
Luminal and Tumor-Associated Gut Microbiome Features Linked to Precancerous Lesions Malignancy Risk: A Compositional Approach
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
- Metabolism of dietary and other components into DNA-damaging compounds—for example, production of deoxycholic acid from cholic acid (one of bile acids) by gut bacteria [9] and of genotoxic hydrogen sulfide (H2S)—by sulfur-metabolizing taxa;
- Maintenance of chronic inflammation known to be one of the driving forces in CRC development [14].
2. Materials and Methods
2.1. Study Design
2.2. Biosamples Preparation and DNA-Sequencing
2.3. Data Preprocessing and Primary Analysis
2.4. Statistical Data Analysis
- 251 healthy controls (Healthy)—subjects with no lesions according to colonoscopy or with up to two small (<5 mm) polyps;
- patients with more than three adenomas with low-grade dysplasia (MP, N = 67);
- patients with stage 0 CRC (S0, N = 73);
- patients with stage I and II CRC (SI_II, N = 111);
- patients with stage III and IV CRC (SIII_IV, N = 74).
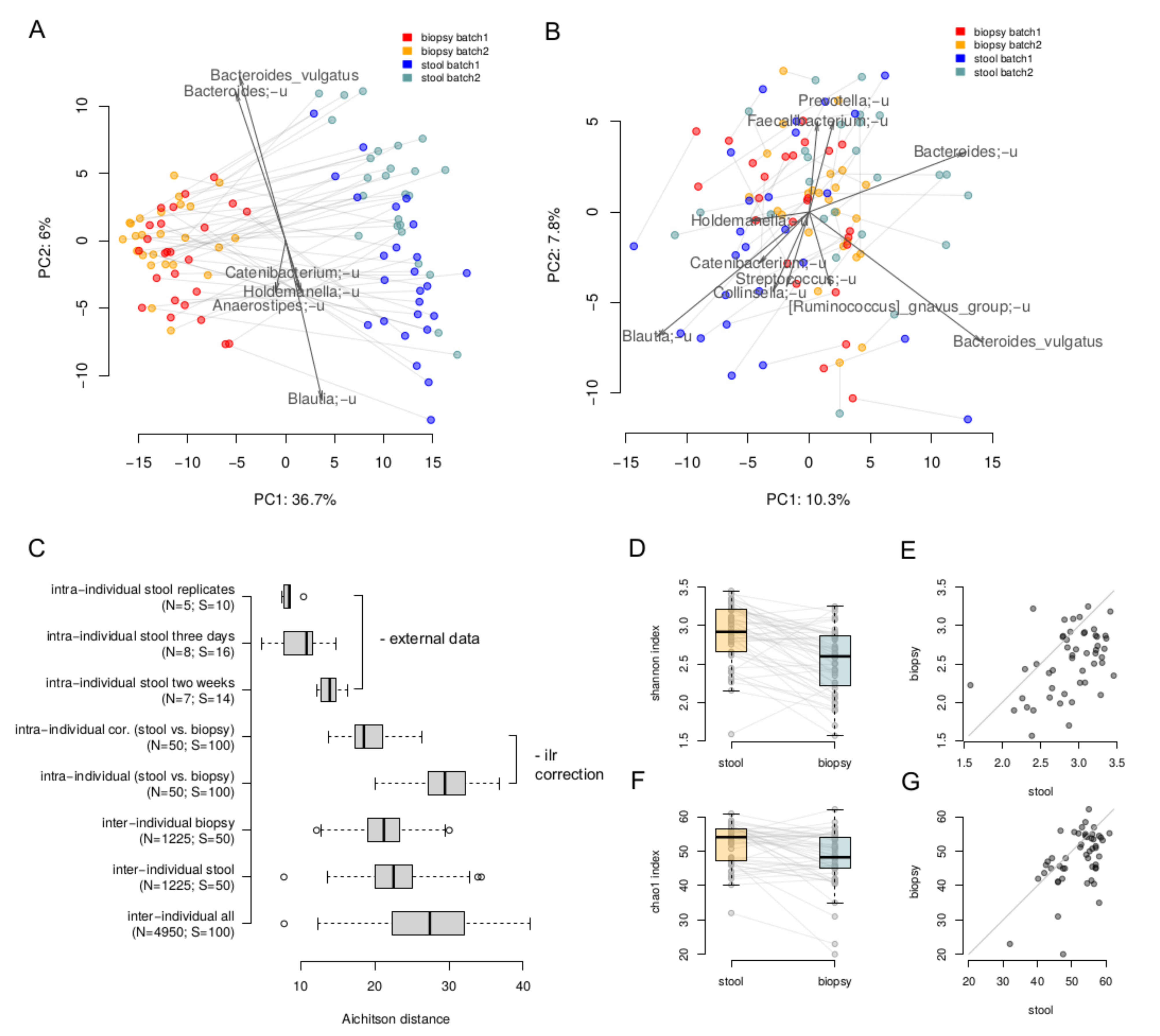
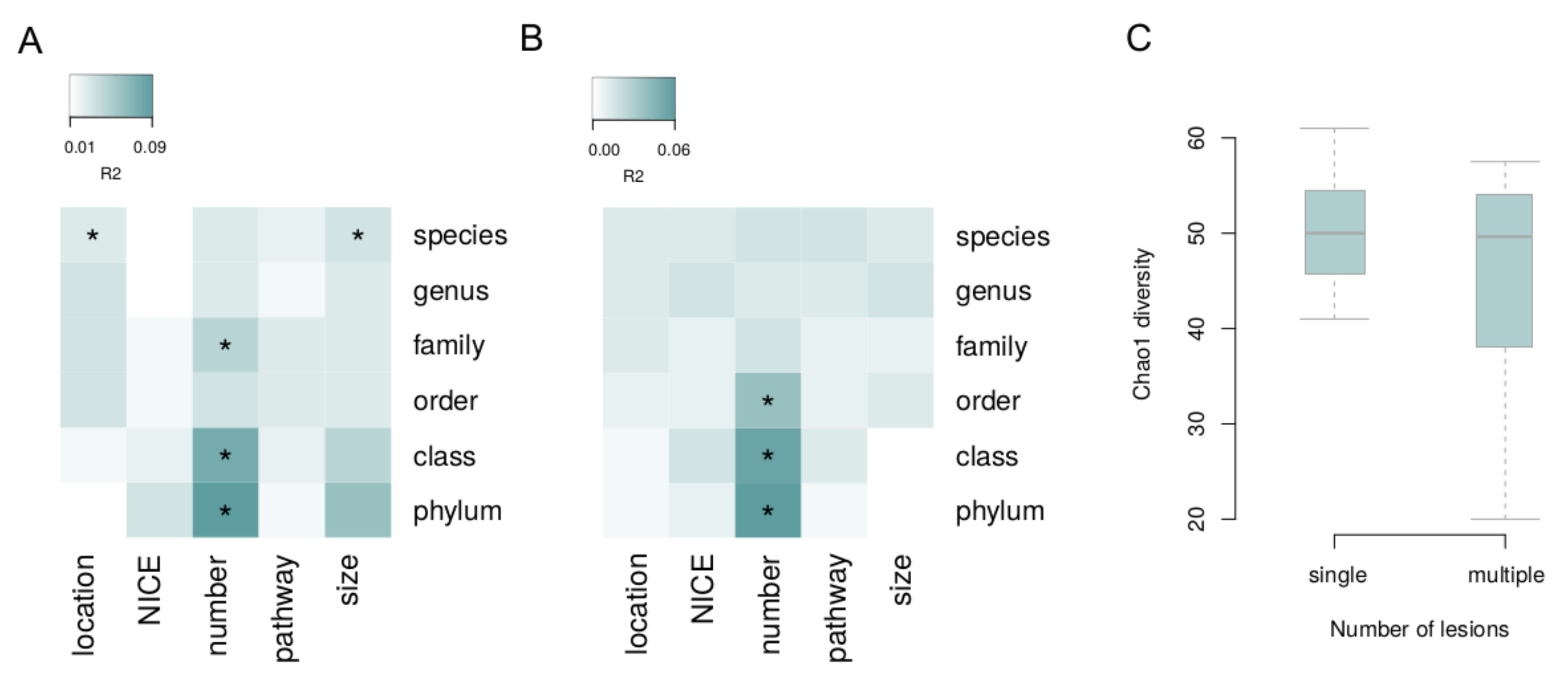
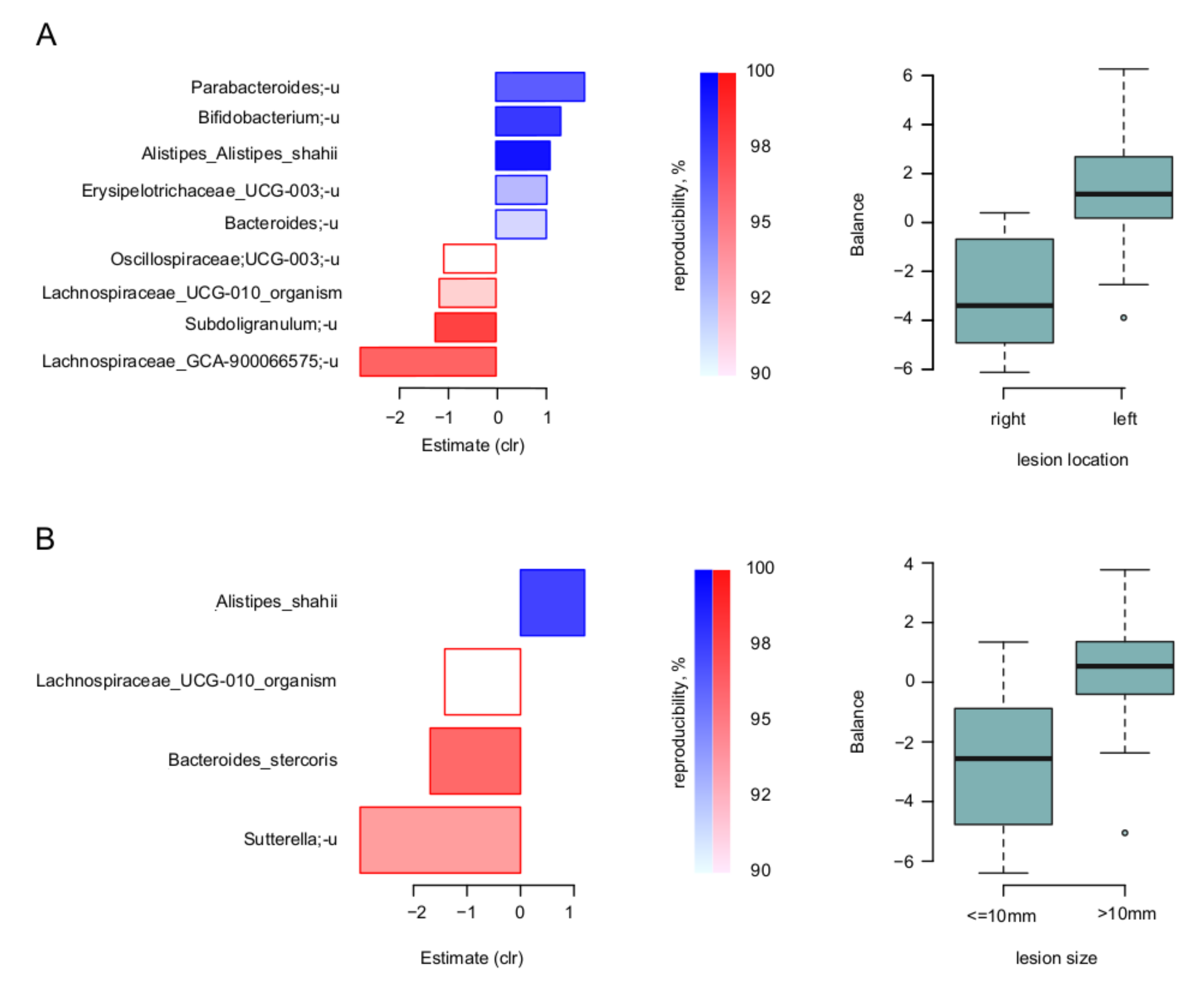
3. Results
4. Discussion
- study of stool samples alone, without mucosal sampling;
- focus on patients with cancer rather than the ones with the precancerous lesions;
- lack of consideration for the lesion type and lesion location;
- use of amplicon sequencing but not the “shotgun” metagenomics.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Worldwide Cancer Data. Available online: https://www.wcrf.org/dietandcancer/worldwide-cancer-data/ (accessed on 16 September 2021).
- Wong, M.C.; Huang, J.; Lok, V.; Wang, J.; Fung, F.; Ding, H.; Zheng, Z.-J. Differences in Incidence and Mortality Trends of Colorectal Cancer Worldwide Based on Sex, Age, and Anatomic Location. Clin. Gastroenterol. Hepatol. 2021, 19, 955–966.e61. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.T.; Fung, E. Polypectomy Techniques. Surg. Clin. N. Am. 2020, 100, 1049–1067. [Google Scholar] [CrossRef] [PubMed]
- Pickhardt, P.J.; Pooler, B.D.; Kim, D.H.; Hassan, C.; Matkowskyj, K.A.; Halberg, R.B. The Natural History of Colorectal Polyps: Overview of Predictive Static and Dynamic Features. Gastroenterol. Clin. N. Am. 2018, 47, 515–536. [Google Scholar]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal Cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Kang, X.; Zhang, R.; Ny Kwong, T.; Lui, R.N.; Wu, W.K.; Sung, J.J.; Yu, J.; Wong, S.H. Serrated neoplasia in the colorectum: Gut microbiota and molecular pathways. Gut Microbes 2021, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; E Powers, S.; Zhu, W.; Hannun, Y.A. Substantial contribution of extrinsic risk factors to cancer development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Crockett, S.D.; Nagtegaal, I. Terminology, Molecular Features, Epidemiology, and Management of Serrated Colorectal Neoplasia. Gastroenterology 2019, 157, 949–966.e4. [Google Scholar] [CrossRef] [Green Version]
- Janney, A.; Powrie, F.; Mann, E.H. Host–microbiota maladaptation in colorectal cancer. Nature 2020, 585, 509–517. [Google Scholar] [CrossRef]
- Sears, C.L.; Garrett, W.S. Microbes, Microbiota, and Colon Cancer. Cell Host Microbe 2014, 15, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Garrett, W.S. The gut microbiota and colon cancer. Science 2019, 364, 1133–1135. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Lupton, J.R. Microbial Degradation Products Influence Colon Cancer Risk: The Butyrate Controversy. J. Nutr. 2004, 134, 479–482. [Google Scholar] [CrossRef] [Green Version]
- DeDecker, L.; Coppedge, B.; Avelar-Barragan, J.; Karnes, W.; Whiteson, K. Microbiome Distinctions between the CRC Car-cinogenic Pathways. Gut Microbes 2021, 13, 1854641. [Google Scholar] [CrossRef] [PubMed]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osman, M.-A.; Neoh, H.-M.; Ab Mutalib, N.-S.; Chin, S.-F.; Jamal, R. 16S rRNA Gene Sequencing for Deciphering the Colo-rectal Cancer Gut Microbiome: Current Protocols and Workflows. Front. Microbiol. 2018, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Coker, O.O.; Nakatsu, G.; Wu, W.K.K.; Zhao, L.; Chen, Z.; Chan, F.K.L.; Kristiansen, K.; Sung, J.J.Y.; Wong, S.H.; et al. Multi-Cohort Analysis of Colorectal Cancer Metagenome Identified Altered Bacteria across Populations and Universal Bacterial Markers. Microbiome 2018, 6, 70. [Google Scholar] [CrossRef]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial Driver–passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef]
- Zwinsová, B.; Petrov, V.A.; Hrivňáková, M.; Smatana, S.; Micenková, L.; Kazdová, N.; Popovici, V.; Hrstka, R.; Šefr, R.; Bencsiková, B.; et al. Colorectal Tumour Mucosa Microbiome Is Enriched in Oral Pathogens and Defines Three Subtypes That Correlate with Markers of Tumour Progression. Cancers 2021, 13, 4799. [Google Scholar] [CrossRef]
- Hugerth, L.W.; Wefer, H.A.; Lundin, S.; Jakobsson, H.E.; Lindberg, M.; Rodin, S.; Engstrand, L.; Andersson, A.F. DegePrime, a Program for Degenerate Primer Design for Broad-Taxonomic-Range PCR in Microbial Ecology Studies. Appl. Environ. Microbiol. 2014, 80, 5116–5123. [Google Scholar] [CrossRef] [Green Version]
- Merkel, A.Y.; Tarnovetskii, I.Y.; Podosokorskaya, O.A.; Toshchakov, S.V. Analysis of 16S rRNA Primer Systems for Profiling of Thermophilic Microbial Communities. Microbiology 2019, 88, 671–680. [Google Scholar] [CrossRef]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efimova, D.; Tyakht, A.; Popenko, A.; Vasilyev, A.; Altukhov, I.; Dovidchenko, N.; Odintsova, V.; Klimenko, N.; Loshkarev, R.; Pashkova, M.; et al. Knomics-Biota—A system for exploratory analysis of human gut microbiota data. BioData Min. 2018, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Chase, J.M.; Kraft, N.J.B.; Smith, K.G.; Vellend, M.; Inouye, B.D. Using null models to disentangle variation in community dissimilarity from variation in α-diversity. Ecosphere 2011, 2, 1–11. [Google Scholar] [CrossRef]
- Odintsova, V.E.; Klimenko, N.S.; Tyakht, A.V. Approximation of a Microbiome Composition Shift by a Change in a Single Balance Between Two Groups of Taxa. mSystems 2022, 7, e00155-22. [Google Scholar] [CrossRef]
- Kurtz, Z.D.; Müller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLoS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef] [Green Version]
- Blondel, V.D.; Guillaume, J.-L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef]
- Egozcue, J.J.; Pawlowsky-Glahn, V.; Mateu-Figueras, G.; Barceló-Vidal, C. Isometric Logratio Transformations for Compositional Data Analysis. Math. Geol. 2003, 35, 279–300. [Google Scholar] [CrossRef]
- Klimenko, N.S.; Tyakht, A.V.; Popenko, A.S.; Vasiliev, A.S.; Altukhov, I.A.; Ischenko, D.S.; Shashkova, T.I.; Efimova, D.A.; Nikogosov, D.A.; Osipenko, D.A.; et al. Microbiome Responses to an Uncontrolled Short-Term Diet Intervention in the Frame of the Citizen Science Project. Nutrients 2018, 10, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernevskaya, E.; Klimenko, N.; Pautova, A.; Buyakova, I.; Tyakht, A.; Beloborodova, N. Host-Microbiome Interactions Mediated by Phenolic Metabolites in Chronically Critically Ill Patients. Metabolites 2021, 11, 122. [Google Scholar] [CrossRef]
- Odintsova, V.; Klimenko, N.; Tyakht, A.; Volokh, O.; Popov, V.; Alexeev, D.; Berezhnaya, Y. Yogurt fortified with vitamins and probiotics impacts the frequency of upper respiratory tract infections but not gut microbiome: A multicenter double-blind placebo controlled randomized study. J. Funct. Foods 2021, 83, 104572. [Google Scholar] [CrossRef]
- Toribio-Mateas, M.A.; Bester, A.; Klimenko, N. Impact of Plant-Based Meat Alternatives on the Gut Microbiota of Consumers: A Real-World Study. Foods 2021, 10, 2040. [Google Scholar] [CrossRef]
- Hale, V.L.; Jeraldo, P.; Mundy, M.; Yao, J.; Keeney, G.; Scott, N.; Cheek, E.H.; Davidson, J.; Greene, M.; Martinez, C.; et al. Synthesis of multi-omic data and community metabolic models reveals insights into the role of hydrogen sulfide in colon cancer. Methods 2018, 149, 59–68. [Google Scholar] [CrossRef]
- Carbonero, F.; Benefiel, A.C.; Gaskins, H.R. Contributions of the microbial hydrogen economy to colonic homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 504–518. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Attene-Ramos, M.S.; Nava, G.M.; Muellner, M.G.; Wagner, E.D.; Plewa, M.J.; Gaskins, H.R. DNA damage and toxicogenomic analyses of hydrogen sulfide in human intestinal epithelial FHs 74 Int cells. Environ. Mol. Mutagen. 2010, 51, 304–314. [Google Scholar] [CrossRef]
- Oliveira, A.; Freire, P.; Souto, P.; Ferreira, M.; Mendes, S.; Lérias, C.; Amaro, P.; Portela, F.; Sofia, C. Association between the location of colon polyps at baseline and surveillance colonoscopy—A retrospective study. Rev. Esp. Enferm. Dig. 2016, 108, 563–567. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Parameter | Categories before Aggregation (Number of Patients with This Value) | Categories after Aggregation (Number of Patients) | Use in the Analysis |
|---|---|---|---|
| Lesion location | Cecum (3) Ascending colon (10) Transverse colon (4) | Right (17) | Statistical analysis |
| Descending colon (5) Sigmoid colon (21) Rectum (7) | Left (33) | ||
| NICE category | 1 (10) | 1 (10) | |
| 2 (36) 3 (4) | >1 (40) | ||
| Number of lesions | 1 (34) 2 (14) 3(1) 5 (1) | 1 (34) >1 (16) | |
| Size of lesions | <5 mm (11) 6–9 mm (16) | <10 mm (27) | |
| 10–14 mm (10) 15–19 mm (7) >20 mm (6) | ≥10 mm (23) | ||
| Macroscopic characteristic | 0–1 s (33) 0–1 p (4) 0–1 sp (13) | For malignization pathway identification only | |
| BRAF mutation | Yes (10) No (15) | ||
| NRAS mutation | Yes (3) No (22) | ||
| Histology | Hyperplastic polyp (9) Sessile serrated lesion (SSL) with low grade dysplasia (13) Adenoma with low grade dysplasia (3) Adenoma with high grade dysplasia (20) Highly differentiated adenocarcinoma (4) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanov, V.A.; Karasev, I.A.; Klimenko, N.S.; Koshechkin, S.I.; Tyakht, A.V.; Malikhova, O.A. Luminal and Tumor-Associated Gut Microbiome Features Linked to Precancerous Lesions Malignancy Risk: A Compositional Approach. Cancers 2022, 14, 5207. https://doi.org/10.3390/cancers14215207
Romanov VA, Karasev IA, Klimenko NS, Koshechkin SI, Tyakht AV, Malikhova OA. Luminal and Tumor-Associated Gut Microbiome Features Linked to Precancerous Lesions Malignancy Risk: A Compositional Approach. Cancers. 2022; 14(21):5207. https://doi.org/10.3390/cancers14215207
Chicago/Turabian StyleRomanov, Vladimir A., Ivan A. Karasev, Natalia S. Klimenko, Stanislav I. Koshechkin, Alexander V. Tyakht, and Olga A. Malikhova. 2022. "Luminal and Tumor-Associated Gut Microbiome Features Linked to Precancerous Lesions Malignancy Risk: A Compositional Approach" Cancers 14, no. 21: 5207. https://doi.org/10.3390/cancers14215207
APA StyleRomanov, V. A., Karasev, I. A., Klimenko, N. S., Koshechkin, S. I., Tyakht, A. V., & Malikhova, O. A. (2022). Luminal and Tumor-Associated Gut Microbiome Features Linked to Precancerous Lesions Malignancy Risk: A Compositional Approach. Cancers, 14(21), 5207. https://doi.org/10.3390/cancers14215207