
Breast Cancer Stem Cells: Signaling Pathways, Cellular Interactions, and Therapeutic Implications

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
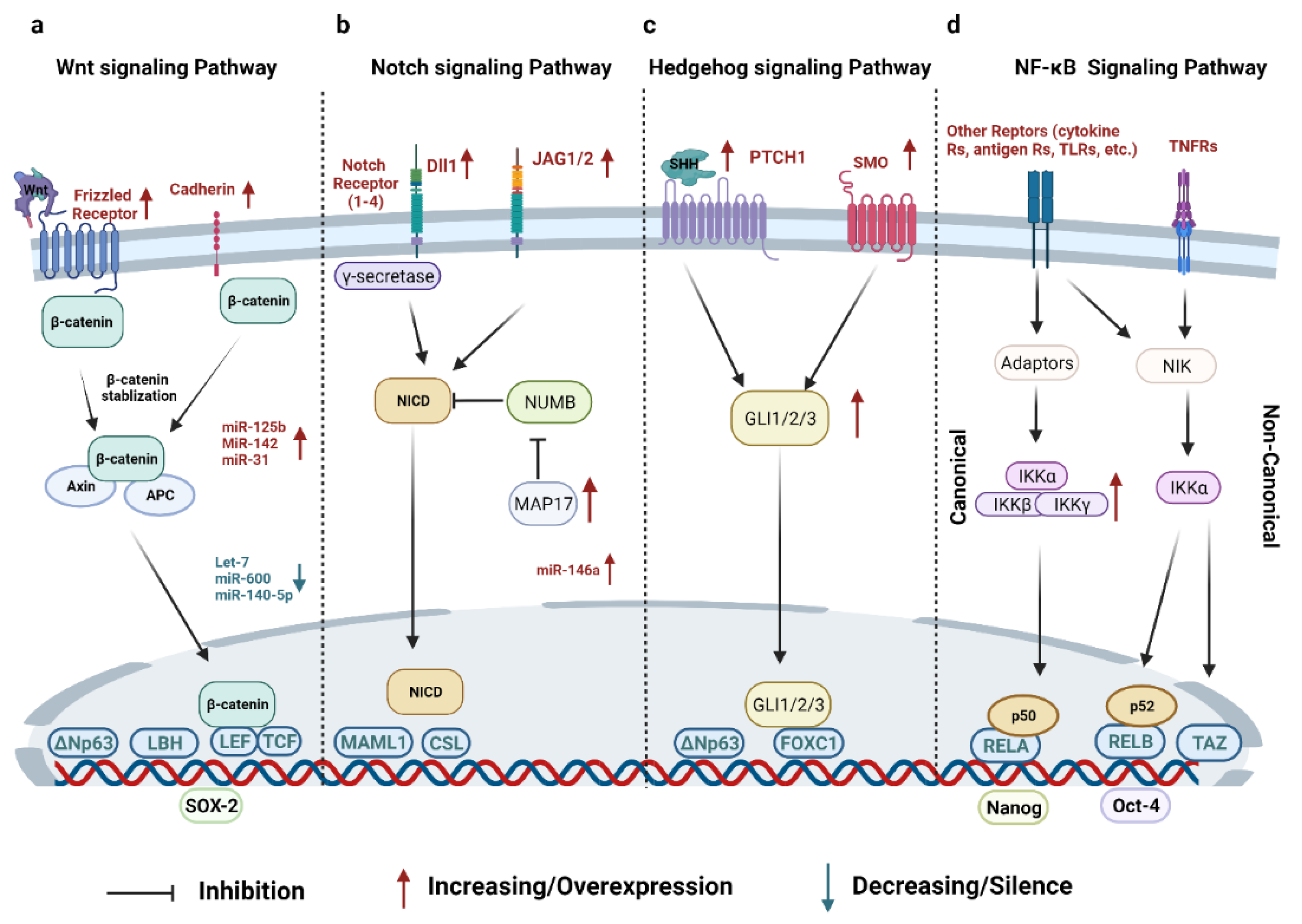
2. Intrinsic Molecular Activities of BCSCs
2.1. Signaling Pathways
2.1.1. Wnt Signaling Pathway
2.1.2. Notch Signaling Pathway
2.1.3. Hedgehog (HH) Signaling Pathway
2.1.4. NF-κB Signaling Pathway
2.1.5. Other Signaling Pathways
2.2. Other Transcription Factors
3. Cellular Crosstalk between BCSCs and Different Cell Populations in the TME
3.1. Adipocytes
3.2. Fibroblasts
3.3. Endothelial Cells and Vasculogenic Mimicry
3.4. Immune Cells
4. Developing Therapeutics for Eliminating BCSCs in Clinical Trials
4.1. Targeting Surface Markers
4.2. Targeting the Crucial Signaling Pathway of BCSCs
4.2.1. Targeting Wnt Signaling Pathway
4.2.2. Targeting Notch Signaling Pathway
4.2.3. Targeting Hedgehog (HH) Signaling Pathway
4.3. Targeting the Components in the BCSC Microenvironment—VEGF
4.4. Immunotherapy
4.4.1. Vaccines
4.4.2. CAR-T Cell Therapy
4.4.3. Inhibition of Immune Signaling Receptors
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Riggio, A.I.; Varley, K.E.; Welm, A.L. The lingering mysteries of metastatic recurrence in breast cancer. Br. J. Cancer 2021, 124, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.K.; Guan, J.-L. Breast cancer: Multiple subtypes within a tumor? Trends Cancer 2017, 3, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.; Fulton, R.; McLellan, M.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.; Fulton, L.; Dooling, D.; Ding, L.; Mardis, E. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Fougner, C.; Bergholtz, H.; Norum, J.H.; Sorlie, T. Re-definition of claudin-low as a breast cancer phenotype. Nat. Commun. 2020, 11, 1787. [Google Scholar] [CrossRef] [Green Version]
- Pommier, R.M.; Sanlaville, A.; Tonon, L.; Kielbassa, J.; Thomas, E.; Ferrari, A.; Sertier, A.-S.; Hollande, F.; Martinez, P.; Tissier, A. Comprehensive characterization of claudin-low breast tumors reflects the impact of the cell-of-origin on cancer evolution. Nat. Commun. 2020, 11, 3431. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sotiriou, C. Luminal breast cancer: From biology to treatment. Nat. Rev. Clin. Oncol. 2013, 10, 494–506. [Google Scholar] [CrossRef]
- Dent, S.; Ammendolea, C.; Christofides, A.; Edwards, S.; Incekol, D.; Pourmirza, B.; Kfoury, S.; Poirier, B. A multidisciplinary perspective on the subcutaneous administration of trastuzumab in HER2-positive breast cancer. Curr. Oncol. 2019, 26, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Nunes, M.R.; Stearns, V. CDK4/6 inhibitors: Game changers in the management of hormone receptor–positive advanced breast cancer? Oncology 2018, 32, 216. [Google Scholar]
- Lee, J.J.; Loh, K.; Yap, Y.-S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol. Med. 2015, 12, 342. [Google Scholar]
- Butti, R.; Das, S.; Gunasekaran, V.P.; Yadav, A.S.; Kumar, D.; Kundu, G.C. Receptor tyrosine kinases (RTKs) in breast cancer: Signaling, therapeutic implications and challenges. Mol. Cancer 2018, 17, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keenan, T.E.; Guerriero, J.L.; Barroso-Sousa, R.; Li, T.; O’Meara, T.; Giobbie-Hurder, A.; Tayob, N.; Hu, J.; Severgnini, M.; Agudo, J. Molecular correlates of response to eribulin and pembrolizumab in hormone receptor-positive metastatic breast cancer. Nat. Commun. 2021, 12, 5563. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Farzaneh, M. Signaling pathways governing breast cancer stem cells behavior. Stem Cell Res. Ther. 2021, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, I.; Miele, L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013, 341, 41–45. [Google Scholar] [CrossRef]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Reavie, L.; Couto, J.P.; De Silva, D.; Stadler, M.B.; Roloff, T.; Britschgi, A.; Eichlisberger, T.; Kohler, H.; Aina, O. PIK3CAH1047R induces multipotency and multi-lineage mammary tumours. Nature 2015, 525, 114–118. [Google Scholar] [CrossRef]
- Poli, V.; Fagnocchi, L.; Fasciani, A.; Cherubini, A.; Mazzoleni, S.; Ferrillo, S.; Miluzio, A.; Gaudioso, G.; Vaira, V.; Turdo, A. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat. Commun. 2018, 9, 1024. [Google Scholar] [CrossRef] [Green Version]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—what challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.; Labots, M. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honeth, G.; Bendahl, P.-O.; Ringnér, M.; Saal, L.H.; Gruvberger-Saal, S.K.; Lövgren, K.; Grabau, D.; Fernö, M.; Borg, Å.; Hegardt, C. The CD44+/CD24-phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008, 10, R53. [Google Scholar] [CrossRef] [Green Version]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitão, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011, 64, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Idowu, M.O.; Kmieciak, M.; Dumur, C.; Burton, R.S.; Grimes, M.M.; Powers, C.N.; Manjili, M.H. CD44+/CD24−/low cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum. Pathol. 2012, 43, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Xie, Y.; Baer, M.R.; Ross, D.D. Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem. Pharmacol. 2012, 83, 1084–1103. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Ye, Y.; Yearsley, K.; Jones, S.; Barsky, S.H. The lymphovascular embolus of inflammatory breast cancer expresses a stem cell-like phenotype. Am. J. Pathol. 2008, 173, 561–574. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Zhong, X.; Qiu, Y.; Yang, L.; Wei, B.; Zhang, Z.; Bu, H. CD49f can act as a biomarker for local or distant recurrence in breast cancer. J. Breast Cancer 2017, 20, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Tang, H.; Kong, Y.; Xie, X.; Chen, J.; Song, C.; Liu, X.; Ye, F.; Li, N.; Wang, N. LGR5 promotes breast cancer progression and maintains stem-like cells through activation of Wnt/β-catenin signaling. Stem Cells 2015, 33, 2913–2924. [Google Scholar] [CrossRef]
- Cheung, S.K.; Chuang, P.-K.; Huang, H.-W.; Hwang-Verslues, W.W.; Cho, C.H.-H.; Yang, W.-B.; Shen, C.-N.; Hsiao, M.; Hsu, T.-L.; Chang, C.-F. Stage-specific embryonic antigen-3 (SSEA-3) and β3GalT5 are cancer specific and significant markers for breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2016, 113, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yin, B.; Yi, Z.; Liu, X.; Hu, Z.; Gao, W.; Yu, H.; Li, Q. Breast cancer stem cells characterized by CD70 expression preferentially metastasize to the lungs. Breast Cancer 2018, 25, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, X.; Liu, C.; Jia, Y.; Bai, Y.; Cai, C.; Wang, J.; Bai, L.; Yang, R.; Lin, C. Protein C receptor is a therapeutic stem cell target in a distinct group of breast cancers. Cell Res. 2019, 29, 832–845. [Google Scholar] [CrossRef] [PubMed]
- Siddharth, S.; Goutam, K.; Das, S.; Nayak, A.; Nayak, D.; Sethy, C.; Wyatt, M.D.; Kundu, C.N. Nectin-4 is a breast cancer stem cell marker that induces WNT/β-catenin signaling via Pi3k/Akt axis. Int. J. Biochem. Cell Biol. 2017, 89, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Ito, S.; Nakamura, H. Ep CAM expression in breast cancer cells is associated with enhanced bone metastasis formation. Int. J. Cancer 2016, 138, 1698–1708. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Fröse, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- Vassilopoulos, A.; Chisholm, C.; Lahusen, T.; Zheng, H.; Deng, C. A critical role of CD29 and CD49f in mediating metastasis for cancer-initiating cells isolated from a Brca1-associated mouse model of breast cancer. Oncogene 2014, 33, 5477–5482. [Google Scholar] [CrossRef]
- Vaillant, F.; Asselin-Labat, M.-L.; Shackleton, M.; Forrest, N.C.; Lindeman, G.J.; Visvader, J.E. The mammary progenitor marker CD61/β3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. 2008, 68, 7711–7717. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.A.; Nusse, R. Wnt proteins are self-renewal factors for mammary stem cells and promote their long-term expansion in culture. Cell Stem Cell 2010, 6, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Welm, B.; Podsypanina, K.; Huang, S.; Chamorro, M.; Zhang, X.; Rowlands, T.; Egeblad, M.; Cowin, P.; Werb, Z.; et al. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc. Natl. Acad. Sci. USA 2003, 100, 15853–15858. [Google Scholar] [CrossRef] [Green Version]
- Isobe, T.; Hisamori, S.; Hogan, D.J.; Zabala, M.; Hendrickson, D.G.; Dalerba, P.; Cai, S.; Scheeren, F.; Kuo, A.H.; Sikandar, S.S. miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. eLife 2014, 3, e01977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, G.-B.; Kim, J.-Y.; Cho, S.-D.; Park, K.-S.; Jung, J.-Y.; Lee, H.-Y.; Hong, I.-S.; Nam, J.-S. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Lu, P.; Zhang, H.; Xu, H.; Gao, N.; Li, M.; Liu, C. Nestin positively regulates the Wnt/beta-catenin pathway and the proliferation, survival and invasiveness of breast cancer stem cells. Breast Cancer Res. 2014, 16, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Zhang, M.; Zhang, Y.; Zhou, W.; Zhu, T.; Ruan, Q.; Chen, H.; Fang, J.; Zhou, F.; Sun, J.; et al. WNT5B governs the phenotype of basal-like breast cancer by activating WNT signaling. Cell Commun. Signal. 2019, 17, 109. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Prosperi, J.R.; Choudhury, N.; Olopade, O.I.; Goss, K.H. β-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS ONE 2015, 10, e0117097. [Google Scholar] [CrossRef] [Green Version]
- DiMeo, T.A.; Anderson, K.; Phadke, P.; Feng, C.; Perou, C.M.; Naber, S.; Kuperwasser, C. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009, 69, 5364–5373. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, R.; Wei, Y.; Hwang, J.; Hang, X.; Andres Blanco, M.; Choudhury, A.; Tiede, B.; Romano, R.-A.; DeCoste, C.; Mercatali, L. ΔNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat. Cell Biol. 2014, 16, 1004–1015. [Google Scholar] [CrossRef]
- Garikapati, K.; Ashad-Bishop, K.; Hong, S.; Qureshi, R.; Rieger, M.E.; Lindley, L.E.; Wang, B.; Azzam, D.J.; Khanlari, M.; Nadji, M. LBH is a cancer stem cell-and metastasis-promoting oncogene essential for WNT stem cell function in breast cancer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ashad-Bishop, K.; Garikapati, K.; Lindley, L.E.; Jorda, M.; Briegel, K.J. Loss of Limb-Bud-and-Heart (LBH) attenuates mammary hyperplasia and tumor development in MMTV-Wnt1 transgenic mice. Biochem. Biophys. Res. Commun. 2019, 508, 536–542. [Google Scholar] [CrossRef]
- Satriyo, P.B.; Bamodu, O.A.; Chen, J.-H.; Aryandono, T.; Haryana, S.M.; Yeh, C.-T.; Chao, T.-Y. Cadherin 11 inhibition downregulates β-catenin, deactivates the canonical WNT signalling pathway and suppresses the cancer stem cell-like phenotype of triple negative breast cancer. J. Clin. Med. 2019, 8, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, T.; Jiang, C.; Dai, T.; Xu, R.; Zhou, X.; Su, X.; Zhao, X. Knockdown of XB130 restrains cancer stem cell-like phenotype through inhibition of Wnt/β-Catenin signaling in breast cancer. Mol. Carcinog. 2019, 58, 1832–1845. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Deng, C.; Wang, J.; Xiao, J.; Gatalica, Z.; Recker, R.R.; Xiao, G.G. Let-7 family miRNAs regulate estrogen receptor alpha signaling in estrogen receptor positive breast cancer. Breast Cancer Res. Treat. 2011, 127, 69–80. [Google Scholar] [CrossRef]
- Sun, X.; Xu, C.; Tang, S.-C.; Wang, J.; Wang, H.; Wang, P.; Du, N.; Qin, S.; Li, G.; Xu, S. Let-7c blocks estrogen-activated Wnt signaling in induction of self-renewal of breast cancer stem cells. Cancer Gene Ther. 2016, 23, 83–89. [Google Scholar] [CrossRef]
- El Helou, R.; Pinna, G.; Cabaud, O.; Wicinski, J.; Bhajun, R.; Guyon, L.; Rioualen, C.; Finetti, P.; Gros, A.; Mari, B. miR-600 acts as a bimodal switch that regulates breast cancer stem cell fate through WNT signaling. Cell Rep. 2017, 18, 2256–2268. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Zhang, J.; Lu, Y.; Bo, S.; Li, L.; Wang, L.; Zhang, Q.; Mao, J. miR-140-5p inhibits the proliferation and enhances the efficacy of doxorubicin to breast cancer stem cells by targeting Wnt1. Cancer Gene Ther. 2019, 26, 74–82. [Google Scholar] [CrossRef]
- Wolfson, B.; Eades, G.; Zhou, Q. Roles of microRNA-140 in stem cell-associated early stage breast cancer. World J. Stem Cells 2014, 6, 591. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, H.; Desai, S.; Schmitt, D.C.; Zhou, M.; Khong, H.T.; Klos, K.S.; McClellan, S.; Fodstad, O.; Tan, M. miR-125b functions as a key mediator for snail-induced stem cell propagation and chemoresistance. J. Biol. Chem. 2013, 288, 4334–4345. [Google Scholar] [CrossRef] [Green Version]
- Lv, C.; Li, F.; Li, X.; Tian, Y.; Zhang, Y.; Sheng, X.; Song, Y.; Meng, Q.; Yuan, S.; Luan, L. MiR-31 promotes mammary stem cell expansion and breast tumorigenesis by suppressing Wnt signaling antagonists. Nat. Commun. 2017, 8, 1036. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Singh, S.R.; Hou, S.X. JAK–STAT is restrained by Notch to control cell proliferation of the Drosophila intestinal stem cells. J. Cell. Biochem. 2010, 109, 992–999. [Google Scholar] [PubMed] [Green Version]
- Ranganathan, P.; Weaver, K.L.; Capobianco, A.J. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat. Rev. Cancer 2011, 11, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Kontomanolis, E.N.; Kalagasidou, S.; Pouliliou, S.; Anthoulaki, X.; Georgiou, N.; Papamanolis, V.; Fasoulakis, Z.N. The notch pathway in breast cancer progression. Sci. World J. 2018, 2018, 2415489. [Google Scholar] [CrossRef] [Green Version]
- Sales-Dias, J.; Ferreira, A.; Lamy, M.; Domenici, G.; Monteiro, S.M.; Pires, A.; Lemos, A.R.; Kucheryava, K.; Nobre, L.S.; Sousa, P.M. Development of antibodies against the notch ligand Delta-Like-1 by phage display with activity against breast cancer cells. New Biotechnol. 2021, 64, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.E.; Nic An tSaoir, C.B.; Blayney, J.K.; Oram, L.C.; Crawford, N.T.; D’Costa, Z.C.; Quinn, J.E.; Kennedy, R.D.; Harkin, D.P.; Mullan, P.B. BRCA1 is a key regulator of breast differentiation through activation of Notch signalling with implications for anti-endocrine treatment of breast cancers. Nucleic Acids Res. 2013, 41, 8601–8614. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Taguchi, Y.; Ito-Kureha, T.; Semba, K.; Yamaguchi, N.; Inoue, J. NF-kappaB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat. Commun. 2013, 4, 2299. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Grivennikov, S.I. Top Notch cancer stem cells by paracrine NF-kappaB signaling in breast cancer. Breast Cancer Res 2013, 15, 316. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Okuda, H.; Watabe, M.; Kobayashi, A.; Pai, S.K.; Liu, W.; Pandey, P.R.; Fukuda, K.; Hirota, S.; Sugai, T. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene 2011, 30, 4075–4086. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Yu, J.; Park, A.; Dubon, M.J.; Do, J.; Kim, Y.; Nam, D.; Noh, J.; Park, K.-S. BMP-4 enhances epithelial mesenchymal transition and cancer stem cell properties of breast cancer cells via Notch signaling. Sci. Rep. 2019, 9, 11724. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Heredia, J.M.; Lucena-Cacace, A.; Verdugo-Sivianes, E.M.; Pérez, M.; Carnero, A. The cargo protein MAP17 (PDZK1IP1) regulates the cancer stem cell pool activating the Notch pathway by abducting NUMB. Clin. Cancer Res. 2017, 23, 3871–3883. [Google Scholar] [CrossRef] [Green Version]
- Shimono, Y.; Mukohyama, J.; Nakamura, S.-I.; Minami, H. MicroRNA regulation of human breast cancer stem cells. J. Clin. Med. 2015, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, S.T.; Gieniec, K.A.; Gregor, C.E.; Faulkner, J.W.; McColl, S.R.; Kochetkova, M. Interplay between CCR7 and Notch1 axes promotes stemness in MMTV-PyMT mammary cancer cells. Mol. Cancer 2017, 16, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhateja, P.; Cherian, M.; Majumder, S.; Ramaswamy, B. The Hedgehog signaling pathway: A viable target in breast cancer? Cancers 2019, 11, 1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.-S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [Green Version]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog—a cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Nakamura, M.; Kameda, C.; Kubo, M.; Sato, N.; Kuroki, S.; Tanaka, M.; Katano, M. The Hedgehog signaling pathway plays an essential role in maintaining the CD44+ CD24-/low subpopulation and the side population of breast cancer cells. Anticancer Res. 2009, 29, 2147–2157. [Google Scholar]
- Memmi, E.M.; Sanarico, A.G.; Giacobbe, A.; Peschiaroli, A.; Frezza, V.; Cicalese, A.; Pisati, F.; Tosoni, D.; Zhou, H.; Tonon, G. p63 Sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 3499–3504. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef] [Green Version]
- Goel, H.L.; Pursell, B.; Chang, C.; Shaw, L.M.; Mao, J.; Simin, K.; Kumar, P.; Kooi, C.W.V.; Shultz, L.D.; Greiner, D.L. GLI1 regulates a novel neuropilin-2/α6β1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO Mol. Med. 2013, 5, 488–508. [Google Scholar] [CrossRef]
- Han, B.; Qu, Y.; Jin, Y.; Yu, Y.; Deng, N.; Wawrowsky, K.; Zhang, X.; Li, N.; Bose, S.; Wang, Q. FOXC1 activates smoothened-independent hedgehog signaling in basal-like breast cancer. Cell Rep. 2015, 13, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nag, S.A.; Zhang, R. Targeting the NFκB signaling pathways for breast cancer prevention and therapy. Curr. Med. Chem. 2015, 22, 264–289. [Google Scholar] [CrossRef]
- Pires, B.R.; Mencalha, A.L.; Ferreira, G.M.; de Souza, W.F.; Morgado-Díaz, J.A.; Maia, A.M.; Corrêa, S.; Abdelhay, E.S. NF-kappaB is involved in the regulation of EMT genes in breast cancer cells. PLoS ONE 2017, 12, e0169622. [Google Scholar]
- Vazquez-Santillan, K.; Melendez-Zajgla, J.; Jimenez-Hernandez, L.E.; Gaytan-Cervantes, J.; Muñoz-Galindo, L.; Piña-Sanchez, P.; Martinez-Ruiz, G.; Torres, J.; Garcia-Lopez, P.; Gonzalez-Torres, C. NF-kappaB-inducing kinase regulates stem cell phenotype in breast cancer. Sci. Rep. 2016, 6, 37340. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Nandi, A.; Singh, S.; Regulapati, R.; Li, N.; Tobias, J.W.; Siebel, C.W.; Blanco, M.A.; Klein-Szanto, A.J.; Lengner, C. Dll1+ quiescent tumor stem cells drive chemoresistance in breast cancer through NF-κB survival pathway. Nat. Commun. 2021, 12, 432. [Google Scholar] [CrossRef]
- Kendellen, M.F.; Bradford, J.W.; Lawrence, C.L.; Clark, K.S.; Baldwin, A.S. Canonical and non-canonical NF-κB signaling promotes breast cancer tumor-initiating cells. Oncogene 2014, 33, 1297–1305. [Google Scholar] [CrossRef] [Green Version]
- Manupati, K.; Paul, R.; Hao, M.; Haas, M.; Bian, Z.C.; Holm, T.M.; Guan, J.-L.; Yeo, S.K. Biglycan Promotes Cancer Stem Cell Properties, NFκB Signaling and Metastatic Potential in Breast Cancer Cells. Cancers 2022, 14, 455. [Google Scholar] [CrossRef]
- Gonzalez-Suarez, E.; Jacob, A.P.; Jones, J.; Miller, R.; Roudier-Meyer, M.P.; Erwert, R.; Pinkas, J.; Branstetter, D.; Dougall, W.C. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010, 468, 103–107. [Google Scholar] [CrossRef]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Tan, W.; Wu, X.; Poustovoitov, M.; Strasner, A.; Li, W.; Borcherding, N.; Ghassemian, M.; Karin, M. A NIK-IKKalpha module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell 2013, 23, 647–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Luo, J.-L.; Karin, M. IκB kinase α kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15852–15857. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Lu, X.; Shi, P.; Yang, G.; Zhou, Z.; Li, W.; Mao, X.; Jiang, D.; Chen, C. TNF-α increases breast cancer stem-like cells through up-regulating TAZ expression via the non-canonical NF-κB pathway. Sci. Rep. 2020, 10, 1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Manupati, K.; Dhoke, N.R.; Debnath, T.; Yeeravalli, R.; Guguloth, K.; Saeidpour, S.; De, U.C.; Debnath, S.; Das, A. Inhibiting epidermal growth factor receptor signalling potentiates mesenchymal–epithelial transition of breast cancer stem cells and their responsiveness to anticancer drugs. FEBS J. 2017, 284, 1830–1854. [Google Scholar] [CrossRef] [Green Version]
- Alanazi, I.O.; Khan, Z. Understanding EGFR signaling in breast cancer and breast cancer stem cells: Overexpression and therapeutic implications. Asian Pac. J. Cancer Prev. 2016, 17, 445–453. [Google Scholar] [CrossRef]
- Chakrabarty, A.; Sánchez, V.; Kuba, M.G.; Rinehart, C.; Arteaga, C.L. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 2718–2723. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Saenz, A.; Dreyer, C.; Campbell, M.R.; Steri, V.; Gulizia, N.; Moasser, M.M. HER2 Amplification in Tumors Activates PI3K/Akt Signaling Independent of HER3HER2-Amplified Tumors Overcome the Requirement for HER3. Cancer Res. 2018, 78, 3645–3658. [Google Scholar] [CrossRef] [Green Version]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas III, C.F.; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.; Osipo, C. Cancer stem cells and HER2 positive breast cancer: The story so far. Genes Dis. 2016, 3, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.-J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y. JAK/STAT3-regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 2018, 27, 136–150.e135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruna, A.; Greenwood, W.; Le Quesne, J.; Teschendorff, A.; Miranda-Saavedra, D.; Rueda, O.M.; Sandoval, J.L.; Vidakovic, A.T.; Saadi, A.; Pharoah, P. TGFβ induces the formation of tumour-initiating cells in claudinlow breast cancer. Nat. Commun. 2012, 3, 1055. [Google Scholar] [CrossRef] [Green Version]
- Lo, P.-K.; Kanojia, D.; Liu, X.; Singh, U.P.; Berger, F.G.; Wang, Q.; Chen, H. CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin–TGFβ signaling. Oncogene 2012, 31, 2614–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef]
- Zhang, C.; Gao, H.; Li, C.; Tu, J.; Chen, Z.; Su, W.; Geng, X.; Chen, X.; Wang, J.; Pan, W. TGFβ1 promotes breast cancer local invasion and liver metastasis by increasing the CD44high/CD24− subpopulation. Technol. Cancer Res. Treat. 2018, 17, 1533033818764497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, Z.Z. Mechanisms that mediate stem cell self-renewal and differentiation. J. Cell. Biochem. 2008, 103, 709–718. [Google Scholar] [CrossRef]
- Papp, B.; Plath, K. Reprogramming to pluripotency: Stepwise resetting of the epigenetic landscape. Cell Res. 2011, 21, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Lu, P.; Zhang, H.; Luo, M.; Zhang, X.; Wei, X.; Gao, J.; Zhao, Z.; Liu, C. Oct-4 and Nanog promote the epithelial-mesenchymal transition of breast cancer stem cells and are associated with poor prognosis in breast cancer patients. Oncotarget 2014, 5, 10803. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.-J.; Nam, J.-S. OCT4 expression enhances features of cancer stem cells in a mouse model of breast cancer. Lab. Anim. Res. 2011, 27, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Ghanei, Z.; Jamshidizad, A.; Joupari, M.D.; Shamsara, M. Isolation and characterization of breast cancer stem cell-like phenotype by Oct4 promoter-mediated activity. J. Cell. Physiol. 2020, 235, 7840–7848. [Google Scholar] [CrossRef] [PubMed]
- Chambers, I.; Colby, D.; Robertson, M.; Nichols, J.; Lee, S.; Tweedie, S.; Smith, A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 2003, 113, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Mazur, S.J.; Lin, T.; Appella, E.; Xu, Y. The pluripotency factor nanog promotes breast cancer tumorigenesis and metastasis. Oncogene 2014, 33, 2655–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leis, O.; Eguiara, A.; Lopez-Arribillaga, E.; Alberdi, M.; Hernandez-Garcia, S.; Elorriaga, K.; Pandiella, A.; Rezola, R.; Martin, A. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 2012, 31, 1354–1365. [Google Scholar] [CrossRef] [Green Version]
- Lengerke, C.; Fehm, T.; Kurth, R.; Neubauer, H.; Scheble, V.; Müller, F.; Schneider, F.; Petersen, K.; Wallwiener, D.; Kanz, L. Expression of the embryonic stem cell marker SOX2 in early-stage breast carcinoma. BMC Cancer 2011, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Xie, C.-M.; Li, H.; Tan, M.; Chen, G.; Schiff, R.; Xiong, X.; Sun, Y. The FBXW2–MSX2–SOX2 axis regulates stem cell property and drug resistance of cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 20528–20538. [Google Scholar] [CrossRef] [Green Version]
- Piva, M.; Domenici, G.; Iriondo, O.; Rábano, M.; Simoes, B.M.; Comaills, V.; Barredo, I.; López-Ruiz, J.A.; Zabalza, I.; Kypta, R. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 2014, 6, 66–79. [Google Scholar] [CrossRef]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef]
- Kolb, R.; Zhang, W. Obesity and breast cancer: A case of inflamed adipose tissue. Cancers 2020, 12, 1686. [Google Scholar] [CrossRef]
- Rausch, L.K.; Netzer, N.C.; Hoegel, J.; Pramsohler, S. The linkage between breast cancer, hypoxia, and adipose tissue. Front. Oncol. 2017, 7, 211. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef] [PubMed]
- Päth, G.n.; Bornstein, S.R.; Gurniak, M.; Chrousos, G.P.; Scherbaum, W.A.; Hauner, H. Human breast adipocytes express interleukin-6 (IL-6) and its receptor system: Increased IL-6 production by β-adrenergic activation and effects of IL-6 on adipocyte function. J. Clin. Endocrinol. Metab. 2001, 86, 2281–2288. [Google Scholar]
- Korkaya, H.; Kim, G.-I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, W.B.; Yao, M.; Brummer, G.; Acevedo, D.; Alhakamy, N.; Berkland, C.; Cheng, N. Targeted gene silencing of CCL2 inhibits triple negative breast cancer progression by blocking cancer stem cell renewal and M2 macrophage recruitment. Oncotarget 2016, 7, 49349. [Google Scholar] [CrossRef]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Mumick, S.; Zhang, C.; Lamb, J.; Dai, H.; Weingarth, D.; Mudgett, J.; Chen, H.; MacNeil, D.J.; Reitman, M.L. Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obes. Res. 2005, 13, 1311–1320. [Google Scholar] [CrossRef]
- Arendt, L.M.; McCready, J.; Keller, P.J.; Baker, D.D.; Naber, S.P.; Seewaldt, V.; Kuperwasser, C. Obesity promotes breast cancer by CCL2-mediated macrophage recruitment and angiogenesis. Cancer Res. 2013, 73, 6080–6093. [Google Scholar] [CrossRef] [Green Version]
- Schäffler, A.; Schölmerich, J.; Buechler, C. Mechanisms of disease: Adipokines and breast cancer—endocrine and paracrine mechanisms that connect adiposity and breast cancer. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 345–354. [Google Scholar] [CrossRef]
- Gui, Y.; Pan, Q.; Chen, X.; Xu, S.; Luo, X.; Chen, L. The association between obesity related adipokines and risk of breast cancer: A meta-analysis. Oncotarget 2017, 8, 75389. [Google Scholar] [CrossRef] [Green Version]
- Bowers, L.W.; Rossi, E.L.; McDonell, S.B.; Doerstling, S.S.; Khatib, S.A.; Lineberger, C.G.; Albright, J.E.; Tang, X.; DeGraffenried, L.A.; Hursting, S.D. Leptin signaling mediates obesity-associated CSC enrichment and EMT in preclinical TNBC models. Mol. Cancer Res. 2018, 16, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Esper, R.M.; Dame, M.; McClintock, S.; Holt, P.R.; Dannenberg, A.J.; Wicha, M.S.; Brenner, D.E. Leptin and adiponectin modulate the self-renewal of normal human breast epithelial stem cells. Cancer Prev. Res. 2015, 8, 1174–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, D.E.; Chen, C.; Punj, V.; Tsukamoto, H.; Machida, K. Pluripotency factor-mediated expression of the leptin receptor (OB-R) links obesity to oncogenesis through tumor-initiating stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 829–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, C.; Vizza, D.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Sisci, D.; De Amicis, F.; Fuqua, S.A.; Catalano, S. Leptin increases HER2 protein levels through a STAT3-mediated up-regulation of Hsp90 in breast cancer cells. Mol. Oncol. 2013, 7, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Banaszak, L.; Fracci, S.; Basali, D.; Dunlap, S.M.; Hursting, S.D.; Rich, J.N.; Hjlemeland, A.B.; Vasanji, A.; Berger, N.A. Leptin receptor maintains cancer stem-like properties in triple negative breast cancer cells. Endocr.-Relat. Cancer 2013, 20, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Shimono, Y.; Funakoshi, Y.; Imamura, Y.; Toyoda, M.; Kiyota, N.; Kono, S.; Takao, S.; Mukohara, T.; Minami, H. Adipose-derived stem cells enhance human breast cancer growth and cancer stem cell-like properties through adipsin. Oncogene 2019, 38, 767–779. [Google Scholar] [CrossRef]
- Mizuno, M.; Khaledian, B.; Maeda, M.; Hayashi, T.; Mizuno, S.; Munetsuna, E.; Watanabe, T.; Kono, S.; Okada, S.; Suzuki, M. Adipsin-dependent secretion of hepatocyte growth factor regulates the adipocyte-cancer stem cell interaction. Cancers 2021, 13, 4238. [Google Scholar] [CrossRef]
- Zhang, F.; Song, C.; Ma, Y.; Tang, L.; Xu, Y.; Wang, H. Effect of fibroblasts on breast cancer cell mammosphere formation and regulation of stem cell-related gene expression. Int. J. Mol. Med. 2011, 28, 365–371. [Google Scholar]
- Wang, B.; Xi, C.; Liu, M.; Sun, H.; Liu, S.; Song, L.; Kang, H. Breast fibroblasts in both cancer and normal tissues induce phenotypic transformation of breast cancer stem cells: A preliminary study. PeerJ 2018, 6, e4805. [Google Scholar] [CrossRef]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.-L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, J.; Sun, X.; Li, M. EMMPRIN down-regulating miR-106a/b modifies breast cancer stem-like cell properties via interaction with fibroblasts through STAT3 and HIF-1α. Sci. Rep. 2016, 6, 28329. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F. CD10+ GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 2018, 172, 841–856.e816. [Google Scholar] [CrossRef]
- Boesch, M.; Onder, L.; Cheng, H.-W.; Novkovic, M.; Mörbe, U.; Sopper, S.; Gastl, G.; Jochum, W.; Ruhstaller, T.; Knauer, M. Interleukin 7-expressing fibroblasts promote breast cancer growth through sustenance of tumor cell stemness. Oncoimmunology 2018, 7, e1414129. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Berishaj, M.; Rajasekhar, V.K.; Ceccarelli, C.; Chang, Q.; Strillacci, A.; Savini, C.; Shapiro, L.; Bowman, R.L.; Mastroleo, C. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res. 2017, 77, 1927–1941. [Google Scholar] [CrossRef] [Green Version]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Rafii, S.; Rafii, A. Endothelial cells provide a notch-dependent pro-tumoral niche for enhancing breast cancer survival, stemness and pro-metastatic properties. PLoS ONE 2014, 9, e112424. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhou, C.; Zhang, Z.; Wang, Q.; Wei, H.; Shi, W.; Li, J.; Wang, Z.; Ou, Y.; Wang, W. Jagged1-Notch1-deployed tumor perivascular niche promotes breast cancer stem cell phenotype through Zeb1. Nat. Commun. 2020, 11, 5129. [Google Scholar] [CrossRef] [PubMed]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Halabi, N.; Guerrouahen, B.S.; Rafii, S.; Rafii, A. Breast cancer cells promote a notch-dependent mesenchymal phenotype in endothelial cells participating to a pro-tumoral niche. J. Transl. Med. 2015, 13, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Guo, Y.; Hao, J.; Guenter, R.; Lathia, J.; Beck, A.W.; Hattaway, R.; Hurst, D.; Wang, Q.J.; Liu, Y. Development of an arteriolar niche and self-renewal of breast cancer stem cells by lysophosphatidic acid/protein kinase D signaling. Commun. Biol. 2021, 4, 780. [Google Scholar] [CrossRef]
- Bussolati, B.; Grange, C.; Sapino, A.; Camussi, G. Endothelial cell differentiation of human breast tumour stem/progenitor cells. J. Cell. Mol. Med. 2009, 13, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Izawa, Y.; Kashii-Magaribuchi, K.; Yoshida, K.; Nosaka, M.; Tsuji, N.; Yamamoto, A.; Kuroyanagi, K.; Tono, K.; Tanihata, M.; Imanishi, M. Stem-like human breast cancer cells initiate vasculogenic mimicry on matrigel. Acta Histochem. ET Cytochem. 2018, 51, 18041. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Zhu, L.; Huang, Z.; Luo, C.; Zhou, T.; Li, L.; Wang, G.; Yang, Z.; Qi, W.; Yang, X. Stem-like tumor cells involved in heterogeneous vasculogenesis in breast cancer. Endocr. -Relat. Cancer 2020, 27, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Yu, F.; Yao, H.; Cui, X.; Jiao, Y.; Lin, L.; Chen, J.; Yin, D.; Song, E.; Liu, Q. miR-27a regulates endothelial differentiation of breast cancer stem like cells. Oncogene 2014, 33, 2629–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, E.M.; Kendsersky, N.D.; Daniel, C.J.; Kuziel, G.M.; Pelz, C.; Murphy, K.M.; Capecchi, M.R.; Sears, R.C. ZEB1-repressed microRNAs inhibit autocrine signaling that promotes vascular mimicry of breast cancer cells. Oncogene 2018, 37, 1005–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Yang, Z.; Chen, F.; Jiang, Y.; Fu, C.; Wang, Y.; Lu, R.; Wu, H. Autophagy is essential for the endothelial differentiation of breast cancer stem-like cells. Int. J. Mol. Med. 2020, 45, 255–264. [Google Scholar] [CrossRef]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [Google Scholar] [CrossRef]
- Elaimy, A.L.; Guru, S.; Chang, C.; Ou, J.; Amante, J.J.; Zhu, L.J.; Goel, H.L.; Mercurio, A.M. VEGF–neuropilin-2 signaling promotes stem-like traits in breast cancer cells by TAZ-mediated repression of the Rac GAP β2-chimaerin. Sci. Signal. 2018, 11, eaao6897. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, H.; Li, C.; Zhao, Y.; Wu, L.; Du, X.; Han, Z. VEGF-A/Neuropilin 1 pathway confers Cancer Stemness via activating Wnt/β-catenin Axis in breast Cancer cells. Cell. Physiol. Biochem. 2017, 44, 1251–1262. [Google Scholar] [CrossRef] [Green Version]
- Maroufi, N.F.; Rashidi, M.; Vahedian, V.; Jahanbazi, R.; Mostafaei, S.; Akbarzadeh, M.; Kazemzadeh, H.; Nejabati, H.-R.; Isazadeh, A.; Rashidi, M.-R. Effect of Apatinib plus melatonin on vasculogenic mimicry formation by cancer stem cells from breast cancer cell line. Breast Cancer 2021, 29, 260–273. [Google Scholar] [CrossRef]
- Liu, T.; Sun, B.; Zhao, X.; Zhao, X.; Sun, T.; Gu, Q.; Yao, Z.; Dong, X.; Zhao, N.; Liu, N. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013, 32, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Yao, N.; Cheng, S.; Li, L.; Liu, S.; Yang, Z.; Shang, G.; Zhang, D.; Yao, Z. Cancer stem-like cells directly participate in vasculogenic mimicry channels in triple-negative breast cancer. Cancer Biol. Med. 2019, 16, 299. [Google Scholar]
- Ferguson, L.P.; Diaz, E.; Reya, T. The role of the microenvironment and immune system in regulating stem cell fate in cancer. Trends Cancer 2021, 7, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Agudo, J.; Park, E.S.; Rose, S.A.; Alibo, E.; Sweeney, R.; Dhainaut, M.; Kobayashi, K.S.; Sachidanandam, R.; Baccarini, A.; Merad, M. Quiescent tissue stem cells evade immune surveillance. Immunity 2018, 48, 271–285.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, M.M.L.; Lee, T.K.W. Cancer stem cells: Emerging key players in immune evasion of cancers. Front. Cell Dev. Biol. 2021, 9, 1643. [Google Scholar] [CrossRef]
- Jelenčić, V.; Šestan, M.; Kavazović, I.; Lenartić, M.; Marinović, S.; Holmes, T.D.; Prchal-Murphy, M.; Lisnić, B.; Sexl, V.; Bryceson, Y.T. NK cell receptor NKG2D sets activation threshold for the NCR1 receptor early in NK cell development. Nat. Immunol. 2018, 19, 1083–1092. [Google Scholar]
- Wang, B.; Wang, Q.; Wang, Z.; Jiang, J.; Yu, S.-C.; Ping, Y.-F.; Yang, J.; Xu, S.-L.; Ye, X.-Z.; Xu, C. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014, 74, 5746–5757. [Google Scholar] [CrossRef] [Green Version]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; De Stanchina, E.; Massagué, J. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [Green Version]
- Stein, R.G.; Ebert, S.; Schlahsa, L.; Scholz, C.J.; Braun, M.; Hauck, P.; Horn, E.; Monoranu, C.M.; Thiemann, V.J.; Wustrow, M.P.; et al. Cognate Nonlytic Interactions between CD8(+) T Cells and Breast Cancer Cells Induce Cancer Stem Cell-like Properties. Cancer Res 2019, 79, 1507–1519. [Google Scholar] [CrossRef] [Green Version]
- Celià-Terrassa, T.; Liu, D.D.; Choudhury, A.; Hang, X.; Wei, Y.; Zamalloa, J.; Alfaro-Aco, R.; Chakrabarti, R.; Jiang, Y.-Z.; Koh, B.I. Normal and cancerous mammary stem cells evade interferon-induced constraint through the miR-199a–LCOR axis. Nat. Cell Biol. 2017, 19, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chen, M.; Wu, P.; Chen, C.; Xu, Z.P.; Gu, W. Increased PD-L1 expression in breast and colon cancer stem cells. Clin. Exp. Pharmacol. Physiol. 2017, 44, 602–604. [Google Scholar] [CrossRef]
- Hsu, J.-M.; Xia, W.; Hsu, Y.-H.; Chan, L.-C.; Yu, W.-H.; Cha, J.-H.; Chen, C.-T.; Liao, H.-W.; Kuo, C.-W.; Khoo, K.-H. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 2018, 9, 1908. [Google Scholar] [CrossRef]
- Tseng, D.; Volkmer, J.-P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M. Anti-CD47 antibody–mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Fan, J.; Maity, S.; McFadden, G.; Shi, Y.; Kong, W. PD-L1 interacts with Frizzled 6 to activate β-catenin and form a positive feedback loop to promote cancer stem cell expansion. Oncogene 2022, 41, 1100–1113. [Google Scholar] [CrossRef] [PubMed]
- Drukker, M.; Benvenisty, N. The immunogenicity of human embryonic stem-derived cells. Trends Biotechnol. 2004, 22, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.-C.; Kersbergen, A.; Lam, E.Y.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell 2019, 36, 385–401.e388. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Núez, I.; Rozalén, C.; Palomeque, J.Á.; Sangrador, I.; Dalmau, M.; Comerma, L.; Hernández-Prat, A.; Casadevall, D.; Menendez, S.; Liu, D.D. LCOR mediates interferon-independent tumor immunogenicity and responsiveness to immune-checkpoint blockade in triple-negative breast cancer. Nat. Cancer 2022, 3, 355–370. [Google Scholar]
- Xu, Y.; Dong, X.; Qi, P.; Ye, Y.; Shen, W.; Leng, L.; Wang, L.; Li, X.; Luo, X.; Chen, Y. Sox2 communicates with tregs through CCL1 to promote the stemness property of breast cancer cells. Stem Cells 2017, 35, 2351–2365. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Tu, J.; Zhou, L.; Dong, M.; Fan, J.; Chang, Z.; Zhang, L.; Bian, X.; Liu, S. Single-cell transcriptomics reveal the heterogeneity and dynamic of cancer stem-like cells during breast tumor progression. Cell Death Dis. 2021, 12, 979. [Google Scholar] [CrossRef]
- Ouzounova, M.; Lee, E.; Piranlioglu, R.; El Andaloussi, A.; Kolhe, R.; Demirci, M.F.; Marasco, D.; Asm, I.; Chadli, A.; Hassan, K.A. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun. 2017, 8, 14979. [Google Scholar] [CrossRef]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Bai, L.; Cao, F.; Wang, S.; He, H.; Song, M.; Chen, H.; Liu, Y.; Guo, J.; Si, Q. Targeting LIN28B reprograms tumor glucose metabolism and acidic microenvironment to suppress cancer stemness and metastasis. Oncogene 2019, 38, 4527–4539. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Xia, Y.; Wu, Y.; Zhang, Z.; Wang, X.; Lu, L.; Dai, C.; Song, Y.; Xu, K.; Ji, W. Lin28B-high breast cancer cells promote immune suppression in the lung pre-metastatic niche via exosomes and support cancer progression. Nat. Commun. 2022, 13, 897. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Wicha, M.S. Targeting breast cancer stem cells. J. Clin. Oncol. 2010, 28, 4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Liu, C.; Yao, J.; Wan, H.; Wan, G.; Li, Y.; Chen, N. Breast cancer stem cells, heterogeneity, targeting therapies and therapeutic implications. Pharmacol. Res. 2021, 163, 105320. [Google Scholar] [CrossRef]
- Marangoni, E.; Lecomte, N.; Durand, L.; De Pinieux, G.; Decaudin, D.; Chomienne, C.; Smadja-Joffe, F.; Poupon, M. CD44 targeting reduces tumour growth and prevents post-chemotherapy relapse of human breast cancers xenografts. Br. J. Cancer 2009, 100, 918–922. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Sarvestani, S.K.; Moeinzadeh, S.; He, X.; Jabbari, E. Effect of CD44 binding peptide conjugated to an engineered inert matrix on maintenance of breast cancer stem cells and tumorsphere formation. PLoS ONE 2013, 8, e59147. [Google Scholar] [CrossRef] [Green Version]
- McClements, L.; Yakkundi, A.; Papaspyropoulos, A.; Harrison, H.; Ablett, M.P.; Jithesh, P.V.; McKeen, H.D.; Bennett, R.; Donley, C.; Kissenpfennig, A. Targeting treatment-resistant breast cancer stem cells with FKBPL and its peptide derivative, AD-01, via the CD44 pathway. Clin. Cancer Res. 2013, 19, 3881–3893. [Google Scholar] [CrossRef] [Green Version]
- Brugnoli, F.; Grassilli, S.; Al-Qassab, Y.; Capitani, S.; Bertagnolo, V. CD133 in breast cancer cells: More than a stem cell marker. J. Oncol. 2019, 2019, 7512632. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, S.K.; Roger, E.; Toti, U.; Niu, L.; Ohlfest, J.R.; Panyam, J. CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J. Control. Release 2013, 171, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Yang, L.; Li, S.; Ye, D.; Yang, L.; Liu, Q.; Zhao, Z.; Cai, Q.; Tan, J.; Li, X. Quercetin inhibits breast cancer stem cells via downregulation of aldehyde dehydrogenase 1A1 (ALDH1A1), chemokine receptor type 4 (CXCR4), mucin 1 (MUC1), and epithelial cell adhesion molecule (EpCAM). Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesharwani, R.K.; Srivastava, V.; Singh, P.; Rizvi, S.I.; Adeppa, K.; Misra, K. A novel approach for overcoming drug resistance in breast cancer chemotherapy by targeting new synthetic curcumin analogues against aldehyde dehydrogenase 1 (ALDH1A1) and glycogen synthase kinase-3 β (GSK-3β). Appl. Biochem. Biotechnol. 2015, 176, 1996–2017. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Cho, Y.; Oh, E.; Lee, N.; An, H.; Sung, D.; Cho, T.-M.; Seo, J.H. Disulfiram targets cancer stem-like properties and the HER2/Akt signaling pathway in HER2-positive breast cancer. Cancer Lett. 2016, 379, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Sung, D.; Oh, E.; Cho, Y.; Cho, T.-M.; Farrand, L.; Seo, J.H.; Kim, J.Y. Flubendazole overcomes trastuzumab resistance by targeting cancer stem-like properties and HER2 signaling in HER2-positive breast cancer. Cancer Lett. 2018, 412, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Kim, Y.-J.; Park, S.; Park, M.; Farrand, L.; Nguyen, C.-T.; Ann, J.; Nam, G.; Park, H.-J.; Lee, J. A novel HSP90 inhibitor targeting the C-terminal domain attenuates trastuzumab resistance in HER2-positive breast cancer. Mol. Cancer 2020, 19, 161. [Google Scholar] [CrossRef]
- Ithimakin, S.; Day, K.C.; Malik, F.; Zen, Q.; Dawsey, S.J.; Bersano-Begey, T.F.; Quraishi, A.A.; Ignatoski, K.W.; Daignault, S.; Davis, A.; et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of adjuvant trastuzumab. Cancer Res 2013, 73, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Jang, G.-B.; Hong, I.-S.; Kim, R.-J.; Lee, S.-Y.; Park, S.-J.; Lee, E.-S.; Park, J.H.; Yun, C.-H.; Chung, J.-U.; Lee, K.-J. Wnt/β-catenin small-molecule inhibitor CWP232228 preferentially inhibits the growth of breast cancer stem-like cells. Cancer Res. 2015, 75, 1691–1702. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.-Z.; Yan, Y.-Y.; He, M.; Xiao, Q.-H.; Yao, W.-F.; Zhao, L.; Wu, H.-Z.; Yu, Z.-J.; Zhou, M.-Y.; Lv, M.-T. Salinomycin induces selective cytotoxicity to MCF-7 mammosphere cells through targeting the Hedgehog signaling pathway. Oncol. Rep. 2016, 35, 912–922. [Google Scholar] [CrossRef] [Green Version]
- Shan, N.L.; Wahler, J.; Lee, H.J.; Bak, M.J.; Gupta, S.D.; Maehr, H.; Suh, N. Vitamin D compounds inhibit cancer stem-like cells and induce differentiation in triple negative breast cancer. J. Steroid Biochem. Mol. Biol. 2017, 173, 122–129. [Google Scholar] [CrossRef]
- Diamond, J.R.; Becerra, C.; Richards, D.; Mita, A.; Osborne, C.; O’Shaughnessy, J.; Zhang, C.; Henner, R.; Kapoun, A.M.; Xu, L. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res. Treat. 2020, 184, 53–62. [Google Scholar] [CrossRef]
- Safholm, A.; Tuomela, J.; Rosenkvist, J.; Dejmek, J.; Harkonen, P.; Andersson, T. The Wnt-5a-derived hexapeptide Foxy-5 inhibits breast cancer metastasis in vivo by targeting cell motility. Clin Cancer Res 2008, 14, 6556–6563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soerensen, P.G.; Andersson, T.; Buhl, U.; Moelvadgaard, T.; Jensen, P.B.; Brunner, N.; Nielsen, D. Phase I dose-escalating study to evaluate the safety, tolerability, and pharmacokinetic and pharmacodynamic profiles of Foxy-5 in patients with metastatic breast, colorectal, or prostate cancer. J. Clin. Oncol. 2014, 32, TPS1140. [Google Scholar] [CrossRef]
- Choi, M.Y.; Widhopf II, G.F.; Ghia, E.M.; Kidwell, R.L.; Hasan, M.K.; Yu, J.; Rassenti, L.Z.; Chen, L.; Chen, Y.; Pittman, E. Phase I trial: Cirmtuzumab inhibits ROR1 signaling and stemness signatures in patients with chronic lymphocytic leukemia. Cell Stem Cell 2018, 22, 951–959.e953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, A.F.; Landis, M.D.; Dontu, G.; Griffith, K.A.; Layman, R.M.; Krop, I.; Paskett, L.A.; Wong, H.; Dobrolecki, L.E.; Lewis, M.T. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin. Cancer Res. 2013, 19, 1512–1524. [Google Scholar] [CrossRef] [Green Version]
- Krop, I.; Demuth, T.; Guthrie, T.; Wen, P.Y.; Mason, W.P.; Chinnaiyan, P.; Butowski, N.; Groves, M.D.; Kesari, S.; Freedman, S.J. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J. Clin. Oncol. 2012, 30, 2307–2313. [Google Scholar] [CrossRef]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog signaling in the maintenance of cancer stem cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef]
- Azaro, A.; Massard, C.; Tap, W.D.; Cassier, P.A.; Merchan, J.; Italiano, A.; Anderson, B.; Yuen, E.; Yu, D.; Oakley, G. A phase 1b study of the Notch inhibitor crenigacestat (LY3039478) in combination with other anticancer target agents (taladegib, LY3023414, or abemaciclib) in patients with advanced or metastatic solid tumors. Investig. New Drugs 2021, 39, 1089–1098. [Google Scholar] [CrossRef]
- Tsubamoto, H.; Ueda, T.; Inoue, K.; Sakata, K.; Shibahara, H.; Sonoda, T. Repurposing itraconazole as an anticancer agent. Oncol. Lett. 2017, 14, 1240–1246. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Jang, K.; Miller, P.; Picon-Ruiz, M.; Yeasky, T.; El-Ashry, D.; Slingerland, J. VEGFA links self-renewal and metastasis by inducing Sox2 to repress miR-452, driving Slug. Oncogene 2017, 36, 5199–5211. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Roodhart, J.M.; Voest, E.E. Target practice: Lessons from phase III trials with bevacizumab and vatalanib in the treatment of advanced colorectal cancer. Oncologist 2007, 12, 443–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [Green Version]
- Cecil, D.L.; Slota, M.; O’Meara, M.M.; Curtis, B.C.; Gad, E.; Dang, Y.; Herendeen, D.; Rastetter, L.; Disis, M.L. Immunization against HIF-1α inhibits the growth of basal mammary tumors and targets mammary stem cells in vivo. Clin. Cancer Res. 2017, 23, 3396–3404. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Qian, D.Z.; Tan, Y.S.; Lee, K.; Gao, P.; Ren, Y.R.; Rey, S.; Hammers, H.; Chang, D.; Pili, R. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19579–19586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Investig. 2010, 120, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.H.; Roselli, M.; Ferroni, P.; Costarelli, L.; Cavaliere, F.; Taffuri, M.; Palena, C.; Guadagni, F. Brachyury, a vaccine target, is overexpressed in triple negative breast cancer. Endocr. -Relat. Cancer 2016, 23, 783. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.-M.; Zhang, J.-G.; Zhang, X.; Li, Q. Targeting cancer stem cells for reversing therapy resistance: Mechanism, signaling, and prospective agents. Signal Transduct. Target. Ther. 2021, 6, 62. [Google Scholar] [CrossRef]
- Dai, H.; Tong, C.; Shi, D.; Chen, M.; Guo, Y.; Chen, D.; Han, X.; Wang, H.; Wang, Y.; Shen, P. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: A single-arm, open-label, phase II trial. Oncoimmunology 2020, 9, 1846926. [Google Scholar] [CrossRef]
- Feng, K.-C.; Guo, Y.-L.; Liu, Y.; Dai, H.-R.; Wang, Y.; Lv, H.-Y.; Huang, J.-H.; Yang, Q.-M.; Han, W.-D. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Osta, W.A.; Chen, Y.; Mikhitarian, K.; Mitas, M.; Salem, M.; Hannun, Y.A.; Cole, D.J.; Gillanders, W.E. EpCAM is overexpressed in breast cancer and is a potential target for breast cancer gene therapy. Cancer Res. 2004, 64, 5818–5824. [Google Scholar] [CrossRef] [Green Version]
- Stoecklein, N.H.; Siegmund, A.; Scheunemann, P.; Luebke, A.M.; Erbersdobler, A.; Verde, P.E.; Eisenberger, C.F.; Peiper, M.; Rehders, A.; Am Esch, J.S. Ep-CAM expression in squamous cell carcinoma of the esophagus: A potential therapeutic target and prognostic marker. BMC Cancer 2006, 6, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Z.; Wu, Y.; Ma, W.; Zhang, S.; Zhang, Y.-Q. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upton, R.; Banuelos, A.; Feng, D.; Biswas, T.; Kao, K.; McKenna, K.; Willingham, S.; Ho, P.Y.; Rosental, B.; Tal, M.C. Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proc. Natl. Acad. Sci. USA 2021, 118, e2026849118. [Google Scholar] [CrossRef]
- Burguin, A.; Diorio, C.; Durocher, F. Breast Cancer Treatments: Updates and New Challenges. J. Pers. Med. 2021, 11, 808. [Google Scholar] [CrossRef]
- Lancet, T. Breast cancer targeted therapy: Successes and challenges. Lancet 2017, 389, 2350. [Google Scholar] [CrossRef]
- Lobbezoo, D.; Van Kampen, R.; Voogd, A.; Dercksen, M.; Van Den Berkmortel, F.; Smilde, T.; Van De Wouw, A.; Peters, F.; Van Riel, J.; Peters, N. Prognosis of metastatic breast cancer: Are there differences between patients with de novo and recurrent metastatic breast cancer? Br. J. Cancer 2015, 112, 1445–1451. [Google Scholar] [CrossRef]
- Luo, M.; Brooks, M.; S Wicha, M. Epithelial-mesenchymal plasticity of breast cancer stem cells: Implications for metastasis and therapeutic resistance. Curr. Pharm. Des. 2015, 21, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Zhang, X.; Zhang, W.; Chiou, Y.S.; Qian, W.; Liu, X.; Zhang, M.; Yan, H.; Li, S.; Li, T. Cancer stem cell regulated phenotypic plasticity protects metastasized cancer cells from ferroptosis. Nat. Commun. 2022, 13, 1371. [Google Scholar] [CrossRef] [PubMed]
- Jewer, M.; Lee, L.; Leibovitch, M.; Zhang, G.; Liu, J.; Findlay, S.D.; Vincent, K.M.; Tandoc, K.; Dieters-Castator, D.; Quail, D.F. Translational control of breast cancer plasticity. Nat. Commun. 2020, 11, 2498. [Google Scholar] [CrossRef] [PubMed]
- Thankamony, A.P.; Saxena, K.; Murali, R.; Jolly, M.K.; Nair, R. Cancer stem cell plasticity–A deadly deal. Front. Mol. Biosci. 2020, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Hughes, C.J.; Ford, H.L. Cellular plasticity in breast cancer progression and therapy. Front. Mol. Biosci. 2020, 7, 72. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, R.N.; Esen, B.Ö.; Mellemkjær, L.; Christiansen, P.; Ejlertsen, B.; Lash, T.L.; Nørgaard, M.; Cronin-Fenton, D. The incidence of breast cancer recurrence 10–32 years after primary diagnosis. JNCI J. Natl. Cancer Inst. 2022, 114, 391–399. [Google Scholar] [CrossRef]
- Pei, J.; Wang, Y.; Li, Y. Identification of key genes controlling breast cancer stem cell characteristics via stemness indices analysis. J. Transl. Med. 2020, 18, 74. [Google Scholar] [CrossRef] [Green Version]
- Krop, I.E.; Kim, S.-B.; Martin, A.G.; LoRusso, P.M.; Ferrero, J.-M.; Badovinac-Crnjevic, T.; Hoersch, S.; Smitt, M.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): Final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol. 2017, 18, 743–754. [Google Scholar] [CrossRef]
- Qiu, Y.; Yang, L.; Liu, H.; Luo, X. Cancer stem cell-targeted therapeutic approaches for overcoming trastuzumab resistance in HER2-positive breast cancer. Stem Cells 2021, 39, 1125–1136. [Google Scholar] [CrossRef]
- Koziel, J.E.; Herbert, B.-S. The telomerase inhibitor imetelstat alone, and in combination with trastuzumab, decreases the cancer stem cell population and self-renewal of HER2+ breast cancer cells. Breast Cancer Res. Treat. 2015, 149, 607–618. [Google Scholar] [CrossRef] [Green Version]
- Dillon, R.L.; Chooniedass, S.; Premsukh, A.; Adams, G.P.; Entwistle, J.; MacDonald, G.C.; Cizeau, J. Trastuzumab-deBouganin conjugate overcomes multiple mechanisms of T-DM1 drug resistance. J. Immunother. 2016, 39, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Zhao, J.; Hu, Y.; Zhou, Y.; Guo, R.; Bai, J.; Zhang, S.; Zhang, H.; Zhang, J. The combination of NVP-BKM120 with trastuzumab or RAD001 synergistically inhibits the growth of breast cancer stem cells in vivo. Oncol. Rep. 2016, 36, 356–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.C.; Joalland, N.; Bridgeman, J.S.; Alchami, F.S.; Jarry, U.; Khan, M.W.A.; Piggott, L.; Shanneik, Y.; Li, J.; Herold, M.J. Synergistic targeting of breast cancer stem-like cells by human γδ T cells and CD8+ T cells. Immunol. Cell Biol. 2017, 95, 620–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Human | CD44+ [13] |
| Aldehyde dehydrogenase 1 (ADLH1) [22] | |
| ATP-binding cassette subfamily G member 2 (ABCG2) [27] | |
| CD133 [28] | |
| CD49f [29] | |
| Leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5) [30] | |
| Stage-specific embryonic antigen 3 (SSEA-3) [31] | |
| CD70 [32] | |
| Protein C receptor (PROCR) [33] | |
| Nectin-4 [34] | |
| EpCAM [35] | |
| CD90 [36] | |
| Mouse | CD29 [37] |
| CD61 [38] |
| BCSCs Marker | Agent/Intervention | Study Phase | Clinicaltrials.gov Identifier | Study Status | Type/Stage of Breast Cancer |
|---|---|---|---|---|---|
| CD44 | Bivatuzumab Mertansine (CD44v6) | I | NCT02254005 | Completed | Breast Neoplasms |
| CD133 | Other: Immunohistochemistry Staining Method | N/A | NCT04873154 | Recruiting | Breast Cancer |
| ALDH1 | Other: Immunohistochemistry Staining Method and Laboratory Biomarker Analysis | N/A | NCT00949013 | Completed | Early-Stage Breast Cancer |
| Other: Immunohistochemistry Staining Method and Laboratory Biomarker Analysis | N/A | NCT01424865 | Unknown | Breast Cancer | |
| Doxorubicin-Cyclophosphamide Regimen | N/A | NCT04581967 | Recruiting | Breast Cancer | |
| EGFR/HER2 | Trastuzumab | N/A | NCT01424865 | Unknown | Breast Cancer |
| Targeted Signaling Pathway | Agent | Study Phase | Clinicaltrials.gov Identifier | Study Status | Type/Stage of Breast Cancer |
|---|---|---|---|---|---|
| Wnt Signaling Pathway | Vantictumab | I | NCT01973309 | Completed | Metastatic HER2-Negative Breast Cancer |
| Foxy-5 | I | NCT02020291 | Completed | Metastatic Breast Cancer | |
| Cirmtuzumab with Paclitaxel I | I | NCT02776917 | Active, not Recruiting | Breast Neoplasms | |
| LGK974 | I | NCT01351103 | Recruiting | TNBC | |
| Notch Signaling Pathway | MK-0752 (GSI) | I | NCT00106145 | Completed | Advanced Breast Cancer |
| MK-0752 (GSI) with Docetaxel | I/II | NCT00645333 | Completed | Metastatic Breast Cancer | |
| PF-03084014 | II | NCT02299635 | Terminated | Advanced-Stage TNBC | |
| PF-03084014 with Docetaxel 260 | I | NCT01876251 | Terminated | Advanced-Stage TNBC | |
| Melatonin with Vitamin D | II | NCT01965522 | Completed | Early-Stage Breast Cancer | |
| Hedgehog Signaling pathway | Vismodegib with Neoadjuvant Paclitaxel, Cyclophosphamide, and Epirubicin | II | NCT02694224 | Recruiting | TNBC |
| Taladegib | I | NCT02784795 | Completed | Metastatic Breast Cancer | |
| Itraconazole | Unknown | NCT00798135 | Completed | Metastatic/Non-Metastatic Breast Cancer | |
| Itraconazole with Capivasertib | I | NCT04712396 | Completed | Metastatic TNBC/HR2-Positive Breast Cancer |
| Components | Agent/Intervention | Study Phase | Clinicaltrials.gov Identifier | Study Status | Type/Stage of Breast Cancer |
|---|---|---|---|---|---|
| Vascular Endothelial Growth Factor (VEGF) | Bevacizumab | II | NCT00016549 | Completed | Breast Cancer |
| NCT01190345 | Completed | ||||
| Bevacizumab with Herceptin | I/II | NCT00095706 | Completed | Breast Cancer |
| Strategies | Agent | Study Phase | Clinicaltrials.gov Identifier | Study Status | Type/Stage of Breast Cancer |
|---|---|---|---|---|---|
| Vaccine | Digoxin | II | NCT01763931 | Completed | Breast Cancer |
| MVA-brachyury-TRICOM | I | NCT02179515 | Completed | HER2-Positive; TNBC; Metastatic Breast Cancer | |
| NCT04296942 | Terminated | ||||
| NCT04134312 | Completed | ||||
| NCT03384316 | Completed | ||||
| Multiantigen DNA Plasmid-based Vaccine (CD105, Yb-1, SOX2, CDH3 and MDM2) | I | NCT02157051 | Recruiting | HER2-Negative Breast Carcinoma; Recurrent Breast Carcinoma; Stage III/IIIA/IIIB/IIIC/IV III Breast Cancer | |
| DCs Pulsed with the Lysate of Aldefluor-Positive Cells | I/II | NCT02063893 | Completed | Breast Neoplasms | |
| Chimeric Antigen Receptor (CAR) T cells | Anti-CD133-CAR Vector-Transduced T cells | I/II | NCT02541370 | Completed | Breast Cancer |
| Anti-EPCAM | I | NCT02915445 | Recruiting | Recurrent Breast Cancer | |
| Chemokine Inhibitors | Reparixin and Paclitaxel | I | NCT02001974 | Completed | HER2-Negative Metastatic Breast Cancer |
| Reparixin and Paclitaxel with Placebo | II | NCT02370238 | Completed | Metastatic TNBC | |
| Anti-CD47 | BI 765063 and BI 754091 | I | NCT03990233 | Recruiting | Solid Breast Cancer |
| HX009 | I | NCT04097769 | Active, Not Recruiting | Advanced Malignancies | |
| IMM2902 | I | NCT05076591 | Active, Not Recruiting | Advanced Breast Cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Jin, Z.; Master, R.P.; Maharjan, C.K.; Carelock, M.E.; Reccoppa, T.B.A.; Kim, M.-C.; Kolb, R.; Zhang, W. Breast Cancer Stem Cells: Signaling Pathways, Cellular Interactions, and Therapeutic Implications. Cancers 2022, 14, 3287. https://doi.org/10.3390/cancers14133287
Wang L, Jin Z, Master RP, Maharjan CK, Carelock ME, Reccoppa TBA, Kim M-C, Kolb R, Zhang W. Breast Cancer Stem Cells: Signaling Pathways, Cellular Interactions, and Therapeutic Implications. Cancers. 2022; 14(13):3287. https://doi.org/10.3390/cancers14133287
Chicago/Turabian StyleWang, Lei, Zeng Jin, Rohan P. Master, Chandra K. Maharjan, Madison E. Carelock, Tiffany B. A. Reccoppa, Myung-Chul Kim, Ryan Kolb, and Weizhou Zhang. 2022. "Breast Cancer Stem Cells: Signaling Pathways, Cellular Interactions, and Therapeutic Implications" Cancers 14, no. 13: 3287. https://doi.org/10.3390/cancers14133287
APA StyleWang, L., Jin, Z., Master, R. P., Maharjan, C. K., Carelock, M. E., Reccoppa, T. B. A., Kim, M.-C., Kolb, R., & Zhang, W. (2022). Breast Cancer Stem Cells: Signaling Pathways, Cellular Interactions, and Therapeutic Implications. Cancers, 14(13), 3287. https://doi.org/10.3390/cancers14133287