
Novel Drug Candidate Prediction for Intrahepatic Cholangiocarcinoma via Hub Gene Network Analysis and Connectivity Mapping

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
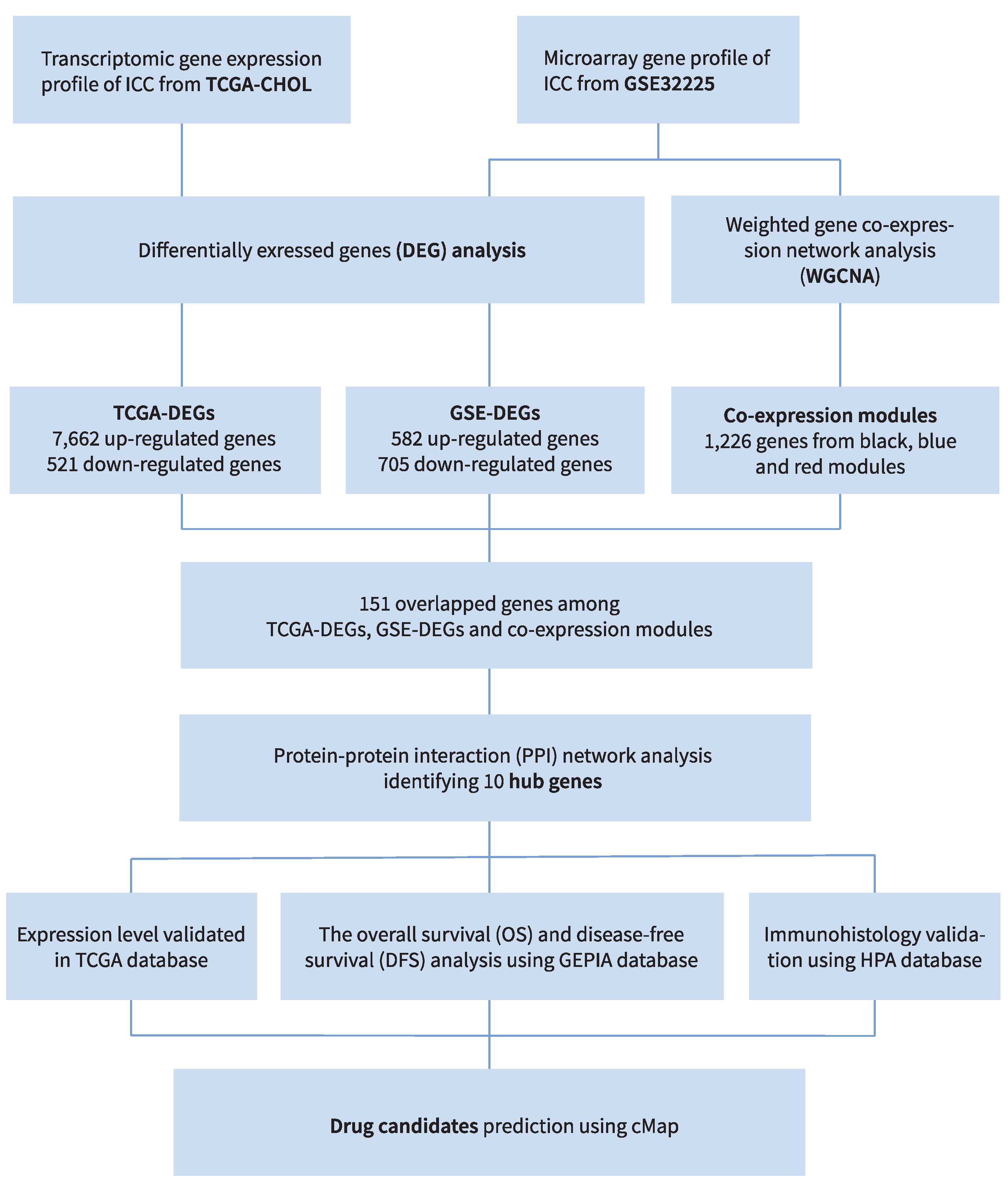
2. Materials and Methods
2.1. Database Selection and Preprocessing
2.2. Differentially Expressed Gene, Function Annotation and Enrichment Analysis
2.3. Co-Expression Modules by WGCNA
2.4. Key Gene Selection, Protein–Protein Interaction Analysis, and Hub Gene Screening
2.5. Hub Gene Survival Analysis and Identification of Potential Drug Candidates
3. Results
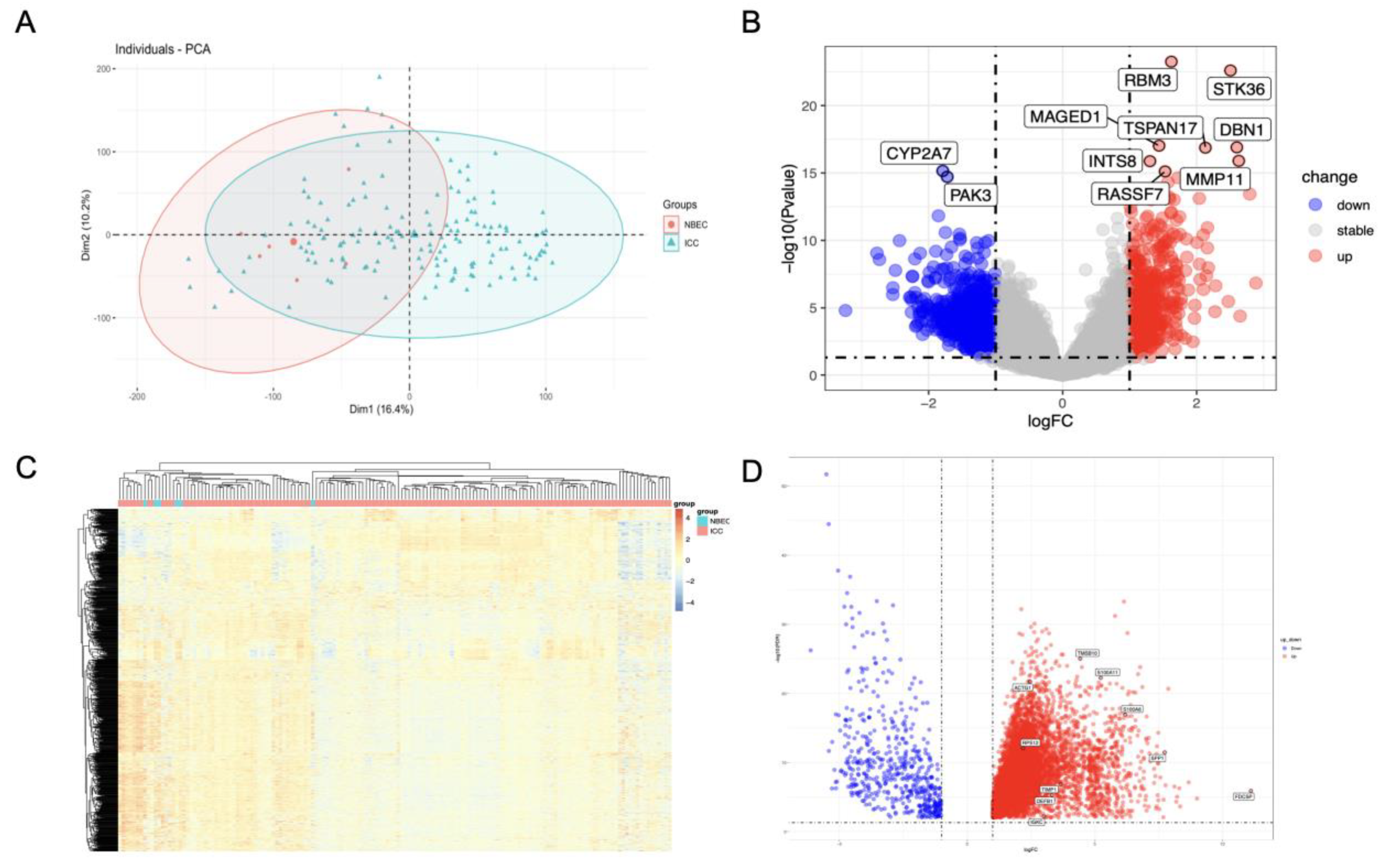
3.1. Identification of Differentially Expressed Genes in GSE32225 and TCGA-CHOL

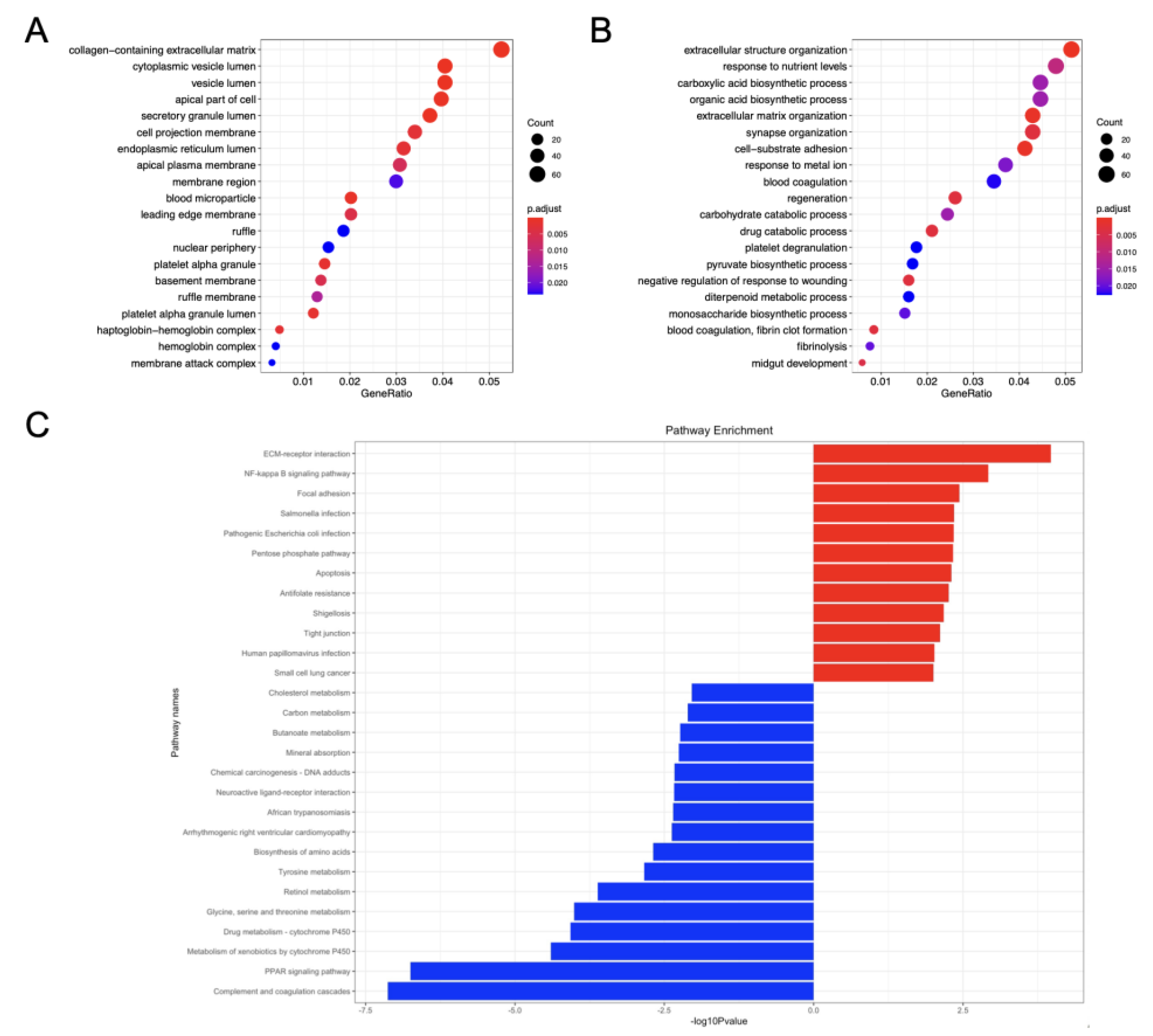
3.2. Function Annotation and Enrichment Analysis of GSE-DEGs

3.3. Construction of Weighted Gene Co-Expression Modules
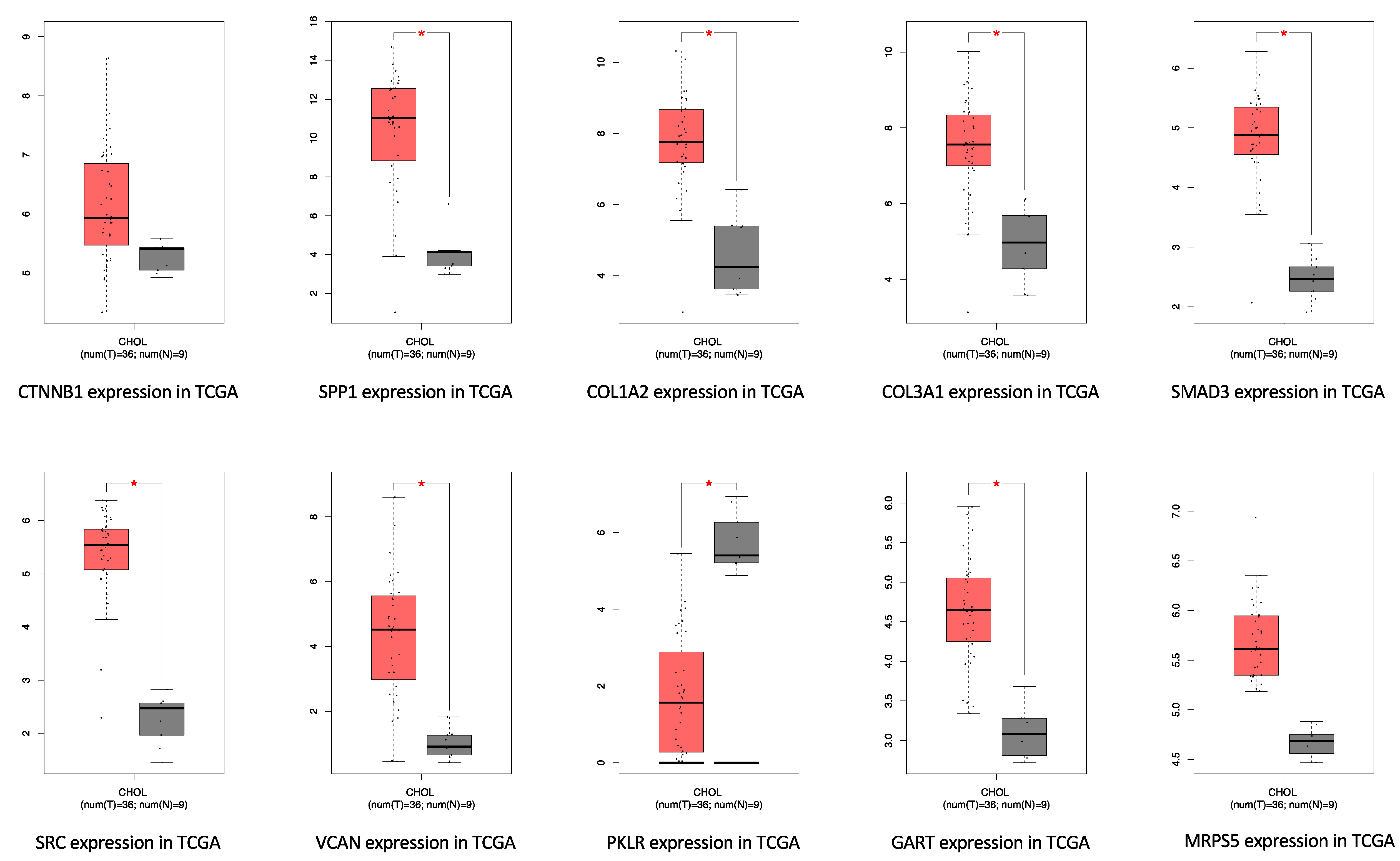
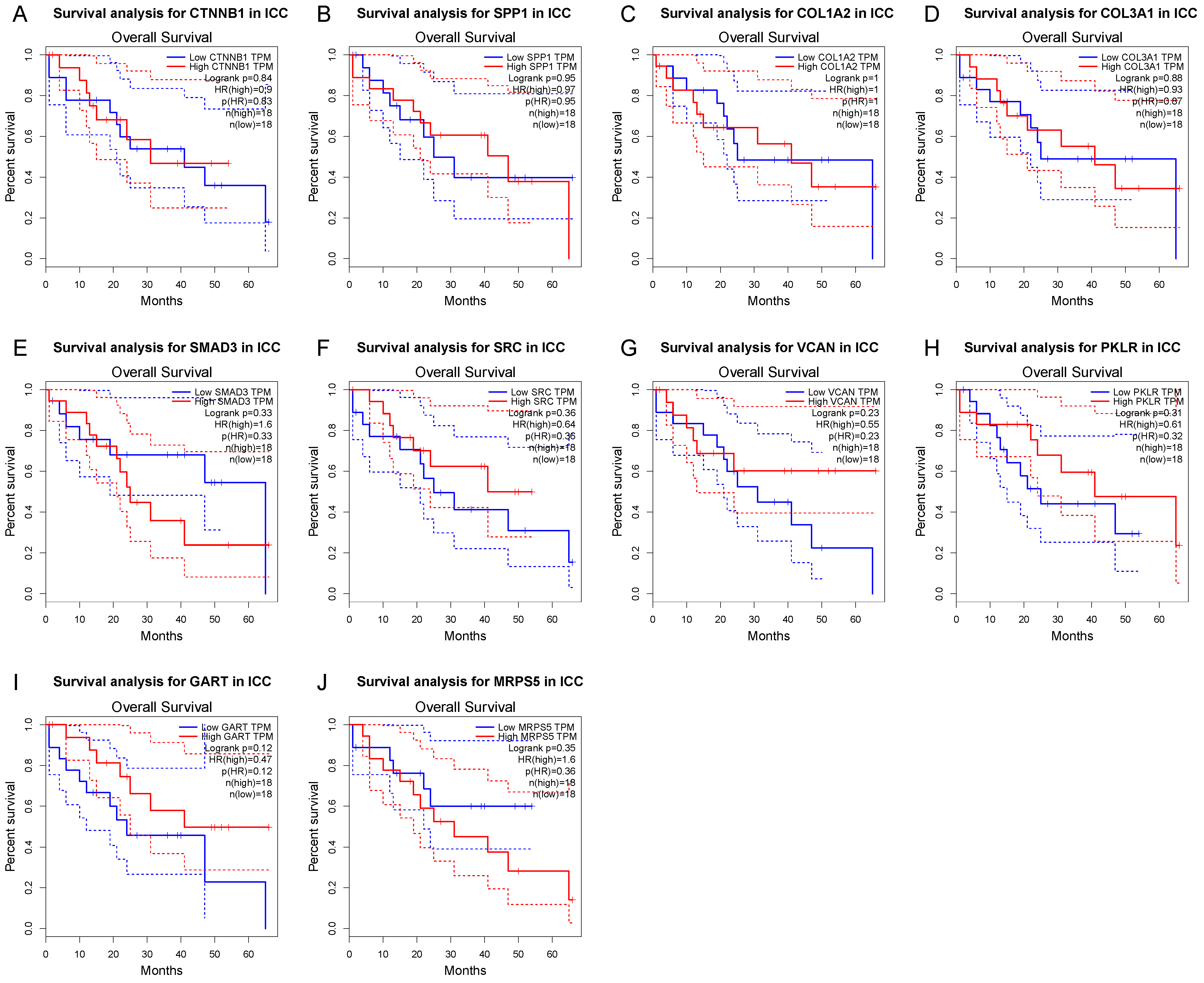
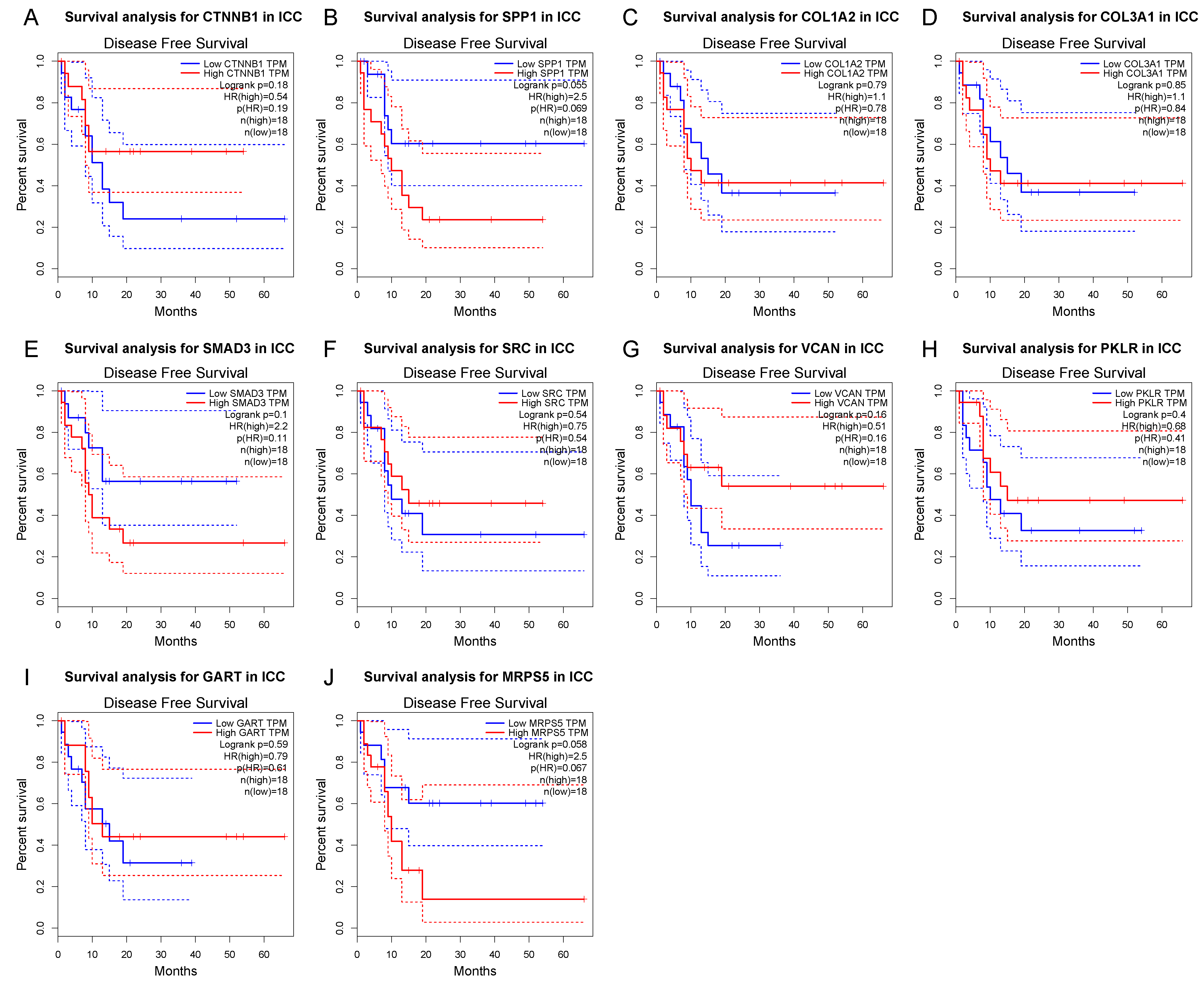
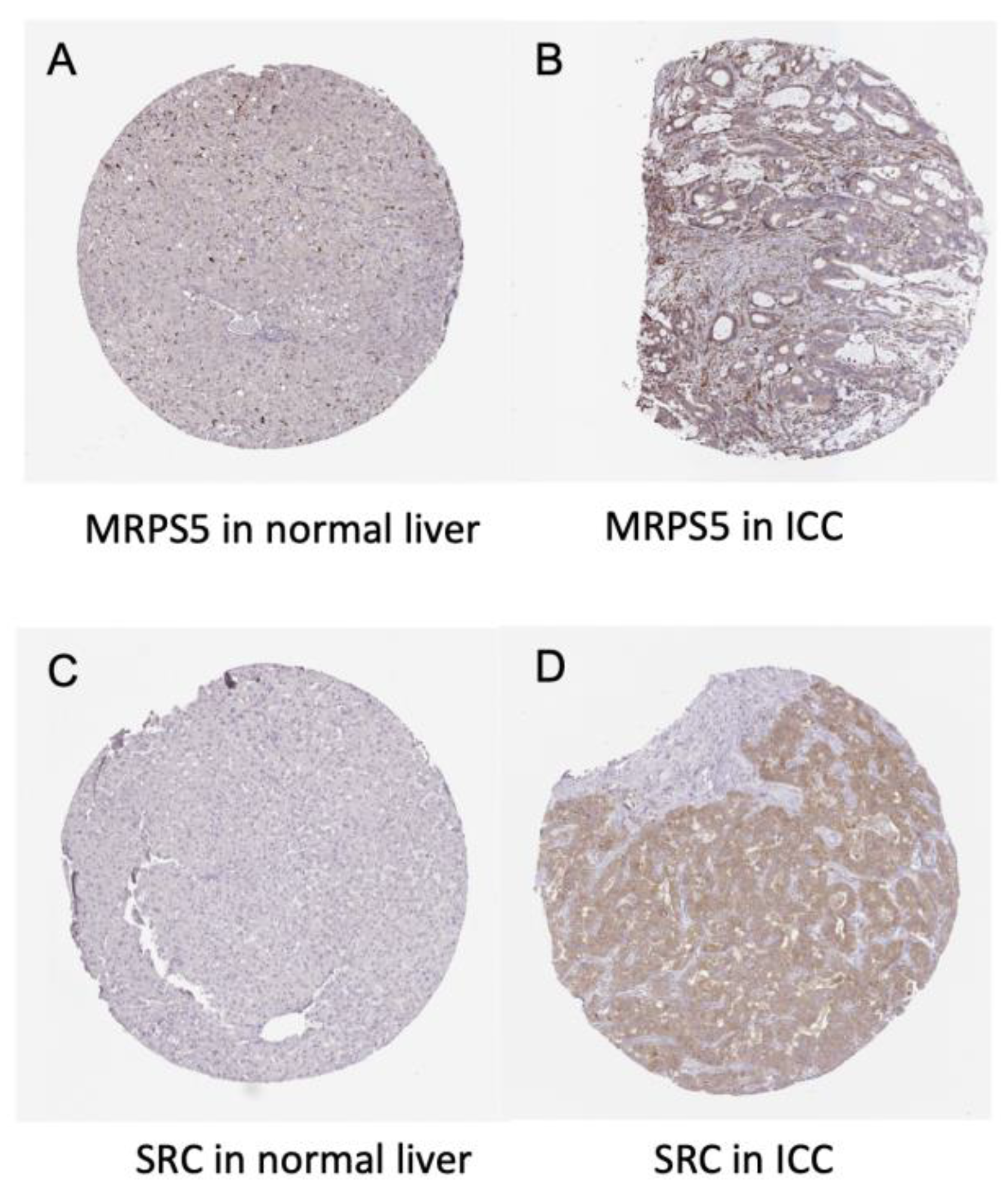
3.4. Survival Gene Screening, PPI Analysis, and Hub Gene Selection
3.5. Potential Drug Candidates of ICC-Related Gene Profile
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buettner, S.; van Vugt, J.L.; Ijzermans, J.N.; Koerkamp, B.G. Intrahepatic cholangiocarcinoma: Current perspectives. OncoTargets Ther. 2017, 10, 1131–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ejaz, A.; Cloyd, J.M.; Pawlik, T.M. Advances in the Diagnosis and Treatment of Patients with Intrahepatic Cholangiocarci-noma. Ann. Surg. Oncol. 2020, 27, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, I.T.; Koerkamp, B.G.; Do, R.; Gönen, M.; Fong, Y.; Allen, P.J.; D’Angelica, M.I.; Kingham, T.P.; DeMatteo, R.P.; Klimstra, D.S.; et al. Unresectable intrahepatic cholangiocarcinoma: Systemic plus hepatic arterial infusion chemotherapy is associated with longer survival in comparison with systemic chemotherapy alone. Cancer 2016, 122, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Amini, N.; Ejaz, A.; Spolverato, G.; Kim, Y.; Herman, J.M.; Pawlik, T.M. Temporal trends in liver-directed therapy of patients with intrahepatic cholangiocarcinoma in the United States: A population-based analysis. J. Surg. Oncol. 2014, 110, 163–170. [Google Scholar] [CrossRef]
- Cloyd, J.M.; Ejaz, A.; Pawlik, T.M. The Landmark Series: Intrahepatic Cholangiocarcinoma. Ann. Surg. Oncol. 2020, 27, 2859–2865. [Google Scholar] [CrossRef]
- Zhu, A.X.; Borger, D.R.; Kim, Y.; Cosgrove, D.; Ejaz, A.; Alexandrescu, S.; Groeschl, R.T.; Deshpande, V.; Lindberg, J.M.; Ferrone, C.; et al. Genomic profiling of intrahepatic cholangiocarcinoma: Refining prognosis and identifying therapeutic targets. Ann. Surg. Oncol. 2014, 21, 3827–3834. [Google Scholar] [CrossRef] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 2012, 46, i11. [Google Scholar] [CrossRef] [Green Version]
- Li, C.Y.; Cai, J.H.; Tsai, J.J.P.; Wang, C.C.N. Identification of Hub Genes Associated With Development of Head and Neck Squamous Cell Carcinoma by Inte-grated Bioinformatics Analysis. Front Oncol. 2020, 10, 681. [Google Scholar] [CrossRef]
- Xing, J.; Shi, Q.; Zhao, J.; Yu, Z. Identifying drug candidates for hepatocellular carcinoma based on differentially expressed genes. Am. J. Transl. Res. 2020, 12, 2664–2674. [Google Scholar]
- Chen, Y.-T.; Xie, J.-Y.; Sun, Q.; Mo, W.-J. Novel drug candidates for treating esophageal carcinoma: A study on differentially expressed genes, using connectivity mapping and molecular docking. Int. J. Oncol. 2019, 54, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Montal, R.; Sia, D.; Montironi, C.; Leow, W.Q.; Esteban-Fabró, R.; Pinyol, R.; Torres-Martin, M.; Bassaganyas, L.; Moeini, A.; Peix, J.; et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 315–327. [Google Scholar] [CrossRef]
- Xiao, Y.; Xu, G.; Cloyd, J.M.; Du, S.; Mao, Y.; Pawlik, T.M. Predicting Novel Drug Candidates for Pancreatic Neuroendocrine Tumors via Gene Signature Comparison and Connectivity Mapping. J. Gastrointest. Surg. 2022, 1–9. [Google Scholar] [CrossRef]
- Isik, Z.; Baldow, C.; Cannistraci, C.V.; Schroeder, M. Drug target prioritization by perturbed gene expression and network information. Sci. Rep. 2015, 5, 17417. [Google Scholar] [CrossRef]
- Tsilimigras, D.I.; Sahara, K.; Wu, L.; Moris, D.; Bagante, F.; Guglielmi, A.; Aldrighetti, L.; Weiss, M.; Bauer, T.W.; Alexandrescu, S.; et al. Very Early Recurrence After Liver Resection for Intrahepatic Cholangiocarcinoma: Considering Alternative Treatment Approaches. JAMA Surg. 2020, 155, 823–831. [Google Scholar] [CrossRef]
- Spolverato, G.; Vitale, A.; Cucchetti, A.; Popescu, I.; Marques, H.P.; Aldrighetti, L.; Gamblin, T.C.; Maithel, S.K.; Sandroussi, C.; Bauer, T.W.; et al. Can hepatic resection provide a long-term cure for patients with intrahepatic cholangiocarcinoma? Cancer 2015, 121, 3998–4006. [Google Scholar] [CrossRef]
- Edeline, J.; Benabdelghani, M.; Bertaut, A.; Watelet, J.; Hammel, P.; Joly, J.-P.; Boudjema, K.; Fartoux, L.; Bouhier-Leporrier, K.; Jouve, J.-L.; et al. Gemcitabine and oxaliplatin chemotherapy or surveillance in resected biliary tract cancer (PRODIGE 12-ACCORD 18-UNICANCER GI): A randomized phase III study. J. Clin. Oncol. 2019, 37, 658–667. [Google Scholar] [CrossRef]
- Altman, A.M.; Kizy, S.; Marmor, S.; Hui, J.Y.; Tuttle, T.M.; Jensen, E.H.; Denbo, J.W. Adjuvant chemotherapy for intrahepatic cholangiocarcinoma: Approaching clinical practice consensus? Hepatobiliary Surg. Nutr. 2020, 9, 577–586. [Google Scholar] [CrossRef]
- Akateh, C.; Ejaz, A.M.; Pawlik, T.M.; Cloyd, J.M. Neoadjuvant treatment strategies for intrahepatic cholangiocarcinoma. World J. Hepatol. 2020, 12, 693–708. [Google Scholar] [CrossRef]
- Primrose, J.N.; Fox, R.P.; Palmer, D.H.; Malik, H.Z.; Prasad, R.; Mirza, D.; Anthony, A.; Corrie, P.; Falk, S.; Finch-Jones, M.; et al. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): A randomised, controlled, multicentre, phase 3 study. Lancet Oncol. 2019, 20, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer-Fabrega, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, W.; Jiang, Y.; Liu, K.; Ran, L.; Song, F. Identification of functional lncRNAs in gastric cancer by integrative analysis of GEO and TCGA data. J. Cell. Biochem. 2019, 120, 17898–17911. [Google Scholar] [CrossRef]
- Sun, J.; Huang, J.; Lan, J.; Zhou, K.; Gao, Y.; Song, Z.; Deng, Y.; Liu, L.; Dong, Y.; Liu, X. Overexpression of CENPF correlates with poor prognosis and tumor bone metastasis in breast cancer. Cancer Cell Int. 2019, 19, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Li, C.Y.; Cai, J.-H.; Sheu, P.C.-Y.; Tsai, J.J.; Wu, M.-Y.; Hou, M.-F. Identification of Prognostic Candidate Genes in Breast Cancer by Integrated Bioinformatic Analysis. J. Clin. Med. 2019, 8, 1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Emadossadaty, S.; Ladep, N.G.; Thomas, H.C.; Elliott, P.; Taylor-Robinson, S.D.; Toledano, M.B. Rising trends in cholangiocarcinoma: Is the ICD classification system misleading us? J. Hepatol. 2012, 56, 848–854. [Google Scholar] [CrossRef]
- Plentz, R.R.; Malek, N.P. Clinical presentation, risk factors and staging systems of cholangiocarcinoma. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 245–252. [Google Scholar] [CrossRef]
- Thinkhamrop, K.; Khuntikeo, N.; Phonjitt, P.; Chamadol, N.; Thinkhamrop, B.; Moore, M.A.; Promthet, S. Association between Diabetes Mellitus and Fatty Liver Based on Ultrasonography Screening in the World’s Highest Cholangiocarcinoma Incidence Region, Northeast Thailand. Asian Pac. J. Cancer Prev. 2015, 16, 3931–3936. [Google Scholar] [CrossRef] [Green Version]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.-W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [Green Version]
- Mavros, M.N.; Economopoulos, K.P.; Alexiou, V.G.; Pawlik, T.M. Treatment and Prognosis for Patients With Intrahepatic Cholangiocarcinoma: Systematic Review and Me-ta-analysis. JAMA Surg. 2014, 149, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Maithel, S.K.; Gamblin, T.C.; Kamel, I.; Corona-Villalobos, C.P.; Thomas, M.; Pawlik, T.M. Multidisciplinary approaches to intrahepatic cholangiocarcinoma. Cancer 2013, 119, 3929–3942. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Suda, R.; Sakai, N.; Matsushita, K.; Ishige, T.; Kawasaki, Y.; Shiko, Y.; Furukawa, K.; Mishima, T.; Nakadai, E.; Ohtsuka, M. Prediction of mismatch repair deficient biliary tract cancer: Role of morphological features and host immune re-sponse detected by routine hematoxylin-eosin staining. J. Hepatobiliary Pancreat Sci. 2021, 28, 680–691. [Google Scholar] [CrossRef]
- Cortes, J.E.; Saglio, G.; Kantarjian, H.M.; Baccarani, M.; Mayer, J.; Boqué, C.; Shah, N.P.; Chuah, C.; Casanova, L.; Bradley-Garelik, B.; et al. Final 5-Year Study Results of DASISION: The Dasatinib Versus Imatinib Study in Treatment-Naïve Chronic Myeloid Leukemia Patients Trial. J. Clin. Oncol. 2016, 34, 2333–2340. [Google Scholar] [CrossRef]
- Saha, S.K.; Gordan, J.D.; Kleinstiver, B.P.; Vu, P.; Najem, M.S.; Yeo, J.-C.; Shi, L.; Kato, Y.; Levin, R.S.; Webber, J.T.; et al. Isocitrate Dehydrogenase Mutations Confer Dasatinib Hypersensitivity and SRC Dependence in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2016, 6, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Best, J.; Schotten, C.; Lohmann, G.; Gerken, G.; Dechêne, A. Tivantinib for the treatment of hepatocellular carcinoma. Expert Opin. Pharmacother. 2017, 18, 727–733. [Google Scholar] [CrossRef]
- Puzanov, I.; Sosman, J.; Santoro, A.; Saif, M.W.; Goff, L.; Dy, G.K.; Zucali, P.; Means-Powell, J.A.; Ma, W.W.; Simonelli, M.; et al. Phase 1 trial of tivantinib in combination with sorafenib in adult patients with advanced solid tumors. Investig. New Drugs 2015, 33, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, A.; Rimassa, L.; Borbath, I.; Daniele, B.; Salvagni, S.; Van Laethem, J.L.; Van Vlierberghe, H.; Trojan, J.; Kolligs, F.T.; Weiss, A.; et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: A randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013, 14, 55–63. [Google Scholar] [CrossRef]
- Tralongo, P.; Annunziata, M.A.; Santoro, A.; Tirelli, U.; Surbone, A. Beyond semantics: The need to better categorize patients with cancer. J. Clin. Oncol. 2013, 31, 2637–2638. [Google Scholar] [CrossRef] [PubMed]
- Hanuš, L.O.; Meyer, S.M.; Muñoz, E.; Taglialatela-Scafati, O.; Appendino, G. Phytocannabinoids: A unified critical inventory. Nat. Prod. Rep. 2016, 33, 1357–1392. [Google Scholar] [CrossRef] [Green Version]
- Seltzer, E.S.; Watters, A.K.; MacKenzie, J.D., Jr.; Granat, L.M.; Zhang, D. Cannabidiol (CBD) as a Promising Anti-Cancer Drug. Cancers 2020, 12, 3203. [Google Scholar] [CrossRef]
- Preet, A.; Qamri, Z.; Nasser, M.W.; Prasad, A.; Shilo, K.; Zou, X.; Groopman, J.E.; Ganju, R.K. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis. Cancer Prev. Res. 2011, 4, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Kisková, T.; Mungenast, F.; Suváková, M.; Jäger, W.; Thalhammer, T. Future Aspects for Cannabinoids in Breast Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 1673. [Google Scholar] [CrossRef] [Green Version]
- Pagano, E.; Venneri, T.; Lucariello, G.; Cicia, D.; Brancaleone, V.; Nanì, M.; Cacciola, N.; Capasso, R.; Izzo, A.; Borrelli, F.; et al. Palmitoylethanolamide Reduces Colon Cancer Cell Proliferation and Migration, Influences Tumor Cell Cycle and Exerts In Vivo Chemopreventive Effects. Cancers 2021, 13, 1923. [Google Scholar] [CrossRef]
- Zhu, B.T.; Han, G.Z.; Shim, J.Y.; Wen, Y.; Jiang, X.R. Quantitative structure-activity relationship of various endogenous estrogen metabolites for human estrogen receptor alpha and beta subtypes: Insights into the structural determinants favoring a differential subtype binding. Endocrinology 2006, 147, 4132–4150. [Google Scholar] [CrossRef]
- Folkerd, E.J.; Dowsett, M. Influence of Sex Hormones on Cancer Progression. J. Clin. Oncol. 2010, 28, 4038–4044. [Google Scholar] [CrossRef]
- Westin, S.N.; Fellman, B.; Sun, C.C.; Broaddus, R.R.; Woodall, M.L.; Pal, N.; Urbauer, D.L.; Ramondetta, L.M.; Schmeler, K.M.; Soliman, P.T.; et al. Prospective phase II trial of levonorgestrel intrauterine device: Nonsurgical approach for complex atypical hyperplasia and early-stage endometrial cancer. Am. J. Obstet. Gynecol. 2021, 224, 191.e1–191.e15. [Google Scholar] [CrossRef]
- Alvaro, D.; Barbaro, B.; Franchitto, A.; Onori, P.; Glaser, S.S.; Alpini, G.; Francis, H.; Marucci, L.; Sterpetti, P.; Ginanni-Corradini, S.; et al. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am. J. Pathol. 2006, 169, 877–888. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.S.; Adami, H.-O.; Andreotti, G.; Beane-Freeman, L.E.; de González, A.B.; Buring, J.E.; Fraser, G.E.; Freedman, N.D.; Gapstur, S.M.; Gierach, G.; et al. Associations between reproductive factors and biliary tract cancers in women from the Biliary Tract Cancers Pooling Project. J. Hepatol. 2020, 73, 863–872. [Google Scholar] [CrossRef]
- Petrick, J.L.; McMenamin, C.; Zhang, X.; Zeleniuch-Jacquotte, A.; Wactawski-Wende, J.; Simon, T.G.; Sinha, R.; Sesso, H.D.; Schairer, C.; Rosenberg, L.; et al. Exogenous hormone use, reproductive factors and risk of intrahepatic cholangiocarcinoma among women: Results from cohort studies in the Liver Cancer Pooling Project and the UK Biobank. Br. J. Cancer 2020, 123, 316–324. [Google Scholar] [CrossRef]
- Cui, H.; Zhang, M.; Yang, Q.; Li, X.; Liebman, M.; Yu, Y.; Xie, L. The Prediction of Drug-Disease Correlation Based on Gene Expression Data. Biomed. Res. Int. 2018, 2018, 4028473. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Name | Score |
|---|---|---|
| 1 | CTNNB1 | 83 |
| 2 | SPP1 | 66 |
| 3 | COL1A2 | 50 |
| 3 | COL3A1 | 50 |
| 5 | SMAD3 | 39 |
| 6 | SRC | 25 |
| 7 | VCAN | 24 |
| 8 | PKLR | 17 |
| 9 | GART | 11 |
| 10 | MRPS5 | 10 |
| Drug Name | Tested Cell Line | Test Dose | Test Time | Mechanism of Action | Raw Connectivity Score |
|---|---|---|---|---|---|
| Levonorgestrel | A549 | 10 uM | 24 h | Estrogen receptor agonist|Glucocorticoid receptor antagonist|Progesterone receptor agonist|Progesterone receptor antagonist | −0.79 |
| Nicotinamide | HEPG2 | 10 uM | 6 h | Protein synthesis stimulant | −0.75 |
| Palmitoylethanolamide | A549 | 0.04 uM | 24 h | Cannabinoid receptor agonist | −0.74 |
| Arachidonamide | HCC515 | 10 uM | 6 h | Cannabinoid receptor agonist | −0.74 |
| Moxifloxacin | PC3 | 1.11 uM | 24 h | Bacterial DNA inhibitor | −0.74 |
| Anisodamine | HELA | 10 uM | 24 h | Lipid peroxidase inhibitor | −0.74 |
| Dasatinib | SHSY5Y | 0.04 uM | 24 h | KIT inhibitor|Bcr-Abl inhibitor|Ephrin inhibitor|PDGFR inhibitor|Src inhibitor|Tyrosine kinase inhibitor | −0.73 |
| NVP-BHG712 | CD34 | 10 uM | 24 h | Ephrin inhibitor | −0.73 |
| Pyrimethamine | HT29 | 10 uM | 24 h | Dihydrofolate reductase inhibitor | −0.73 |
| CO-101244 | VCAP | 10 uM | 6 h | Ionotropic glutamate receptor antagonist | −0.72 |
| MK-2206 | HME1 | 20 uM | 24 h | AKT inhibitor | −0.71 |
| Phenamil | HA1E | 10 uM | 24 h | TRPV antagonist | −0.71 |
| Tivantinib | A549 | 10 uM | 24 h | Tyrosine kinase inhibitor | −0.71 |
| Amoxicillin | HCC515 | 10 uM | 6 h | Penicillin binding protein inhibitor | −0.71 |
| MDL-28170 | HCC515 | 10 uM | 6 h | Calpain inhibitor | −0.71 |
| Pyrimethamine | MCF7 | 10 uM | 24 h | Dihydrofolate reductase inhibitor | −0.71 |
| Salvinorin-A | HT29 | 0.25 uM | 24 h | Opioid receptor agonist | −0.71 |
| Varenicline | NPC | 0.74 uM | 24 h | Acetylcholine receptor agonist | −0.71 |
| FR-180204 | A549 | 10 uM | 24 h | MAP kinase inhibitor | −0.71 |
| Flunisolide | A549 | 10 uM | 24 h | Cytochrome P450 inhibitor | −0.71 |
| PNU-22394 | HT29 | 10 uM | 6 h | Serotonin receptor agonist | −0.7 |
| Tianeptine | A375 | 10 uM | 6 h | Selective serotonin reuptake inhibitor (SSRI) | −0.7 |
| Dazoxiben | MDAMB231 | 2.22 uM | 24 h | Thromboxane synthase inhibitor | −0.7 |
| CDC | MCF7 | 10 uM | 6 h | Lipoxygenase inhibitor | −0.7 |
| BRL-37344 | NPC | 10 uM | 24 h | Adrenergic receptor agonist | −0.7 |
| Ursolic-acid | A375 | 70 uM | 24 h | 11-beta-HSD1 inhibitor|Acetylcholinesterase inhibitor|ATPase inhibitor|Caspase inhibitor|HIV protease inhibitor|Lipid peroxidase inhibitor|NFKB inhibitor|Quorum sensing signaling modulator|STAT inhibitor|Steryl sulfatase inhibitor|Tyrosine phosphatase inhibitor | −0.7 |
| Meglitinide | YAPC | 0.08 uM | 24 h | Potassium channel antagonist | −0.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Y.; Zhang, B.; Cloyd, J.M.; Alaimo, L.; Xu, G.; Du, S.; Mao, Y.; Pawlik, T.M. Novel Drug Candidate Prediction for Intrahepatic Cholangiocarcinoma via Hub Gene Network Analysis and Connectivity Mapping. Cancers 2022, 14, 3284. https://doi.org/10.3390/cancers14133284
Xiao Y, Zhang B, Cloyd JM, Alaimo L, Xu G, Du S, Mao Y, Pawlik TM. Novel Drug Candidate Prediction for Intrahepatic Cholangiocarcinoma via Hub Gene Network Analysis and Connectivity Mapping. Cancers. 2022; 14(13):3284. https://doi.org/10.3390/cancers14133284
Chicago/Turabian StyleXiao, Yao, Baoluhe Zhang, Jordan M. Cloyd, Laura Alaimo, Gang Xu, Shunda Du, Yilei Mao, and Timothy M. Pawlik. 2022. "Novel Drug Candidate Prediction for Intrahepatic Cholangiocarcinoma via Hub Gene Network Analysis and Connectivity Mapping" Cancers 14, no. 13: 3284. https://doi.org/10.3390/cancers14133284
APA StyleXiao, Y., Zhang, B., Cloyd, J. M., Alaimo, L., Xu, G., Du, S., Mao, Y., & Pawlik, T. M. (2022). Novel Drug Candidate Prediction for Intrahepatic Cholangiocarcinoma via Hub Gene Network Analysis and Connectivity Mapping. Cancers, 14(13), 3284. https://doi.org/10.3390/cancers14133284