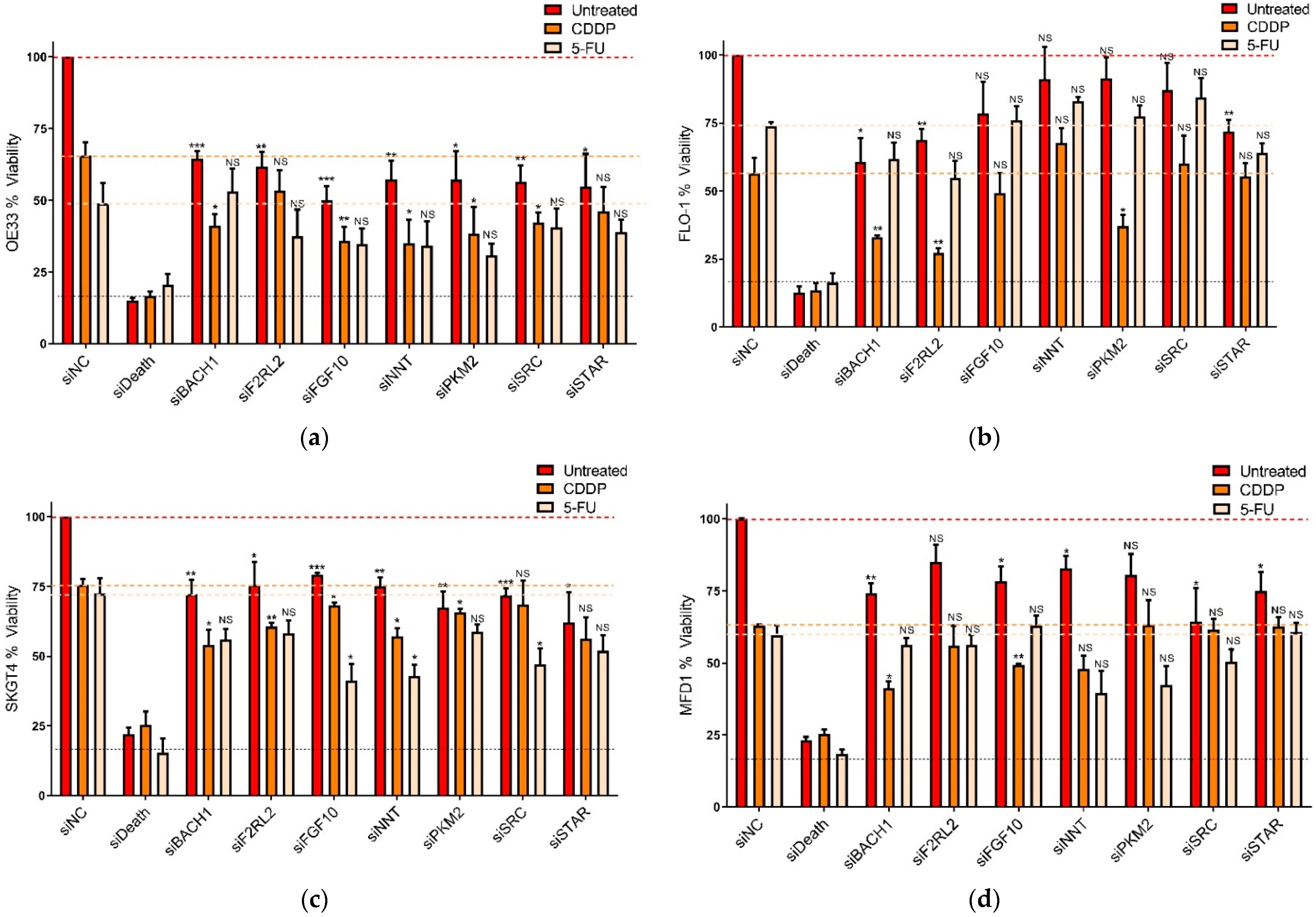
3.2. Functional Assessment of Selected Genes in OAC Cells
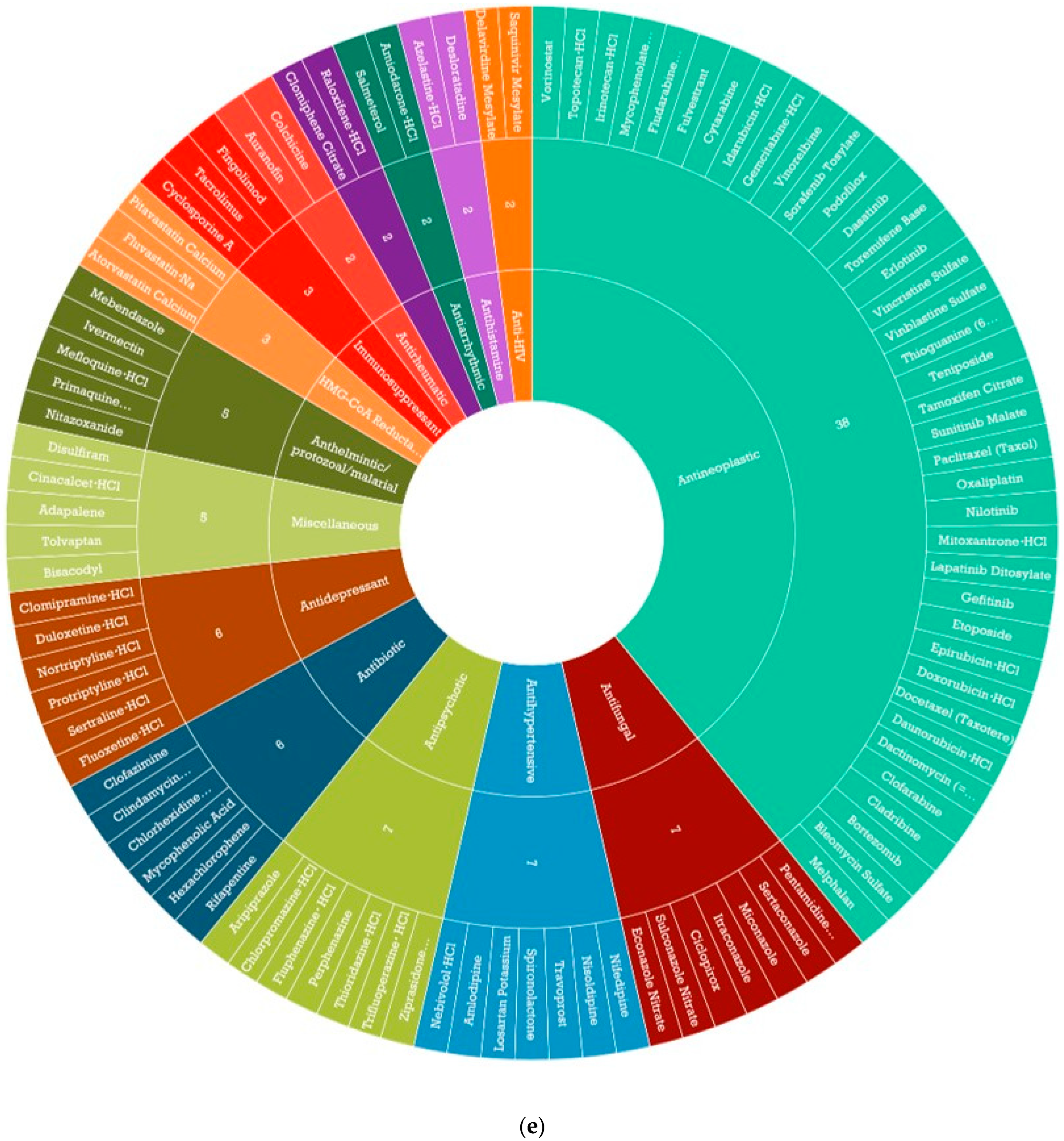
3.3. Identification of CDDP Enhancing Compounds in OAC Cell Line Models
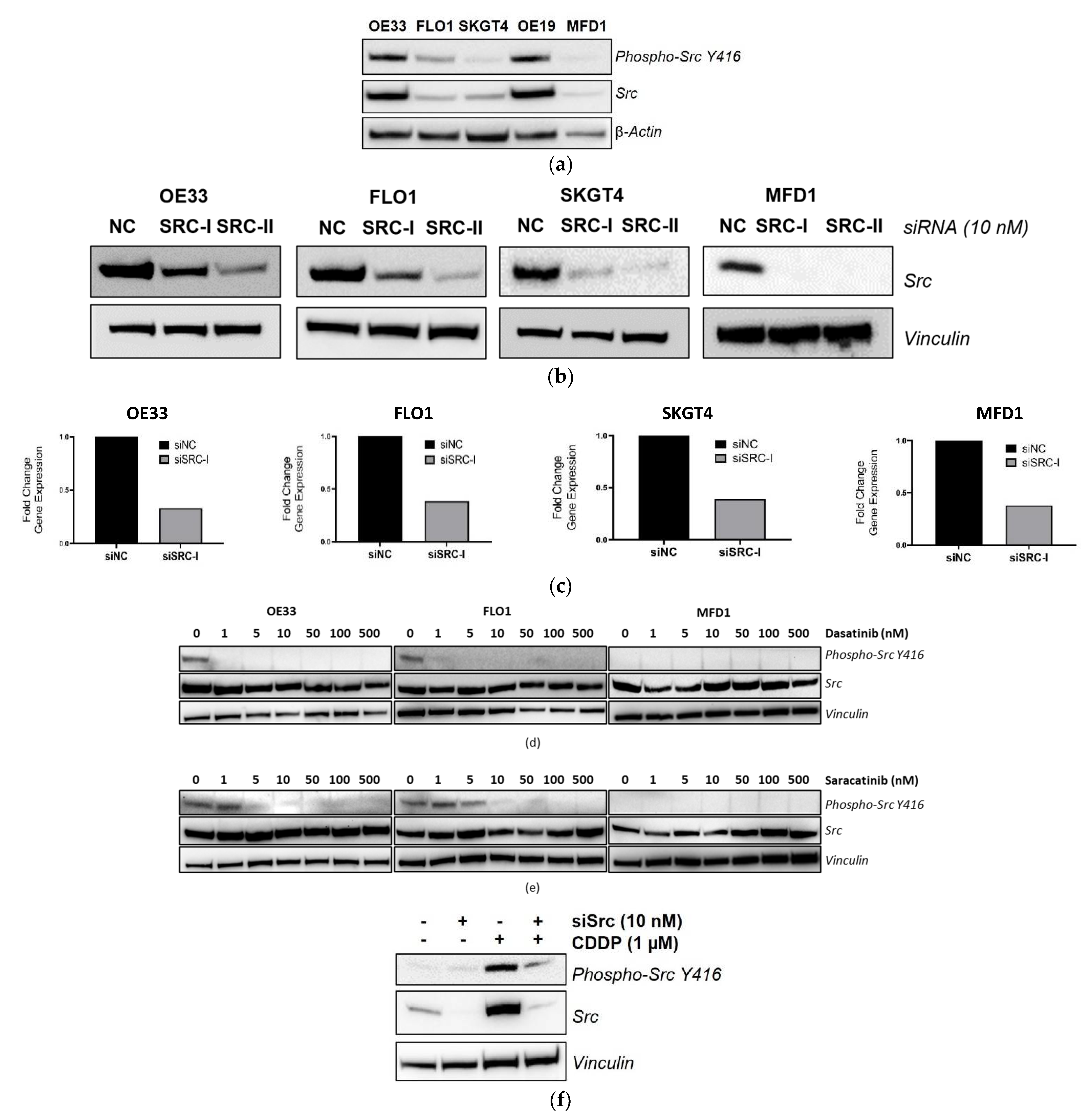
3.4. Validation of SRC Knockdown and Phospho-Src Y416 Inhibition
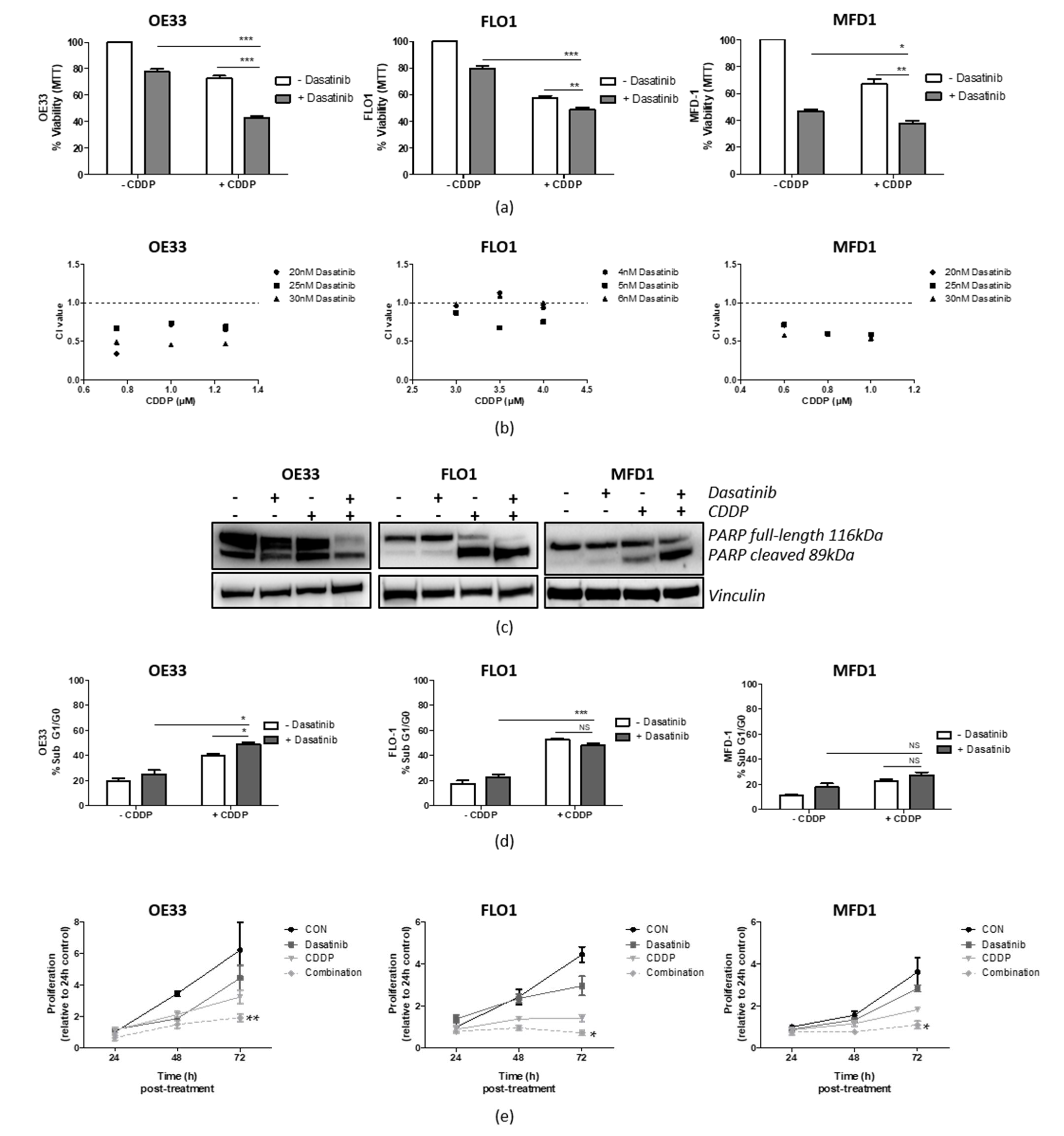
3.5. Src Inhibition Synergizes with CDDP in OAC Cell Lines
3.6. Src Inhibition Synergizes with 5-FU in OAC Cell Lines
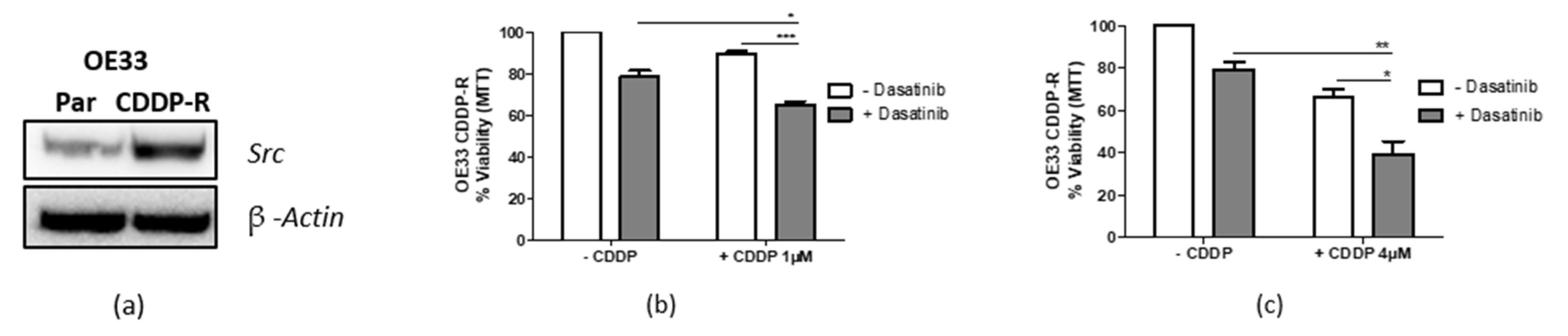
3.7. Targeting Src Re-Sensitizes CDDP-Resistant Cells
3.8. Src Expression Is Elevated in Gastric Adenocarcinoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rustgi, A.K.; El-Serag, H.B. Esophageal Carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Cancer Research, UK. Oesophageal Cancer Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/oesophageal-cancer (accessed on 4 March 2022).
- Noble, F.; Lloyd, M.A.; Turkington, R.; Griffiths, E.; O’Donovan, M.; O’Neill, J.R.; Mercer, S.; Parsons, S.L.; Fitzgerald, R.C.; Underwood, T.J.; et al. Multicentre Cohort Study to Define and Validate Pathological Assessment of Response to Neoadjuvant Therapy in Oesophagogastric Adenocarcinoma. Br. J. Surg. 2017, 104, 1816–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allum, W.H.; Stenning, S.P.; Bancewicz, J.; Clark, P.I.; Langley, R.E. Long-Term Results of a Randomized Trial of Surgery with or without Preoperative Chemotherapy in Esophageal Cancer. J. Clin. Oncol. 2009, 27, 5062–5067. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Allum, W.H.; Stenning, S.P.; Thompson, J.N.; van de Velde, C.J.H.; Nicolson, M.; Scarffe, J.H.; Lofts, F.J.; Falk, S.J.; Iveson, T.J.; et al. Perioperative Chemotherapy versus Surgery Alone for Resectable Gastroesophageal Cancer. N. Engl. J. Med. 2006, 2213–2224. [Google Scholar] [CrossRef] [Green Version]
- Turkington, R.C.; Knight, L.A.; Blayney, J.K.; Secrier, M.; Douglas, R.; Parkes, E.E.; Sutton, E.K.; Stevenson, L.; McManus, D.; Halliday, S.; et al. Immune Activation by DNA Damage Predicts Response to Chemotherapy and Survival in Oesophageal Adenocarcinoma. Gut 2019, 68, 1918–1927. [Google Scholar] [CrossRef]
- Garcia, E.; Hayden, A.; Birts, C.; Britton, E.; Cowie, A.; Pickard, K.; Mellone, M.; Choh, C.; Derouet, M.; Duriez, P.; et al. Authentication and Characterisation of a New Oesophageal Adenocarcinoma Cell Line: MFD-1. Sci. Rep. 2016, 6, 32417. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-C.; Talalay, P. Quantitative Analysis of Dose-Effect Relationships: The Combined Effects of Multiple Drugs or Enzyme Inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Chou, T.C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef]
- JH Zhang, TD Chung, K. O. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [CrossRef]
- Gene Set Enrichment Analysis (GSEA) Broad Institute. Available online: https://www.gsea-msigdb.org/gsea/msigdb/index.jsp (accessed on 9 October 2017).
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Kuehn, H.; Gould, J.; Tamayo, P.; Mesirov, J.P. GSEA-P: A Desktop Application for Gene Set Enrichment Analysis. Bioinformatics 2007, 23, 3251–3253. [Google Scholar] [CrossRef]
- Cavallo, F.; De Giovanni, C.; Nanni, P.; Forni, G.; Lollini, P.L. 2011: The Immune Hallmarks of Cancer. Cancer Immunol. Immunother. 2011, 60, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Araujo, J.; Logothetis, C. Dasatinib: A Potent SRC Inhibitor in Clinical Development for the Treatment of Solid Tumors. Cancer Treat. Rev. 2010, 36, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Martellucci, S.; Clementi, L.; Sabetta, S.; Mattei, V.; Botta, L.; Angelucci, A. Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned so Far. Cancers 2020, 12, 1448. [Google Scholar] [CrossRef]
- Kim, L.C.; Song, L.; Haura, E.B. Src Kinases as Therapeutic Targets for Cancer. Nat. Rev. Clin. Oncol. 2009, 6, 587–595. [Google Scholar] [CrossRef]
- Li, Q.; Ingram, L.; Kim, S.; Beharry, Z.; Cooper, J.A.; Cai, H. Paracrine Fibroblast Growth Factor Initiates Oncogenic Synergy with Epithelial FGFR/Src Transformation in Prostate Tumor Progression. Neoplasia 2018, 20, 233–243. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Aldape, K.; Lu, Z.; Company, X.B.; Province, Z. Nuclear PKM2 Regulates β-Catenin Transactivation upon EGFR Activation. Nature 2012, 480, 118–122. [Google Scholar] [CrossRef]
- Chaitanya, G.V.; Alexander, J.S.; Babu, P.P. PARP-1 Cleavage Fragments: Signatures of Cell-Death Proteases in Neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000197122-SRC/pathology (accessed on 14 April 2022).
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated Genomic Characterization of Oesophageal Carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Nowicki-Osuch, K.; Zhuang, L.; Jammula, S.; Bleaney, C.W.; Mahbubani, K.T.; Devonshire, G.; Katz-Summercorn, A.; Eling, N.; Wilbrey-Clark, A.; Madissoon, E.; et al. Molecular Phenotyping Reveals the Identity of Barrett’s Esophagus and Its Malignant Transition. Science 2021, 373, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.H.; Kantarjian, H.M.; Cortes, J.E. The Role of Src in Solid and Hematologic Malignancies: Development of New-Generation Src Inhibitors. Cancer 2006, 107, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Kumble, S.; Omary, M.B.; Cartwright, C.A.; Triadafilopoulos, G. Src Activation in Malignant and Premalignant Epithelia of Barrett’s Esophagus. Gastroenterology 1997, 112, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Inge, L.J.; Fowler, A.J.; Paquette, K.M.; Richer, A.L.; Tran, N.; Bremner, R.M. Dasatinib, a Small Molecule Inhibitor of the Src Kinase, Reduces the Growth and Activates Apoptosis in Pre-Neoplastic Barrett’s Esophagus Cell Lines: Evidence for a Noninvasive Treatment of High-Grade Dysplasia. J. Thorac. Cardiovasc. Surg. 2013, 145, 531–538. [Google Scholar] [CrossRef] [Green Version]
- MacKay, H.J.; Au, H.J.; McWhirter, E.; Alcindor, T.; Jarvi, A.; MacAlpine, K.; Wang, L.; Wright, J.J.; Oza, A.M. A Phase II Trial of the Src Kinase Inhibitor Saracatinib (AZD0530) in Patients with Metastatic or Locally Advanced Gastric or Gastro Esophageal Junction (GEJ) Adenocarcinoma: A Trial of the PMH Phase II Consortium. Investig. New Drugs 2012, 30, 1158–1163. [Google Scholar] [CrossRef]
- Hong, Y.S.; Kim, J.; Pectasides, E.; Fox, C.; Hong, S.W.; Ma, Q.; Wong, G.S.; Peng, S.; Stachler, M.D.; Thorner, A.R.; et al. Src Mutation Induces Acquired Lapatinib Resistance in ERBB2-Amplified Human Gastroesophageal Adenocarcinoma Models. PLoS ONE 2014, 9, e109440. [Google Scholar] [CrossRef] [Green Version]
- Northern Ireland Biobank. Available online: https://www.nibiobank.org.uk/ (accessed on 5 April 2012).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GSEA Pathway Names | |
|---|---|
| Non- Responders (C2) | 1 AMIT_EGF_RESPONSE_120_MCF10 2 BIOCARTA_RHO_PATHWAY 3 REACTOME_METAL_ION_PC12_SLC_TRANSPORTERS |
| Non- Responders (C5) | 1 GO_ENERGY_COUPLED_PROTON_TRANSPORT_DOWN_ELECTROCHEMICAL_GRADIENT 2 GO_ATP_BIOSYNTHETIC_PROCESS 3 GO_PCG_PROTEIN_COMPLEX 4 GO_MITOCHONDRIAL_ATP_SYNTHESIS_COUPLED_PROTON_TRANSPORT 5 GO_OXIDOREDUCTASE_ACTIVITY_OXIDISING_METAL_IONS 6 GO_POSPHATIDYL_ACYL_CHAIN_REMODELLING 7 GO_MITOCHONDRIAL_MEMBRANE_PART 8 GO_ORGANELLAR_LARGE_RIBOSOMAL_SUBUNIT 9 GO_INTRINSIC_COMPONENT_OF_MITOCHONDRIAL_MEMBRANE 10 GO_POSITIVE_REGULATION_OF_RESPONSE_TO_EXTRACELLULAR_STIMULUS 11 GO_MITOCHONDRIAL_TRANSPORT 12 GO_PROTEIN_TARGETING_TO_MEMBRANE 13 GO_MULTICELLULAR_ORGANISMAL_HOMEOSTASIS 14 GO_VACUOLAR_TRANSPORT 15 GO_MACROAUTOPHAGY |
| Responders (C2) | 1 PID_IL2_STAT5_PATHWAY 2 REACTOME_ANTIGEN_ACTIVATES_B_CELL RECEPTOR 3 PLASARI_TGFB1_SIGNALLING_VIA_NFIC_1HR_UP 4 REACTOME_AMINO_ACID_TRANSPORT_ACROSS_THE_PLASMA_MEMBRANE 5 BIOCARTA_DC_PATHWAY 6 BANDRES_RESPONSE_TO_CARMUSTIN_MGMT_48HR_UP 7 BROWNE_HCMV_INFECTION_30MIN_UP 8 REACTOME_IL_3_5_AND_GM_SCF_SIGNALLING |
| Responders (C5) | 1 GO_PHOSPHOLIPASE_C_ACTIVITY 2 GO_CELLULAR_RESPONSE_TO_PROSTAGLANDIN_STIMULUS 3 GO_ALDEHYDE_DEHYDROGENASE_NAD_ACTIVITY 4 GO_REGULATION_OF_FIBROBLAST_MIGRATION 5 GO_RESPONSE_TO_PROSTAGLANDIN 6 GO_HEART_TRABECULA_MORPHOGENESIS 7 GO_CELLULAR_RESPONSE_TO_PROSTAGLANDIN_E_STIMULUS 8 GO_CELLUALAR_TO LITHIUM_ION 9 GO_RESPONSE_TO_PROSTAGLANDIN_E 10 GO_REGULATION_OF_INTERFERON_GAMMA_BIOSYNTHETIC_PROCESS 11 GO_DENDRITIC_SHAFT 12 GO_RNA_POLYMERASE_II_DISTAL_ENHANCER_SEQUENCE_SPECIFIC_BINDING 13 GO_NEUROMUSCULAR_JUNCTION_DEVELOPMENT 14 GO_TRANSCRIPTIONAL_ACTIVATOR_ACTIVITY_RNA_POLYMERASE_II_DISTAL_ENHANCER_SEQUENCE_SPECIFIC_BINDING 15 GO_REGULATION_OF_EXTENT_OF_CELL_GROWTH 16 GO_ESTABLISHMENT_OR_MAINTENANCE_OF_CELL_POLARITY 17 GO_BASOLATERAL_PLASMA_MEMBRANE |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCabe, N.H.; Stevenson, L.; Scanlon, E.; Douglas, R.; Kennedy, S.; Keminer, O.; Windshügel, B.; Zisterer, D.; Kennedy, R.D.; Blayney, J.K.; et al. Identification of Src as a Therapeutic Target in Oesophageal Adenocarcinoma through Functional Genomic and High-Throughput Drug Screening Approaches. Cancers 2022, 14, 3726. https://doi.org/10.3390/cancers14153726
McCabe NH, Stevenson L, Scanlon E, Douglas R, Kennedy S, Keminer O, Windshügel B, Zisterer D, Kennedy RD, Blayney JK, et al. Identification of Src as a Therapeutic Target in Oesophageal Adenocarcinoma through Functional Genomic and High-Throughput Drug Screening Approaches. Cancers. 2022; 14(15):3726. https://doi.org/10.3390/cancers14153726
Chicago/Turabian StyleMcCabe, Niamh H., Leanne Stevenson, Enya Scanlon, Rosalie Douglas, Susanna Kennedy, Oliver Keminer, Björn Windshügel, Daniela Zisterer, Richard D. Kennedy, Jaine K. Blayney, and et al. 2022. "Identification of Src as a Therapeutic Target in Oesophageal Adenocarcinoma through Functional Genomic and High-Throughput Drug Screening Approaches" Cancers 14, no. 15: 3726. https://doi.org/10.3390/cancers14153726
APA StyleMcCabe, N. H., Stevenson, L., Scanlon, E., Douglas, R., Kennedy, S., Keminer, O., Windshügel, B., Zisterer, D., Kennedy, R. D., Blayney, J. K., & Turkington, R. C. (2022). Identification of Src as a Therapeutic Target in Oesophageal Adenocarcinoma through Functional Genomic and High-Throughput Drug Screening Approaches. Cancers, 14(15), 3726. https://doi.org/10.3390/cancers14153726