
Overcoming Temozolomide Resistance in Glioblastoma via Enhanced NAD+ Bioavailability and Inhibition of Poly-ADP-Ribose Glycohydrolase


 ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture Conditions
2.2. Cell Extract for Immunoblot Analysis
2.3. Lentivirus Production and Cell Transduction
2.4. MSH6 Knockdown by Expression of shRNA
2.5. MSH6 Knockout by CRISPR/Cas9
2.6. LN428 Cells Modified to Express MGMT
2.7. Cell Growth Analysis
2.8. NAD+ Measurements
2.9. Laser Micro-Irradiation
2.10. CometChip Analysis
2.11. Statistical Analysis
3. Results
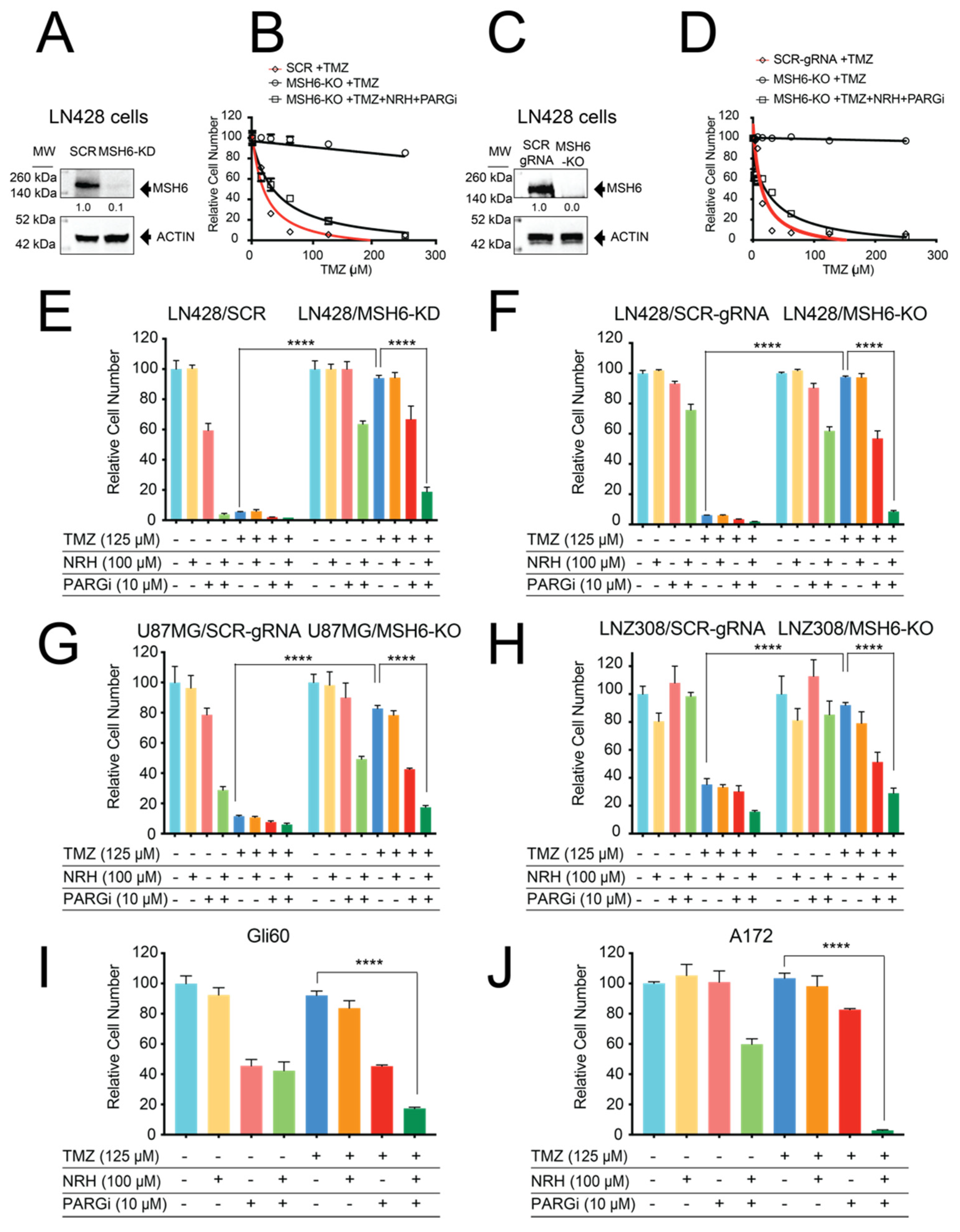
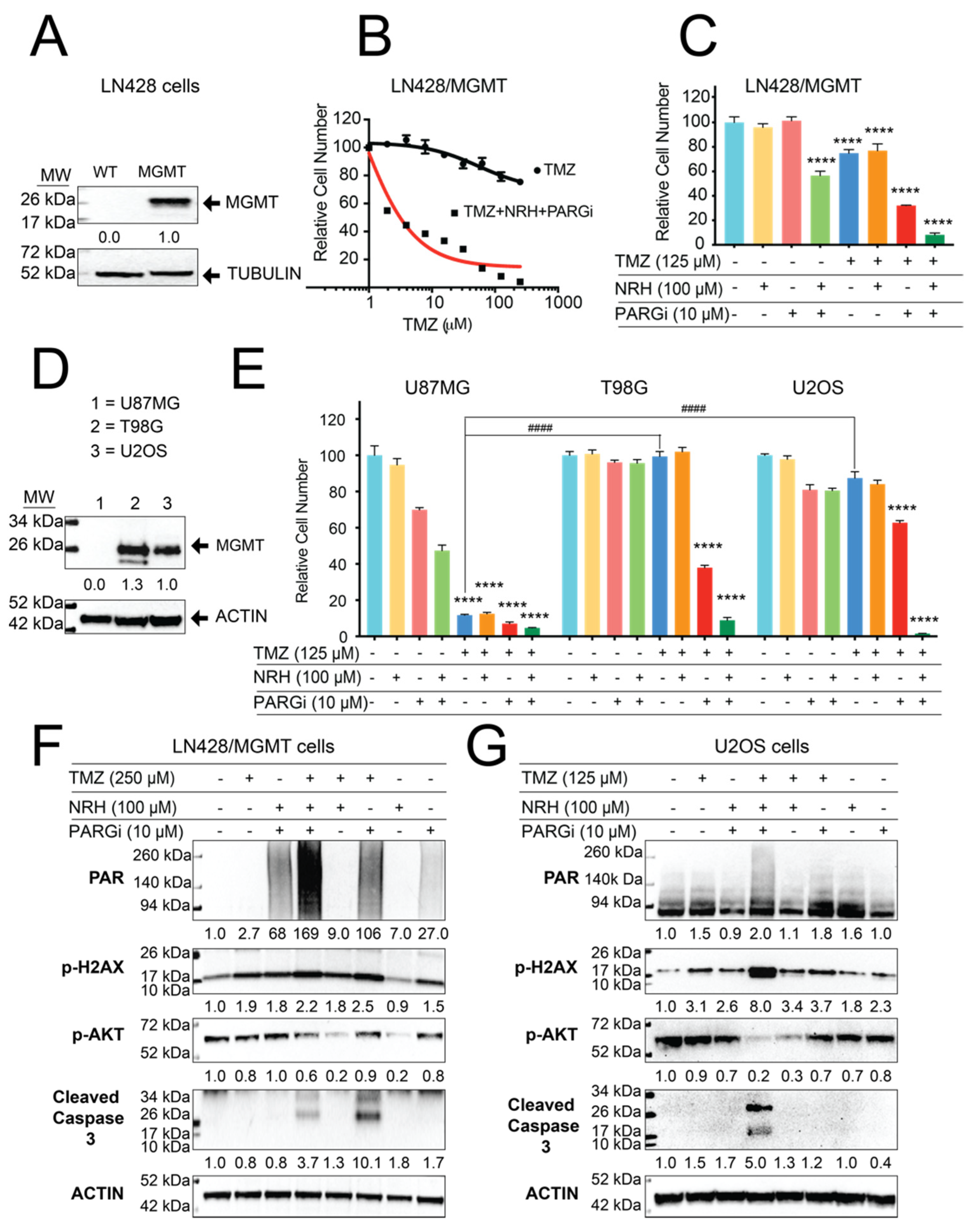
3.1. Loss of MSH6 in GBM Cells Results in TMZ Resistance That Can Be Overcome by Co-Treatment with NRH and PARGi
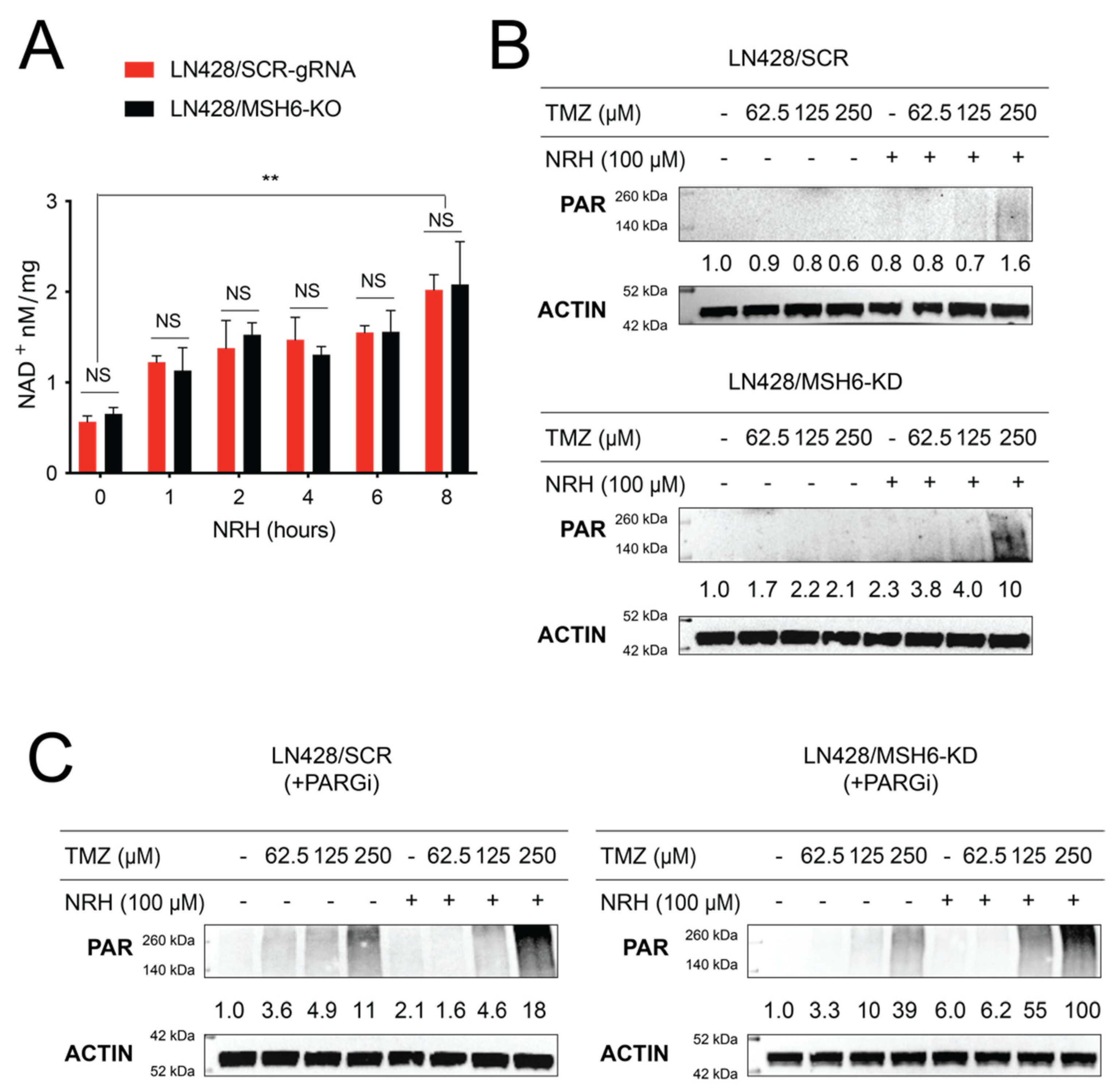
3.2. NRH Increased Cellular NAD+ Levels and Enhanced PARP1 Activity upon TMZ Treatment
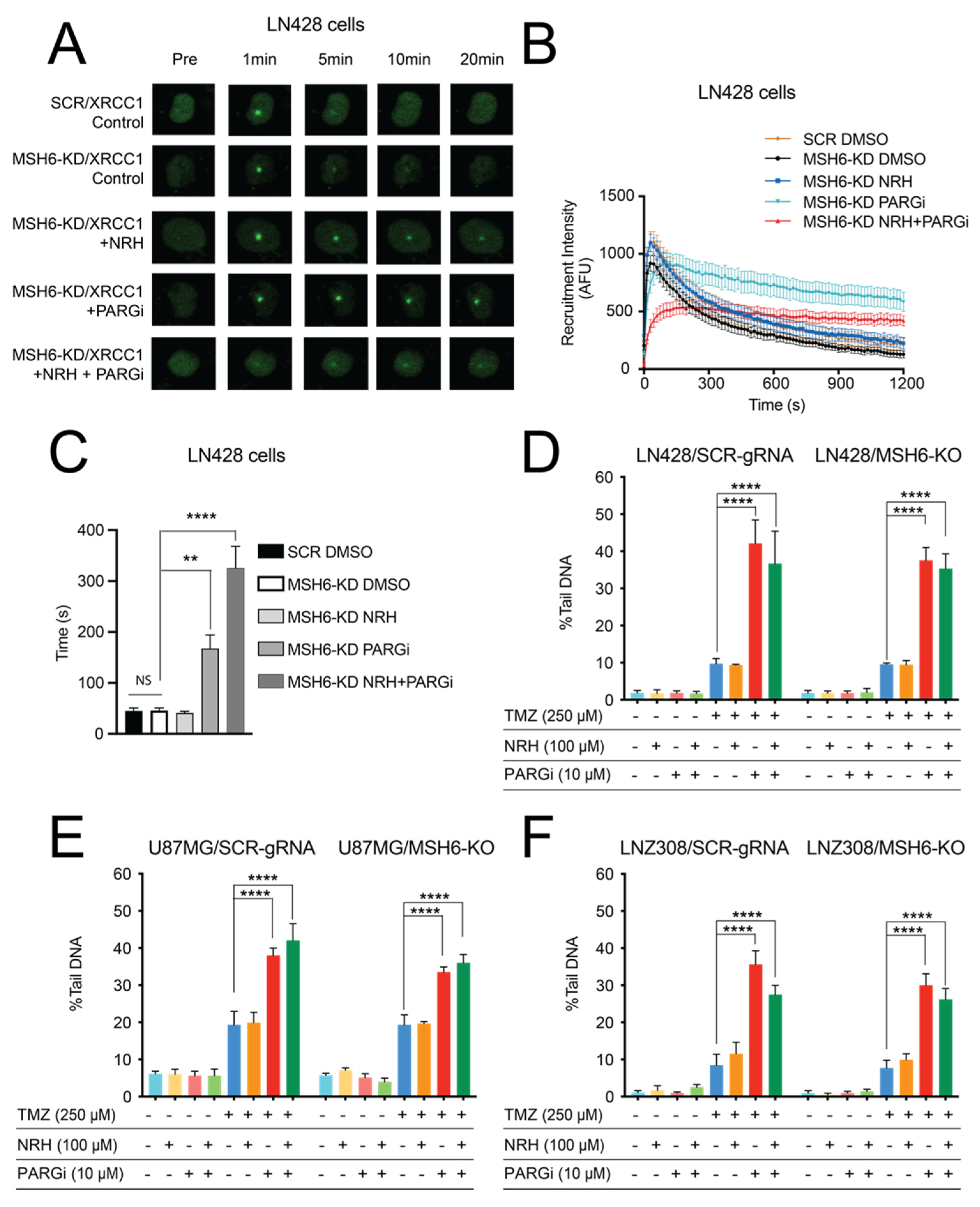
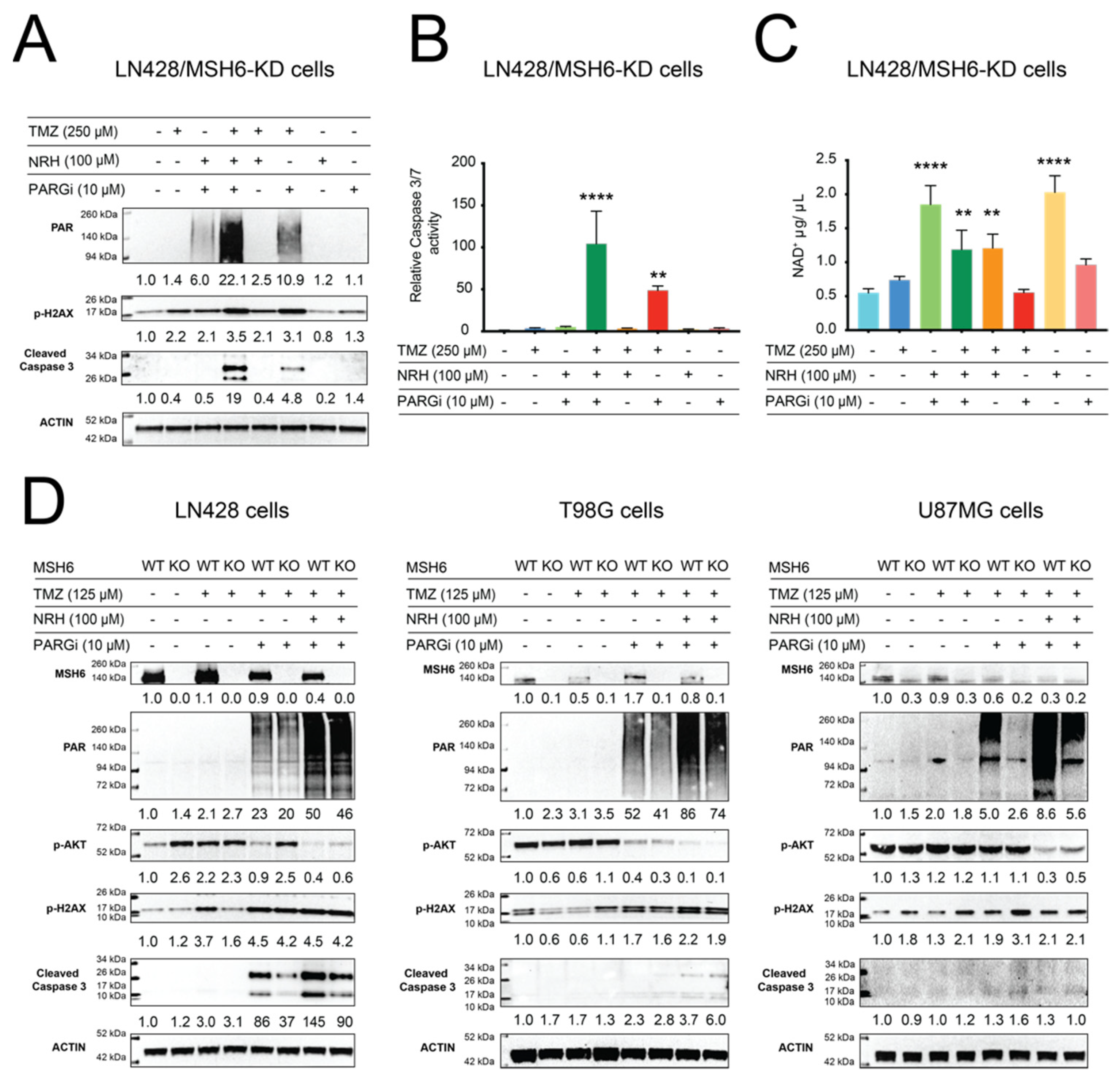
3.3. NAD+ Bioavailability Modulated by NRH Together with PARGi Interferes with the Dynamics of BER Protein Complex Assembly/Disassembly and Inhibits the Repair of DNA Lesions from TMZ Treatment, Suppresses Survival Signaling, and Induces Apoptosis Signaling
3.4. Resistance to TMZ Treatment Due to MGMT Activity Was Overcome by Co-Treatment with TMZ, NRH, and PARGi
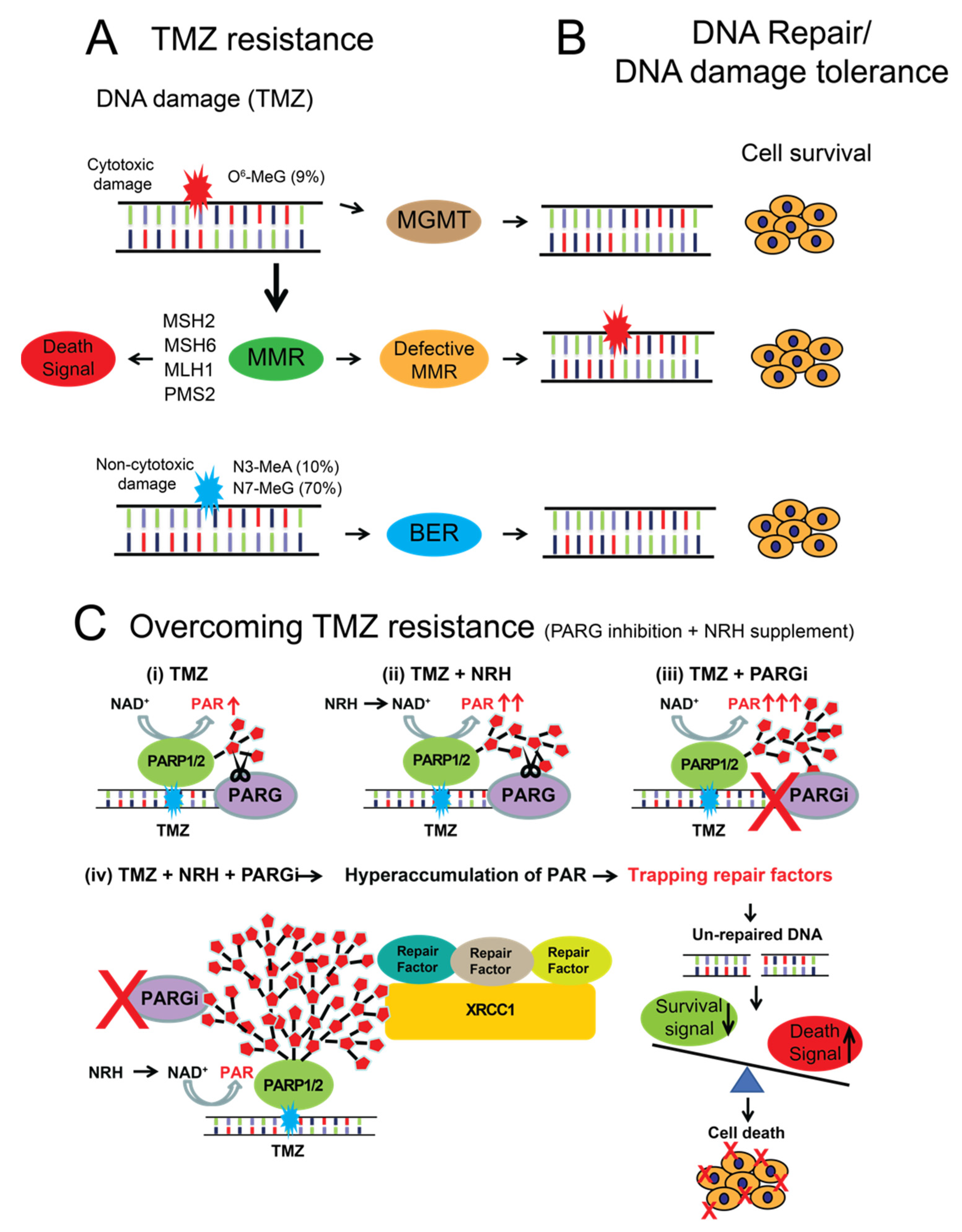
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Johnson, J.R.; Pazdur, R. Food and Drug Administration Drug approval summary: Temozolomide plus radiation therapy for the treatment of newly diagnosed glioblastoma multiforme. Clin. Cancer Res. 2005, 11, 6767–6771. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, K.; Mizoguchi, M.; Hata, N.; Murata, H.; Hatae, R.; Amano, T.; Nakamizo, A.; Sasaki, T. Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma. Front. Oncol. 2012, 2, 186. [Google Scholar] [CrossRef]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.N.; Almeida, K.H.; Fornsaglio, J.L.; Schamus, S.; Sobol, R.W. The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res. 2005, 65, 6394–6400. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.B.; Svilar, D.; Trivedi, R.N.; Wang, X.H.; Goellner, E.M.; Moore, B.; Hamilton, R.L.; Banze, L.A.; Brown, A.R.; Sobol, R.W. N-methylpurine DNA glycosylase and DNA polymerase beta modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro-Oncology 2011, 13, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Domoradzki, J.; Pegg, A.E.; Dolan, M.E.; Maher, V.M.; McCormick, J.J. Depletion of O6-methylguanine-DNA-methyltransferase in human fibroblasts increases the mutagenic response to N-methyl-N′-nitro-N-nitrosoguanidine. Carcinogenesis 1985, 6, 1823–1826. [Google Scholar] [CrossRef]
- Duckett, D.R.; Drummond, J.T.; Murchie, A.I.; Reardon, J.T.; Sancar, A.; Lilley, D.M.; Modrich, P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc. Natl. Acad. Sci. USA 1996, 93, 6443–6447. [Google Scholar] [CrossRef]
- Noonan, E.M.; Shah, D.; Yaffe, M.B.; Lauffenburger, D.A.; Samson, L.D. O6-Methylguanine DNA lesions induce an intra-S-phase arrest from which cells exit into apoptosis governed by early and late multi-pathway signaling network activation. Integr. Biol. 2012, 4, 1237–1255. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-Oncology 2009, 11, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Hickman, M.J.; Samson, L.D. Apoptotic signaling in response to a single type of DNA lesion, O(6)-methylguanine. Mol. Cell 2004, 14, 105–116. [Google Scholar] [CrossRef]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair 2016, 38, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; Codd, P.J.; Batchelor, T.T.; Curry, W.T.; Louis, D.N. MSH6 inactivation and emergent temozolomide resistance in human glioblastomas. Clin. Neurosurg. 2008, 55, 165–171. [Google Scholar] [PubMed]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef]
- Fujii, S.; Sobol, R.W.; Fuchs, R.P. Double-strand breaks: When DNA repair events accidentally meet. DNA Repair 2022, 112, 103303. [Google Scholar] [CrossRef]
- Felsberg, J.; Thon, N.; Eigenbrod, S.; Hentschel, B.; Sabel, M.C.; Westphal, M.; Schackert, G.; Kreth, F.W.; Pietsch, T.; Loffler, M.; et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 2011, 129, 659–670. [Google Scholar] [CrossRef]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef]
- Rahman, M.A.; Gras Navarro, A.; Brekke, J.; Engelsen, A.; Bindesboll, C.; Sarowar, S.; Bahador, M.; Bifulco, E.; Goplen, D.; Waha, A.; et al. Bortezomib administered prior to temozolomide depletes MGMT, chemosensitizes glioblastoma with unmethylated MGMT promoter and prolongs animal survival. Br. J. Cancer 2019, 121, 545–555. [Google Scholar] [CrossRef]
- Ranson, M.; Hersey, P.; Thompson, D.; Beith, J.; McArthur, G.A.; Haydon, A.; Davis, I.D.; Kefford, R.F.; Mortimer, P.; Harris, P.A.; et al. Randomized trial of the combination of lomeguatrib and temozolomide compared with temozolomide alone in chemotherapy naive patients with metastatic cutaneous melanoma. J. Clin. Oncol. 2007, 25, 2540–2545. [Google Scholar] [CrossRef]
- Ohba, S.; Yamashiro, K.; Hirose, Y. Inhibition of DNA Repair in Combination with Temozolomide or Dianhydrogalactiol Overcomes Temozolomide-Resistant Glioma Cells. Cancers 2021, 13, 2570. [Google Scholar] [CrossRef] [PubMed]
- Plummer, R.; Stephens, P.; Aissat-Daudigny, L.; Cambois, A.; Moachon, G.; Brown, P.D.; Campone, M. Phase 1 dose-escalation study of the PARP inhibitor CEP-9722 as monotherapy or in combination with temozolomide in patients with solid tumors. Cancer Chemother. Pharm. 2014, 74, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.A.; Su, J.M.; Onar-Thomas, A.; Billups, C.A.; Li, X.N.; Poussaint, T.Y.; Smith, E.R.; Thompson, P.; Adesina, A.; Ansell, P.; et al. A phase I/II study of veliparib (ABT-888) with radiation and temozolomide in newly diagnosed diffuse pontine glioma: A Pediatric Brain Tumor Consortium study. Neuro-Oncology 2020, 22, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Miwa, M.; Tanaka, M.; Matsushima, T.; Sugimura, T. Purification and properties of glycohydrolase from calf thymus splitting ribose-ribose linkages of poly(adenosine diphosphate ribose). J. Biol. Chem. 1974, 249, 3475–3482. [Google Scholar] [CrossRef]
- Li, J.; Saville, K.M.; Ibrahim, M.; Zeng, X.; McClellan, S.; Angajala, A.; Beiser, A.; Andrews, J.F.; Sun, M.; Koczor, C.A.; et al. NAD(+) bioavailability mediates PARG inhibition-induced replication arrest, intra S-phase checkpoint and apoptosis in glioma stem cells. NAR Cancer 2021, 3, zcab044. [Google Scholar] [CrossRef]
- Tentori, L.; Leonetti, C.; Scarsella, M.; Muzi, A.; Vergati, M.; Forini, O.; Lacal, P.M.; Ruffini, F.; Gold, B.; Li, W.; et al. Poly(ADP-ribose) glycohydrolase inhibitor as chemosensitiser of malignant melanoma for temozolomide. Eur. J. Cancer 2005, 41, 2948–2957. [Google Scholar] [CrossRef]
- Matanes, E.; Lopez-Ozuna, V.M.; Octeau, D.; Baloch, T.; Racovitan, F.; Dhillon, A.K.; Kessous, R.; Raban, O.; Kogan, L.; Salvador, S.; et al. Inhibition of Poly ADP-Ribose Glycohydrolase Sensitizes Ovarian Cancer Cells to Poly ADP-Ribose Polymerase Inhibitors and Platinum Agents. Front. Oncol. 2021, 11, 745981. [Google Scholar] [CrossRef]
- Agostini, L.C.; Jain, A.; Shupp, A.; Nevler, A.; McCarthy, G.; Bussard, K.M.; Yeo, C.J.; Brody, J.R. Combined Targeting of PARG and Wee1 Causes Decreased Cell Survival and DNA Damage in an S-Phase-Dependent Manner. Mol. Cancer Res. 2021, 19, 207–214. [Google Scholar] [CrossRef]
- Nagashima, H.; Lee, C.K.; Tateishi, K.; Higuchi, F.; Subramanian, M.; Rafferty, S.; Melamed, L.; Miller, J.J.; Wakimoto, H.; Cahill, D.P. Poly(ADP-ribose) Glycohydrolase Inhibition Sequesters NAD(+) to Potentiate the Metabolic Lethality of Alkylating Chemotherapy in IDH-Mutant Tumor Cells. Cancer Discov. 2020, 10, 1672–1689. [Google Scholar] [CrossRef]
- Giroud-Gerbetant, J.; Joffraud, M.; Giner, M.P.; Cercillieux, A.; Bartova, S.; Makarov, M.V.; Zapata-Perez, R.; Sanchez-Garcia, J.L.; Houtkooper, R.H.; Migaud, M.E.; et al. A reduced form of nicotinamide riboside defines a new path for NAD(+) biosynthesis and acts as an orally bioavailable NAD(+) precursor. Mol. Metab. 2019, 30, 192–202. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Fouquerel, E.; Goellner, E.M.; Yu, Z.; Gagne, J.P.; de Moura, M.B.; Feinstein, T.; Wheeler, D.; Redpath, P.; Li, J.; Romero, G.; et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014, 8, 1819–1831. [Google Scholar] [CrossRef] [PubMed]
- Koczor, C.A.; Saville, K.M.; Andrews, J.F.; Clark, J.; Fang, Q.; Li, J.; Al-Rahahleh, R.Q.; Ibrahim, M.; McClellan, S.; Makarov, M.V.; et al. Temporal dynamics of base excision/single-strand break repair protein complex assembly/disassembly are modulated by the PARP/NAD(+)/SIRT6 axis. Cell Rep. 2021, 37, 109917. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Witt, K.L.; Revanna, P.; Smith-Roe, S.L.; Dismukes, J.; Lloyd, D.G.; Engelward, B.P.; Sobol, R.W. Next generation high throughput DNA damage detection platform for genotoxic compound screening. Sci. Rep. 2018, 8, 2771. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; Levine, K.K.; Betensky, R.A.; Codd, P.J.; Romany, C.A.; Reavie, L.B.; Batchelor, T.T.; Futreal, P.A.; Stratton, M.R.; Curry, W.T.; et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 2007, 13, 2038–2045. [Google Scholar] [CrossRef]
- Patel, M.; McCully, C.; Godwin, K.; Balis, F.M. Plasma and cerebrospinal fluid pharmacokinetics of intravenous temozolomide in non-human primates. J. Neuro-Oncol. 2003, 61, 203–207. [Google Scholar] [CrossRef]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef]
- Festuccia, C.; Mancini, A.; Colapietro, A.; Gravina, G.L.; Vitale, F.; Marampon, F.; Delle Monache, S.; Pompili, S.; Cristiano, L.; Vetuschi, A.; et al. The first-in-class alkylating deacetylase inhibitor molecule tinostamustine shows antitumor effects and is synergistic with radiotherapy in preclinical models of glioblastoma. J. Hematol. Oncol. 2018, 11, 32. [Google Scholar] [CrossRef]
- Wilk, A.; Hayat, F.; Cunningham, R.; Li, J.; Garavaglia, S.; Zamani, L.; Ferraris, D.M.; Sykora, P.; Andrews, J.; Clark, J.; et al. Extracellular NAD(+) enhances PARP-dependent DNA repair capacity independently of CD73 activity. Sci. Rep. 2020, 10, 651. [Google Scholar] [CrossRef]
- Saville, K.M.; Clark, J.; Wilk, A.; Rogers, G.D.; Andrews, J.F.; Koczor, C.A.; Sobol, R.W. NAD(+)-mediated regulation of mammalian base excision repair. DNA Repair 2020, 93, 102930. [Google Scholar] [CrossRef]
- Liu, Q.; Turner, K.M.; Alfred Yung, W.K.; Chen, K.; Zhang, W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro-Oncology 2014, 16, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Noorbakhsh, S.I.; Sundaram, R.K.; Kalathil, A.N.; Ganesa, S.; Jia, L.; Breslin, H.; Burgenske, D.M.; Gilad, O.; Sarkaria, J.N.; et al. Temozolomide Sensitizes MGMT-Deficient Tumor Cells to ATR Inhibitors. Cancer Res. 2019, 79, 4331–4338. [Google Scholar] [CrossRef]
- Sobol, R.W. Temozolomide. In Encyclopedia of Cancer; Schwab, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 3637–3642. [Google Scholar]
- Goellner, E.M.; Grimme, B.; Brown, A.R.; Lin, Y.C.; Wang, X.H.; Sugrue, K.F.; Mitchell, L.; Trivedi, R.N.; Tang, J.B.; Sobol, R.W. Overcoming temozolomide resistance in glioblastoma via dual inhibition of NAD+ biosynthesis and base excision repair. Cancer Res. 2011, 71, 2308–2317. [Google Scholar] [CrossRef]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Mortusewicz, O.; Fouquerel, E.; Ame, J.C.; Leonhardt, H.; Schreiber, V. PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res. 2011, 39, 5045–5056. [Google Scholar] [CrossRef]
- Chen, S.H.; Yu, X. Targeting dePARylation selectively suppresses DNA repair-defective and PARP inhibitor-resistant malignancies. Sci. Adv. 2019, 5, eaav4340. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Ahmed, S.U.; Carruthers, R.; Gilmour, L.; Yildirim, S.; Watts, C.; Chalmers, A.J. Selective Inhibition of Parallel DNA Damage Response Pathways Optimizes Radiosensitization of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 4416–4428. [Google Scholar] [CrossRef]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.; Gilmour, L.; et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef]
- Morgan, M.A.; Canman, C.E. Replication Stress: An Achilles’ Heel of Glioma Cancer Stem-like Cells. Cancer Res. 2018, 78, 6713–6716. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Lin, C.C.; Dai, H.; Mo, W.; Li, Y.; Stephan, C.; Davies, P.; Lu, Z.; Mills, G.B.; Lee, J.S.; et al. Defective Replication Stress Response Is Inherently Linked to the Cancer Stem Cell Phenotype. Cell Rep. 2018, 23, 2095–2106. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Katayama, M.; Mirzoeva, O.K.; Berger, M.S.; Pieper, R.O. Akt activation suppresses chk2-mediated, methylating agent-induced g2 arrest and protects from temozolomide-induced mitotic catastrophe and cellular senescence. Cancer Res. 2005, 65, 4861–4869. [Google Scholar] [CrossRef]
- Kandel, E.S.; Skeen, J.; Majewski, N.; Di Cristofano, A.; Pandolfi, P.P.; Feliciano, C.S.; Gartel, A.; Hay, N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol. Cell Biol. 2002, 22, 7831–7841. [Google Scholar] [CrossRef]
- Prasad, G.; Sottero, T.; Yang, X.; Mueller, S.; James, C.D.; Weiss, W.A.; Polley, M.Y.; Ozawa, T.; Berger, M.S.; Aftab, D.T.; et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro-Oncology 2011, 13, 384–392. [Google Scholar] [CrossRef]
- Chen, L.; Han, L.; Shi, Z.; Zhang, K.; Liu, Y.; Zheng, Y.; Jiang, T.; Pu, P.; Jiang, C.; Kang, C. LY294002 enhances cytotoxicity of temozolomide in glioma by down-regulation of the PI3K/Akt pathway. Mol. Med. Rep. 2012, 5, 575–579. [Google Scholar] [CrossRef][Green Version]
- Dronca, R.S.; Allred, J.B.; Perez, D.G.; Nevala, W.K.; Lieser, E.A.; Thompson, M.; Maples, W.J.; Creagan, E.T.; Pockaj, B.A.; Kaur, J.S.; et al. Phase II study of temozolomide (TMZ) and everolimus (RAD001) therapy for metastatic melanoma: A North Central Cancer Treatment Group study, N0675. Am. J. Clin. Oncol. 2014, 37, 369–376. [Google Scholar] [CrossRef]
- Tapodi, A.; Bognar, Z.; Szabo, C.; Gallyas, F.; Sumegi, B.; Hocsak, E. PARP inhibition induces Akt-mediated cytoprotective effects through the formation of a mitochondria-targeted phospho-ATM-NEMO-Akt-mTOR signalosome. Biochem. Pharmacol. 2019, 162, 98–108. [Google Scholar] [CrossRef]
- Menendez, D.; Nguyen, T.A.; Freudenberg, J.M.; Mathew, V.J.; Anderson, C.W.; Jothi, R.; Resnick, M.A. Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 2013, 41, 7286–7301. [Google Scholar] [CrossRef]
- Van Meir, E.G.; Kikuchi, T.; Tada, M.; Li, H.; Diserens, A.C.; Wojcik, B.E.; Huang, H.J.; Friedmann, T.; de Tribolet, N.; Cavenee, W.K. Analysis of the p53 gene and its expression in human glioblastoma cells. Cancer Res. 1994, 54, 649–652. [Google Scholar] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Antigen | Company | Catalogue # | Dilution |
|---|---|---|---|
| γ-H2AX | Cell Signaling Technology (Danvers, MA, USA) | 9718s | 1:1000 |
| Phosphorylated Akt (Ser473) | Cell Signaling Technology (Danvers, MA, USA) | 9271S | 1:1000 |
| β-Actin | Cell Signaling Technology (Danvers, MA, USA) | 8457S | 1:2000 |
| MGMT | Cell Signaling Technology (Danvers, MA, USA) | 58121S | 1:1000 |
| MSH6 | Cell Signaling Technology (Danvers, MA, USA) | 3995S | 1:1000 |
| Tubulin | Thermo Fisher Scientific (Waltham, MA, USA) | 62204 | 1:1000 |
| PAR | Gift from Mathias Ziegler(University of Bergen, Bergen, Norway) | N/A | 1:1000 |
| Cleaved Caspase 3 | Cell Signaling Technology (Danvers, MA, USA) | 9661S | 1:1000 |
| Immun-Star Goat anti-mouse-HRP conjugate | Bio-Rad (Hercules, CA, USA) | 170-5047 | 1:5000 |
| Immun-Star Goat anti-rabbit-HRP conjugate | Bio-Rad (Hercules, CA, USA) | 170-5046 | 1:5000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Koczor, C.A.; Saville, K.M.; Hayat, F.; Beiser, A.; McClellan, S.; Migaud, M.E.; Sobol, R.W. Overcoming Temozolomide Resistance in Glioblastoma via Enhanced NAD+ Bioavailability and Inhibition of Poly-ADP-Ribose Glycohydrolase. Cancers 2022, 14, 3572. https://doi.org/10.3390/cancers14153572
Li J, Koczor CA, Saville KM, Hayat F, Beiser A, McClellan S, Migaud ME, Sobol RW. Overcoming Temozolomide Resistance in Glioblastoma via Enhanced NAD+ Bioavailability and Inhibition of Poly-ADP-Ribose Glycohydrolase. Cancers. 2022; 14(15):3572. https://doi.org/10.3390/cancers14153572
Chicago/Turabian StyleLi, Jianfeng, Christopher A. Koczor, Kate M. Saville, Faisal Hayat, Alison Beiser, Steven McClellan, Marie E. Migaud, and Robert W. Sobol. 2022. "Overcoming Temozolomide Resistance in Glioblastoma via Enhanced NAD+ Bioavailability and Inhibition of Poly-ADP-Ribose Glycohydrolase" Cancers 14, no. 15: 3572. https://doi.org/10.3390/cancers14153572
APA StyleLi, J., Koczor, C. A., Saville, K. M., Hayat, F., Beiser, A., McClellan, S., Migaud, M. E., & Sobol, R. W. (2022). Overcoming Temozolomide Resistance in Glioblastoma via Enhanced NAD+ Bioavailability and Inhibition of Poly-ADP-Ribose Glycohydrolase. Cancers, 14(15), 3572. https://doi.org/10.3390/cancers14153572