
Recurrent NOMO1 Gene Deletion Is a Potential Clinical Marker in Early-Onset Colorectal Cancer and Is Involved in the Regulation of Cell Migration

 ,
,  , ,
, ,  , , ,
, , ,  ,
,  , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Tissue Samples
2.2. Cell Lines and Culture Conditions
2.3. DNA Extraction and DNA Quality Evaluation
2.4. Quantitative PCR (qPCR) and Quantitative Reverse Transcription PCR (qRT-PCR)
2.5. NOMO1 Sequencing
2.6. CRISPR/Cas9-Mediated Generation of NOMO1 Knockout Cells
2.7. Western Blotting
2.8. Cell Viability Assay
2.9. Wound Healing Assay
2.10. Transwell Migration Assay
2.11. RNA Extraction, Microarray Data Analysis, and RNA Sequencing
2.12. Proteome Analysis by Liquid Chromatography-Mass Spectrometry (LC-IMS/MS)
2.13. Mouse Strains, Adenovirus Injection, and Histological Analysis
2.14. Statistical Analysis
3. Results
3.1. Quantitative PCR Reveals a Single NOMO Gene
3.2. NOMO1 Is Frequently Inactivated by Deletion or Mutation in EOCRC
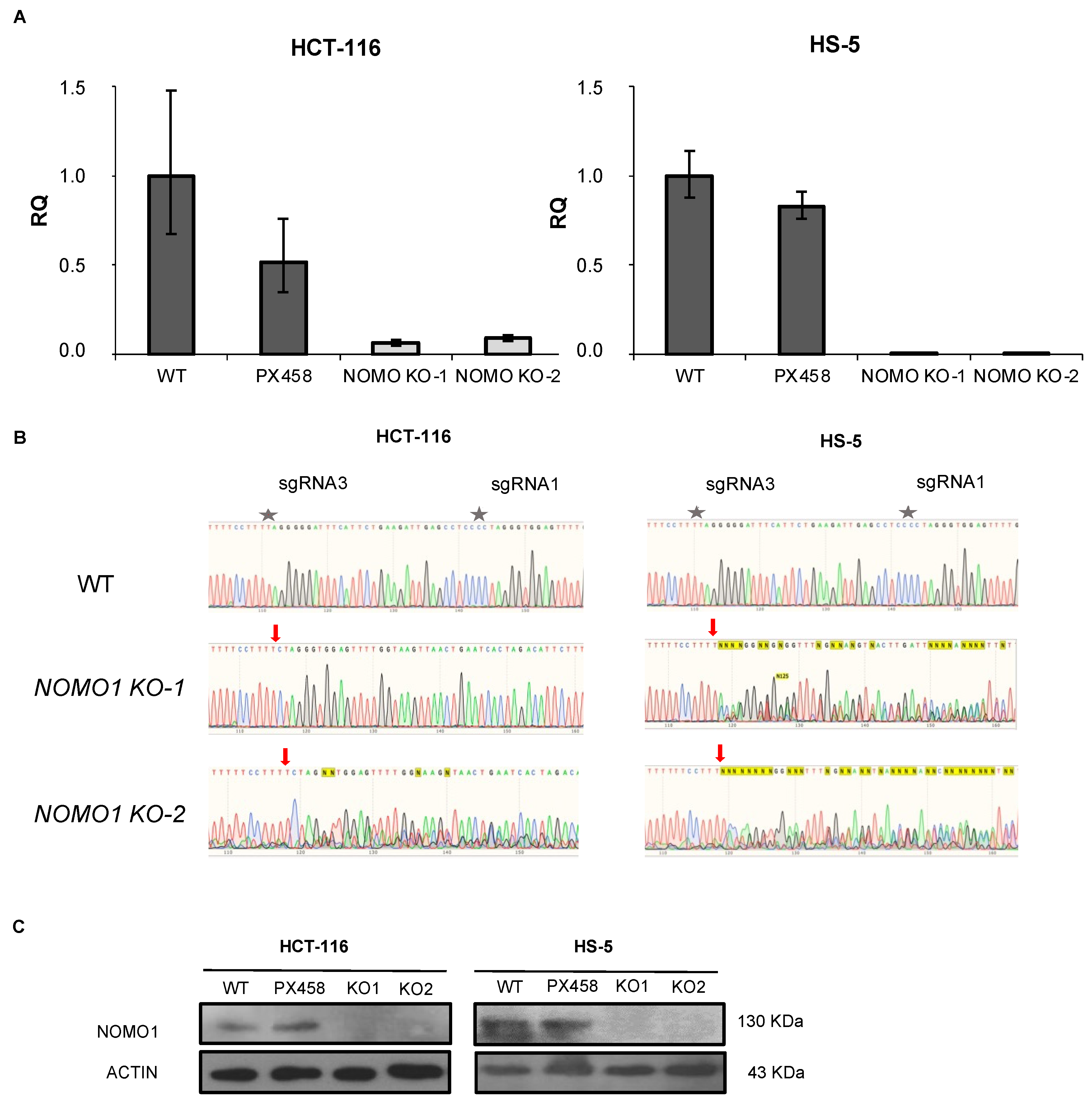
3.3. CRISPR/Cas9 Technology Efficiently Inactivates NOMO1
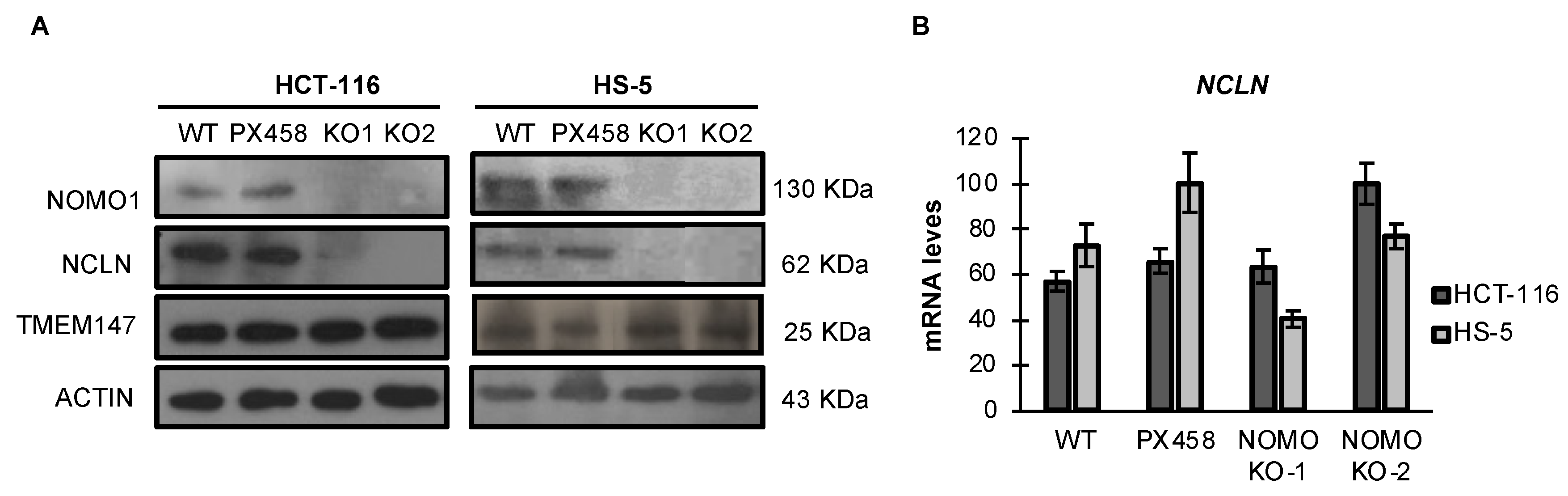
3.4. NOMO1 Inactivation Significantly Reduces the Expression of NCLN in Cell Lines
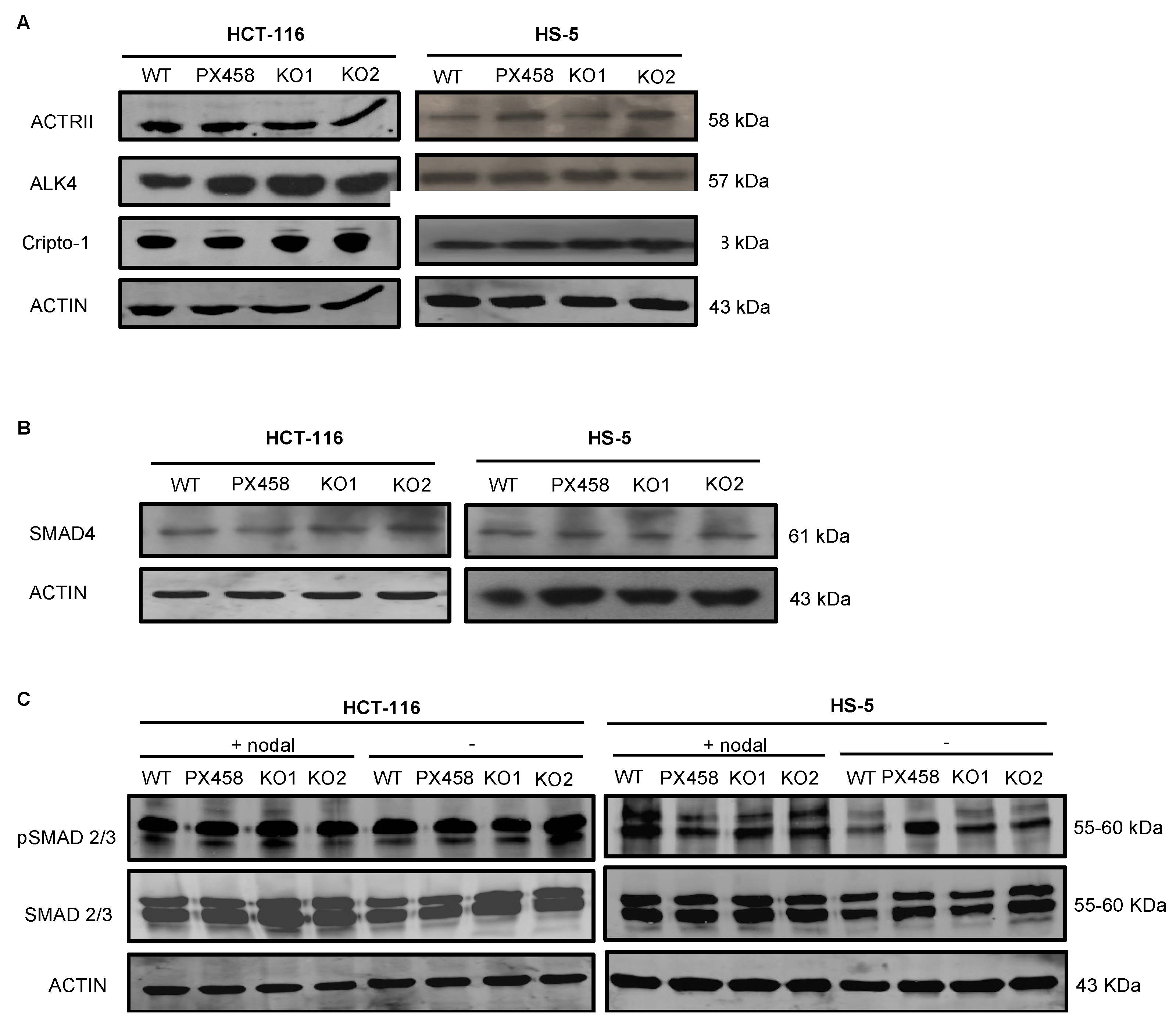
3.5. NOMO1 Inactivation Does Not Affect the Nodal Signaling Pathway Activity or Cell Proliferation
3.6. Gene Expression Profiling in NOMO1-KO Cell Lines
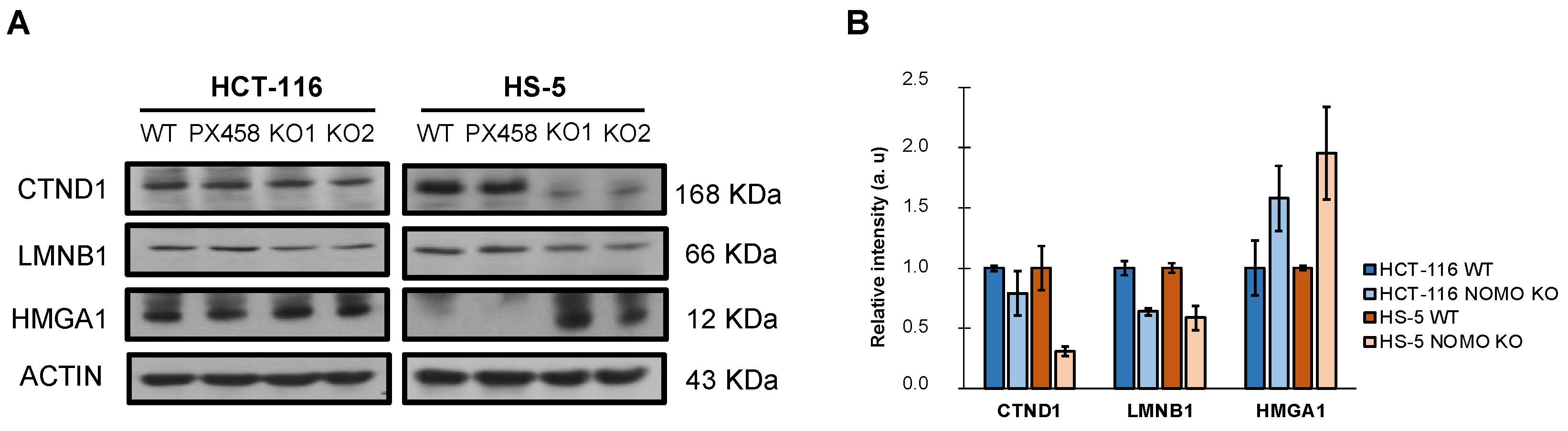
3.7. Protein Expression Profiling in NOMO1-KO Cell Lines
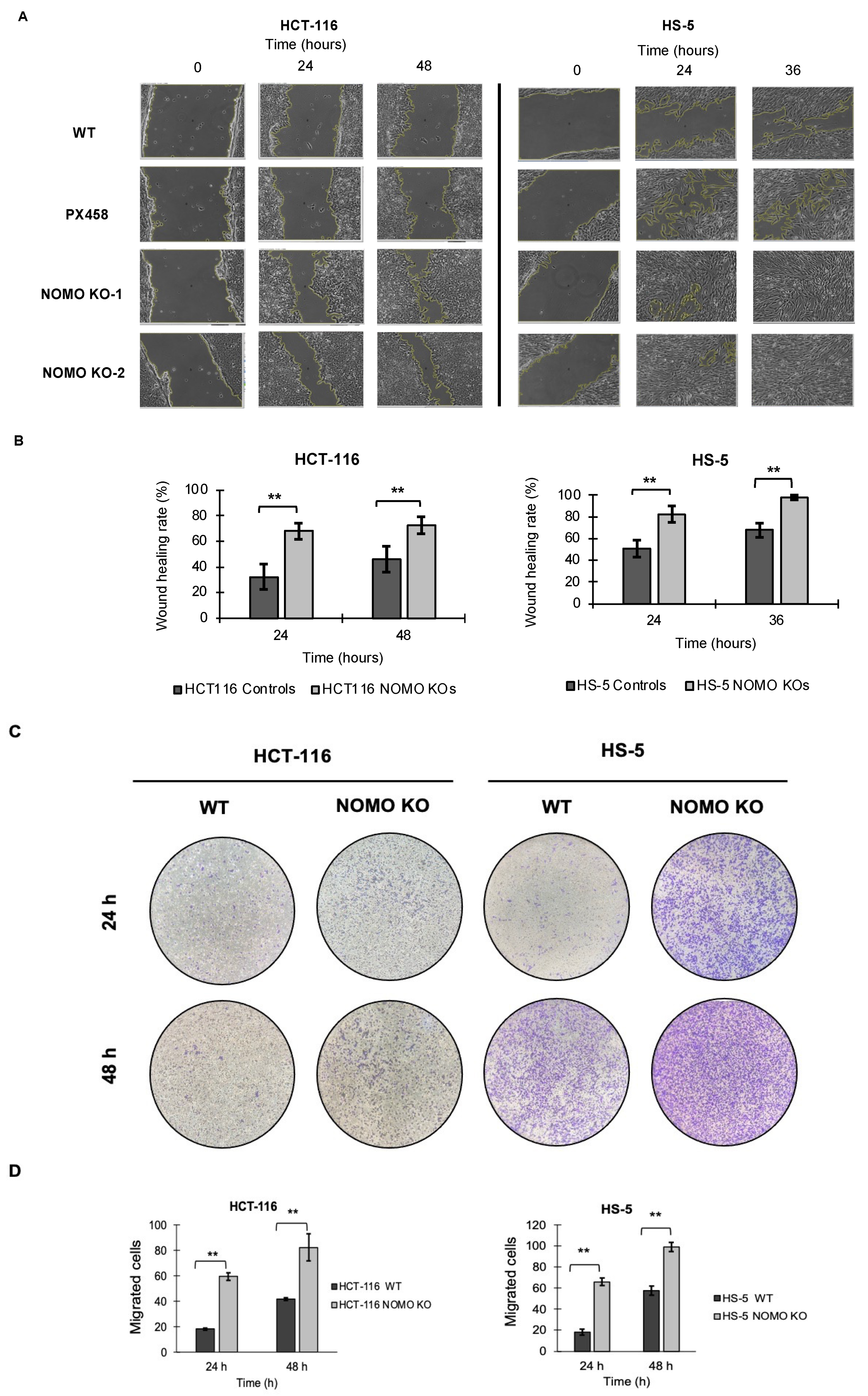
3.8. NOMO1 Inactivation Promotes Cell Migration
3.9. Nomo1 Deficiency in Mouse Colon Cells Does Not Modify the Susceptibility of Developing EOCRC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATCC | American type culture collection |
| CADD | Combined annotation-dependent depletion |
| CNAs | Copy number alterations |
| CRC | Colorectal cancer |
| CSC | Cancer stem cell |
| DMEM | Dulbecco’s modified eagle’s medium |
| DTT | Dithiothreitol |
| EMT | Epithelial-mesenchymal transition |
| EOCRC | Early-onset colorectal cancer |
| FBS | Fetal bovine serum |
| FDR | False discovery rate |
| FFPE | Formalin-fixed paraffin-embedded |
| GSEA | Gene set enrichment analysis |
| HNSCC | Head and neck squamous carcinoma |
| KO | Knockout |
| LC-IMS/MS | Liquid chromatography-mass spectrometry |
| LOCRC | Late-onset colorectal cancer |
| MMR | Missmatch repair |
| MSI | Microsatellite instability |
| MSS | Microsatellite stable |
| NCLN | Nicalin |
| NGS | Next-generation sequencing |
| NOMO1 | Nodal modulator 1 |
| ORA | Over-representation analysis |
| OSCC | Oral squamous cell carcinoma |
| PASEF | Parallel accumulation serial fragmentation |
| PolyPhen | Polymorphism phenotyping |
| qPCR | Quantitative PCR |
| qRT-PCR | Quantitative reverse transcription PCR |
| rhNodal | Recombinant human nodal |
| RMA | Robust multi-array average |
| RNA-seq | RNA sequencing |
| RT-PCR | Real-time PCR |
| SD | Standard deviation |
| Sift | Sorting intolerant from tolerant |
| SPF | Specific pathogen free |
| TMEM147 | Transmembrane protein 147 |
| VUS | Variants of uncertain significance |
| WB | Western blot |
| WebGestalt | WEB-based gene set analysis |
| WT | Wild type |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Patterns and Trends in Colorectal Cancer Incidence and Mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perea, J.; Rodríguez, Y.; Rueda, D.; Marín, J.C.; Díaz-Tasende, J.; Álvaro, E.; Alegre, C.; Osorio, I.; Colina, F.; Lomas, M.; et al. Early-Onset Colorectal Cancer Is an Easy and Effective Tool to Identify Retrospectively Lynch Syndrome. Ann. Surg. Oncol. 2011, 18, 3285–3291. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Sartore-Bianchi, A.; Russo, A.G.; Marsoni, S.; Bardelli, A.; Siena, S. Early-Onset Colorectal Cancer in Young Individuals. Mol. Oncol. 2019, 13, 109–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perea, J.; Rueda, D.; Canal, A.; Rodríguez, Y.; Álvaro, E.; Osorio, I.; Alegre, C.; Rivera, B.; Martínez, J.; Benítez, J.; et al. Age at Onset Should be a Major Criterion for Subclassification of Colorectal Cancer. J. Mol. Diagn. 2014, 16, 116–126. [Google Scholar] [CrossRef]
- Budinska, E.; Popovici, V.; Tejpar, S.; D’Ario, G.; Lapique, N.; Sikora, K.O.; Di Narzo, A.F.; Yan, P.; Graeme Hodgson, J.; Weinrich, S.; et al. Gene Expression Patterns Unveil a New Level of Molecular Heterogeneity in Colorectal Cancer. J. Pathol. 2013, 231, 63–76. [Google Scholar] [CrossRef]
- Jasmine, F.; Rahaman, R.; Dodsworth, C.; Roy, S.; Paul, R.; Raza, M.; Paul-Brutus, R.; Kamal, M.; Ahsan, H.; Kibriya, M.G. A Genome-Wide Study of Cytogenetic Changes in Colorectal Cancer Using SNP Microarrays: Opportunities for Future Personalized Treatment. PLoS ONE 2012, 7, e31968. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Holowatyj, A.N.; Gigic, B.; Herpel, E.; Scalbert, A.; Schneider, M.; Ulrich, C.M.; Achaintre, D.; Brezina, S.; van Duijnhoven, F.J.B.; Gsur, A.; et al. Distinct Molecular Phenotype of Sporadic Colorectal Cancers among Young Patients Based on Multiomics Analysis. Gastroenterology 2020, 158, 1155–1158.e2. [Google Scholar] [CrossRef]
- Arriba, M.; Sánchez, C.; Vivas, A.; Nutu, O.A.; Rueda, D.; Tapial, S.; Rodríguez, Y.; Brandáriz, L.; García, J.L.; García-Olmo, D.; et al. Intermediate-Onset Colorectal Cancer: A Clinical and Familial Boundary between Both Early and Late-Onset Colorectal Cancer. PLoS ONE 2019, 14, e0216472. [Google Scholar] [CrossRef]
- Di Leo, M.; Zuppardo, R.A.; Puzzono, M.; Ditonno, I.; Mannucci, A.; Antoci, G.; Russo Raucci, A.; Patricelli, M.G.; Elmore, U.; Tamburini, A.M.; et al. Risk Factors and Clinical Characteristics of Early-Onset Colorectal Cancer vs. Late-Onset Colorectal Cancer: A Case-Case Study. Eur. J. Gastroenterol. Hepatol. 2021, 33, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Arriba, M.; García, J.L.; Inglada-Pérez, L.; Rueda, D.; Osorio, I.; Rodríguez, Y.; Álvaro, E.; Sánchez, R.; Fernández, T.; Pérez, J.; et al. DNA Copy Number Profiling Reveals Different Patterns of Chromosomal Instability within Colorectal Cancer According to the Age of Onset. Mol. Carcinog. 2016, 55, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.; García, J.L.; Pérez, J.; Rueda, D.; Arriba, M.; Rodríguez, Y.; Urioste, M.; González-Sarmiento, R. NOMO-1 Gene Is Deleted in Early-Onset Colorectal Cancer. Oncotarget 2017, 8, 24429–24436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dettmer, U.; Kuhn, P.H.; Abou-Ajram, C.; Lichtenthaler, S.F.; Krüger, M.; Kremmer, E.; Haass, C.; Haffner, C. Transmembrane Protein 147 (TMEM147) Is a Novel Component of the Nicalin-NOMO Protein Complex. J. Biol. Chem. 2010, 285, 26174–26181. [Google Scholar] [CrossRef] [Green Version]
- Haffner, C.; Frauli, M.; Topp, S.; Irmler, M.; Hofmann, K.; Regula, J.T.; Bally-Cuif, L.; Haass, C. Nicalin and Its Binding Partner Nomo Are Novel Nodal Signaling Antagonists. EMBO J. 2004, 23, 3041–3050. [Google Scholar] [CrossRef] [Green Version]
- Francescangeli, F.; Contavalli, P.; De Angelis, M.L.; Baiocchi, M.; Gambara, G.; Pagliuca, A.; Fiorenzano, A.; Prezioso, C.; Boe, A.; Todaro, M.; et al. Dynamic Regulation of the Cancer Stem Cell Compartment by Cripto-1 in Colorectal Cancer. Cell Death Differ. 2015, 22, 1700–1713. [Google Scholar] [CrossRef] [Green Version]
- Kelber, J.A.; Panopoulos, A.D.; Shani, G.; Booker, E.C.; Belmonte, J.C.; Vale, W.W.; Gray, P.C. Blockade of Cripto Binding to Cell Surface GRP78 Inhibits Oncogenic Cripto Signaling via MAPK/PI3K and Smad2/3 Pathways. Oncogene 2009, 28, 2324–2336. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Guo, Y.; Hai, Y.; Yang, H.; Liu, Y.; Yang, S.; Zhang, Z.; Ma, M.; Liu, L.; Li, Z.; et al. Nodal Promotes the Self-Renewal of Human Colon Cancer Stem Cells via an Autocrine Manner through Smad2/3 Signaling Pathway. Biomed Res. Int. 2014, 2014, 364134. [Google Scholar] [CrossRef]
- Duan, W.; Li, R.; Ma, J.; Lei, J.; Xu, Q.; Jiang, Z.; Nan, L.; Li, X.; Wang, Z.; Huo, X.; et al. Overexpression of Nodal Induces a Metastatic Phenotype in Pancreatic Cancer Cells via the Smad2/3 Pathway. Oncotarget 2015, 6, 1490–1506. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.M. Nodal Signaling: Development Roles and Regulation. Development 2007, 134, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Siegers, G.M.; Jewer, M.; Postovit, L.M. Nodal Signalling in Embryogenesis and Tumourigenesis. Int. J. Biochem. Cell Biol. 2013, 45, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Mancino, M.; Strizzi, L.; Wechselberger, C.; Watanabe, K.; Gonzales, M.; Hamada, S.; Normanno, N.; Salomon, D.S.; Bianco, C. Regulation of Human Cripto-1 Gene Expression by TGF-Β1 and BMP-4 in Embryonal and Colon Cancer Cells. J. Cell. Physiol. 2008, 215, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Bodenstine, T.M.; Chandler, G.S.; Seftor, R.E.B.; Seftor, E.A.; Hendrix, M.J.C. Plasticity Underlies Tumor Progression: Role of Nodal Signaling. Cancer Metastasis Rev. 2016, 35, 21–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveros, J.C.; Franch, M.; Tabas-Madrid, D.; San-León, D.; Montoliu, L.; Cubas, P.; Pazos, F. Breaking-Cas: Off-Targets-Free GRNAs for CRISPR/Cas Technology. Available online: https://bioinfogp.cnb.csic.es/tools/breakingcas/?gset=4x2_GENOMES_EnsemblGenomes_47 (accessed on 29 July 2021).
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome Engineering Using the CRISPR-Cas9 System. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, Normalization, and Summaries of High Density Oligonucleotide Array Probe Level Data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving Gene/Transcript Definitions Significantly Alter the Interpretation of GeneChip Data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential Expression Analysis for Sequence Count Data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene Set Analysis Toolkit with Revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Díez, P.; Droste, C.; Dégano, R.M.; González-Muñoz, M.; Ibarrola, N.; Pérez-Andrés, M.; Garin-Muga, A.; Segura, V.; Marko-Varga, G.; LaBaer, J.; et al. Integration of Proteomics and Transcriptomics Data Sets for the Analysis of a Lymphoma B-Cell Line in the Context of the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2015, 14, 3530–3540. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, R.T.; Searle, B.C.; Llovet, A.; Villén, J. Plug-and-Play Analysis of the Human Phosphoproteome by Targeted High-Resolution Mass Spectrometry. Nat. Methods 2016, 13, 431–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, M.P.; Farnsworth, C.L.; Moritz, A.; Silva, J.C.; Jia, X.; Lee, K.A.; Guo, A.; Polakiewicz, R.D.; Comb, M.J. PTMScan Direct: Identification and Quantification of Peptides from Critical Signaling Proteins by Immunoaffinity Enrichment Coupled with LC-MS/MS. Mol. Cell. Proteom. 2012, 11, 187–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Tuñón, I.; Vuelta, E.; Lozano, L.; Herrero, M.; Méndez, L.; Palomero-Hernandez, J.; Pérez-Caro, M.; Pérez-García, J.; González-Sarmiento, R.; Sánchez-Martín, M. Establishment of a Conditional Nomo1 Mouse Model by CRISPR/Cas9 Technology. Mol. Biol. Rep. 2020, 47, 1381–1391. [Google Scholar] [CrossRef] [Green Version]
- NOMO1 Gene-GeneCards |NOMO1 Protein| NOMO1 Antibody. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=NOMO1 (accessed on 29 July 2021).
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-Time PCR Data by the Comparative CT Method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- National Centre for Biotechnological Information Home-SNP-NCBI. Available online: https://www.ncbi.nlm.nih.gov/snp/ (accessed on 29 July 2021).
- Exome Aggregate Consortium ExAC Browser. Available online: http://exac.broadinstitute.org/ (accessed on 29 July 2021).
- Haffner, C.; Dettmer, U.; Weiler, T.; Haass, C. The Nicastrin-like Protein Nicalin Regulates Assembly and Stability of the Nicalin-Nodal Modulator (NOMO) Membrane Protein Complex. J. Biol. Chem. 2007, 282, 10632–10638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Xu, C.; Wang, Y.; Zhang, L. Quiescin Sulfhydryl Oxidase 1 Regulates the Proliferation, Migration and Invasion of Human Glioblastoma Cells via PI3K/Akt Pathway. Onco. Targets Ther. 2020, 13, 5721–5729. [Google Scholar] [CrossRef] [PubMed]
- Malinowski, M.; Pietraszek, K.; Perreau, C.; Boguslawski, M.; Decot, V.; Stoltz, J.F.; Vallar, L.; Niewiarowska, J.; Cierniewski, C.; Maquart, F.X.; et al. Effect of Lumican on the Migration of Human Mesenchymal Stem Cells and Endothelial Progenitor Cells: Involvement of Matrix Metalloproteinase-14. PLoS ONE 2012, 7, e50709. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhong, L.; Li, Y.; Xiao, D.; Zhang, R.; Liao, D.; Lv, D.; Wang, X.; Wang, J.; Xie, X.; et al. Up-Regulation of PCOLCE by TWIST1 Promotes Metastasis in Osteosarcoma. Theranostics 2019, 9, 4342–4353. [Google Scholar] [CrossRef]
- Yun, D.P.; Wang, Y.Q.; Meng, D.L.; Ji, Y.Y.; Chen, J.X.; Chen, H.Y.; Lu, D.R. Actin-Capping Protein CapG Is Associated with Prognosis, Proliferation and Metastasis in Human Glioma. Oncol. Rep. 2018, 39, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Dai, W.; Wang, P.; Zhang, X.; Tang, Y.; Liu, L.; Wang, Q.; Li, M.; Tang, C. Periostin Promotes Migration, Proliferation, and Differentiation of Human Periodontal Ligament Mesenchymal Stem Cells. Connect. Tissue Res. 2018, 59, 108–119. [Google Scholar] [CrossRef]
- Löbel, M.; Bauer, S.; Meisel, C.; Eisenreich, A.; Kudernatsch, R.; Tank, J.; Rauch, U.; Kü Hl, U.; Schultheiss, H.P.; Volk, H.D.; et al. CCN1: A Novel Inflammation-Regulated Biphasic Immune Cell Migration Modulator. Cell. Mol. Life Sci. 2012, 69, 3101–3113. [Google Scholar] [CrossRef] [PubMed]
- Kiwanuka, E.; Andersson, L.; Caterson, E.J.; Junker, J.P.E.; Gerdin, B.; Eriksson, E. CCN2 Promotes Keratinocyte Adhesion and Migration via Integrin A5β1. Exp. Cell Res. 2013, 319, 2938–2946. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.S.; Oh, S.; Lee, K.-M.; Yoo, S.A.; Shin, I. CD44 Regulates Cell Proliferation, Migration, and Invasion via Modulation of c-Src Transcription in Human Breast Cancer Cells. Cell. Signal. 2015, 27, 1882–1894. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wang, X.; Xu, J.; Lu, L. SerpinE2, a Poor Biomarker of Endometrial Cancer, Promotes the Proliferation and Mobility of EC Cells. Cancer Biomark. 2017, 19, 271–278. [Google Scholar] [CrossRef]
- Oommen, S.; Gupta, S.K.; Vlahakis, N.E. Vascular Endothelial Growth Factor A (VEGF-A) Induces Endothelial and Cancer Cell Migration through Direct Binding to Integrin A9β1: Identification of a Specific A9β1 Binding Site. J. Biol. Chem. 2011, 286, 1083–1092. [Google Scholar] [CrossRef] [Green Version]
- Akimoto, N.; Ugai, T.; Zhong, R.; Hamada, T.; Fujiyoshi, K.; Giannakis, M.; Wu, K.; Cao, Y.; Ng, K.; Ogino, S. Rising Incidence of Early-Onset Colorectal Cancer—A Call to Action. Nat. Rev. Clin. Oncol. 2021, 18, 230–243. [Google Scholar] [CrossRef]
- Burnett-Hartman, A.N.; Lee, J.K.; Demb, J.; Gupta, S. An Update on the Epidemiology, Molecular Characterization, Diagnosis, and Screening Strategies for Early-Onset Colorectal Cancer. Gastroenterology 2021, 160, 1041–1049. [Google Scholar] [CrossRef]
- Willauer, A.N.; Liu, Y.; Pereira, A.A.L.; Lam, M.; Morris, J.S.; Raghav, K.P.S.; Morris, V.K.; Menter, D.; Broaddus, R.; Meric-Bernstam, F.; et al. Clinical and Molecular Characterization of Early-Onset Colorectal Cancer. Cancer 2019, 125, 2002–2010. [Google Scholar] [CrossRef]
- Holowatyj, A.N.; Perea, J.; Lieu, C.H. Gut Instinct: A Call to Study the Biology of Early-Onset Colorectal Cancer Disparities. Nat. Rev. Cancer 2021, 21, 339–340. [Google Scholar] [CrossRef]
- Elliott, R.L.; Blobe, G.C. Role of Transforming Growth Factor Beta in Human Cancer. J. Clin. Oncol. 2005, 23, 2078–2093. [Google Scholar] [CrossRef]
- Venhuizen, J.H.; Jacobs, F.J.C.; Span, P.N.; Zegers, M.M. P120 and E-Cadherin: Double-Edged Swords in Tumor Metastasis. Semin. Cancer Biol. 2020, 60, 107–120. [Google Scholar] [CrossRef]
- Duñach, M.; Del Valle-Pérez, B.; García de Herreros, A. P120-Catenin in Canonical Wnt Signaling. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Kurley, S.J.; Tischler, V.; Bierie, B.; Novitskiy, S.V.; Noske, A.; Varga, Z.; Zürrer-Härdi, U.; Brandt, S.; Carnahan, R.H.; Cook, R.S.; et al. A Requirement for P120-Catenin in the Metastasis of Invasive Ductal Breast Cancer. J. Cell Sci. 2021, 134, jcs250639. [Google Scholar] [CrossRef] [PubMed]
- Kidacki, M.; Lehman, H.L.; Green, M.V.; Warrick, J.I.; Stairs, D.B. P120-Catenin Downregulation and PIK3CA Mutations Cooperate to Induce Invasion through MMP1 in HNSCC. Mol. Cancer Res. 2017, 15, 1398–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, C.; Zuo, Z.; Ji, Q.; Feng, D. P120ctn May Participate in Epithelial-Mesenchymal Transition in OSCC. Indian J. Cancer 2016, 53, 20–24. [Google Scholar] [CrossRef]
- Jia, Y.; Vong, J.S.L.; Asafova, A.; Garvalov, B.K.; Caputo, L.; Cordero, J.; Singh, A.; Boettger, T.; Günther, S.; Fink, L.; et al. Lamin B1 Loss Promotes Lung Cancer Development and Metastasis by Epigenetic Derepression of RET. J. Exp. Med. 2019, 216, 1377–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, F.; Wang, T.; Wu, Z.; Feng, Y.; Wang, W.; Zhou, S.; Ma, X.; Wang, S. HMGA1 Exacerbates Tumor Growth through Regulating the Cell Cycle and Accelerates Migration/Invasion via Targeting MiR-221/222 in Cervical Cancer Article. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Kim, H.; Kim, M.; Im, S.-K.; Fang, S. Mouse Cre-LoxP System: General Principles to Determine Tissue-Specific Roles of Target Genes. Lab. Anim. Res. 2018, 34, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Loesch, R.; Caruso, S.; Paradis, V.; Godard, C.; Gougelet, A.; Renault, G.; Picard, S.; Tanaka, I.; Renoux-Martin, Y.; Perret, C.; et al. Deleting the β-Catenin Degradation Domain in Mouse Hepatocytes Drives Hepatocellular Carcinoma or Hepatoblastoma-like Tumor Growth. J. Hepatol. 2022, 77, 424–435. [Google Scholar] [CrossRef]
- Huang, X.D.; Zheng, Y.B.; Yang, Y.J.; Yang, C.; Li, H.L.; Cheng, H.R. Mouse Models for Human Colorectal Cancer with Liver Metastasis. Zhonghua Yi Xue Za Zhi 2019, 99, 2701–2705. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GVSc | NOMO1 | NOMO2 | NOMO3 | Poblation ID | Subjects | FREQ. ALE> | FREQ. ALE< |
|---|---|---|---|---|---|---|---|
| c.26AC > T p.(L9P) | C | T | C | ||||
| c.156T > G p.(S52S) | T | G | T | ||||
| c.696C > G p.(N232N) | C | G | C | ExAc | 7964 | C = 0.997 | G = 0.003 |
| c.1185G > A p.(P395P) | G | G | A | ExAc | 121,402 | G = 0.999 | A = 0.001 |
| c.1210 A > G p.(I404V) | A | G | G | ExAc | 121,400 | A = 0.999 | G = 0.001 |
| c.1238 A > G p.(Q413R) | A | G | A | ExAc | 11,862 | A = 0.999 | G = 0.000 |
| c.1260C > G p.(P420P) | C | G | C | ExAc | 25,306 | C = 0.999 | G = 0.001 |
| c.1374 T > G p.(K458N) | T | T | G | ExAc | 27,102 | T = 0.777 | G = 0.223 |
| c.1468A > G p.(N490D) | A | G | G | ExAc | 23,336 | A = 0.943 | G = 0.056 |
| c.1477 A > G p.(M493V) | A | G | G | ExAc | 121,206 | A = 0.999 | G = 0.001 |
| c.1738 A > G p.(M580V) | A | G | G | ExAc | 119,700 | A = 0.989 | G = 0.010 |
| c.2187C > T p.(G729G) | C | T | T | ExAc | 121,396 | C = 0.999 | T = 0.001 |
| c.2211C > T p.(P737P) | C | C | T | ExAc | 814 | T = 0.814 | C = 0.185 |
| c.2388T > C p.(H796H) | T | C | C | ||||
| c.2586G > A p.(A862A) | G | A | A | ||||
| c.2694C > T p.(S898S) | C | T | T | ExAc | 121,402 | C = 0.999 | T = 0.001 |
| c.3216A > G p.(T1072T) | A | G | G | ExAc | 121,310 | A = 0.999 | G = 0.001 |
| c.3318C > T p.(D1106D) | C | T | T | ExAc | 121,406 | C = 0.999 | T = 0.001 |
| c.3583C > G p.(R1195R) | C | G | G | ||||
| c.3666T > Gp.(T1222T) | T | G | G |
| SAMPLE | NOMO1 Status | Variant | Alellic Frecuency (%) | Sift | Polyphen | CADD Score |
|---|---|---|---|---|---|---|
| Sample 1 | Heterozygous | c.2684T > A/p.(Leu895Ter) | 22.5 | - | - | |
| Sample 2 | Heterozygous | c.3637C > T/p.(Gln1213Ter) | 27.3 | - | - | |
| Sample 3 | Homozygous | c.2833C > T/p.(Gln945Ter) | 10.5 | - | - | |
| Sample 4 | Heterozygous | c.2428G > T/p.(Glu810Ter) | 17.2 | - | - | |
| Sample 5 | Germinal | c.2656G > A/p.(Asp886Asn) | 33.3 | deleterious (0.02) | probably damaging (0.835) | 25.8 |
| Sample 6 | Germinal | c.2787G > A/p.(Met929Ile) | 16.2 | deleterious (0.01) | probably damaging (0.783) | 26.4 |
| Sample 7 | Homozygous | c.2794G > A/p.(Glu932Lys) | 32.1 | deleterious (0) | probably damaging (0.977) | 32.0 |
| Sample 1 | Heterozygous | c.2810C > T/p.(Pro937Leu) | 52.9 | deleterious (0) | probably damaging (1) | 29.5 |
| Sample 3 | Homozygous | c.2278G > A/ p.(Gly760Arg) | 11.5 | deleterious (0.01) | probably damaging (0.992) | 29.3 |
| Sample 3 | Homozygous | c.3019G > A/ p.(Gly1007Arg) | 25.0 | deleterious (0) | probably damaging (1) | 26.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-García, J.; Martel-Martel, A.; García-Vallés, P.; Corchete, L.A.; García, J.L.; Gestoso-Uzal, N.; Vidal-Tocino, R.; Blanco, Ó.; Méndez, L.; Sánchez-Martín, M.; et al. Recurrent NOMO1 Gene Deletion Is a Potential Clinical Marker in Early-Onset Colorectal Cancer and Is Involved in the Regulation of Cell Migration. Cancers 2022, 14, 4029. https://doi.org/10.3390/cancers14164029
Pérez-García J, Martel-Martel A, García-Vallés P, Corchete LA, García JL, Gestoso-Uzal N, Vidal-Tocino R, Blanco Ó, Méndez L, Sánchez-Martín M, et al. Recurrent NOMO1 Gene Deletion Is a Potential Clinical Marker in Early-Onset Colorectal Cancer and Is Involved in the Regulation of Cell Migration. Cancers. 2022; 14(16):4029. https://doi.org/10.3390/cancers14164029
Chicago/Turabian StylePérez-García, Jésica, Abel Martel-Martel, Paula García-Vallés, Luis A. Corchete, Juan L. García, Nerea Gestoso-Uzal, Rosario Vidal-Tocino, Óscar Blanco, Lucía Méndez, Manuel Sánchez-Martín, and et al. 2022. "Recurrent NOMO1 Gene Deletion Is a Potential Clinical Marker in Early-Onset Colorectal Cancer and Is Involved in the Regulation of Cell Migration" Cancers 14, no. 16: 4029. https://doi.org/10.3390/cancers14164029
APA StylePérez-García, J., Martel-Martel, A., García-Vallés, P., Corchete, L. A., García, J. L., Gestoso-Uzal, N., Vidal-Tocino, R., Blanco, Ó., Méndez, L., Sánchez-Martín, M., Fuentes, M., Herrero, A. B., Holowatyj, A. N., Perea, J., & González-Sarmiento, R. (2022). Recurrent NOMO1 Gene Deletion Is a Potential Clinical Marker in Early-Onset Colorectal Cancer and Is Involved in the Regulation of Cell Migration. Cancers, 14(16), 4029. https://doi.org/10.3390/cancers14164029