
The RSL3 Induction of KLK Lung Adenocarcinoma Cell Ferroptosis by Inhibition of USP11 Activity and the NRF2-GSH Axis

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and methods
2.1. Cell Lines and Culture
2.2. NRF2 siRNA, USP11 Vector, and Cell Infection to Establish Stable LUAD Cell Sublines
2.3. RNA Isolation and RNA-Seq
2.4. Quantitative Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR)
2.5. Western Blot
2.6. Cell Viability Assay
2.7. Measurement of GSH (Glutathione) and GSSG (Glutathione Oxidized) Levels in Cells
2.8. Lipid Peroxidation Assay
2.9. Animal Experiment
2.10. Detection of Malondialdehyde (MDA) Level
2.11. Immunohistochemistry
2.12. Immunoprecipitation (IP)-Western Blot
2.13. Molecule-Docking Analysis
2.14. Purification of Wild Type and Mutant USP11 Proteins
2.15. Affinity Binding Assay
2.16. Statistical Analysis
3. Results
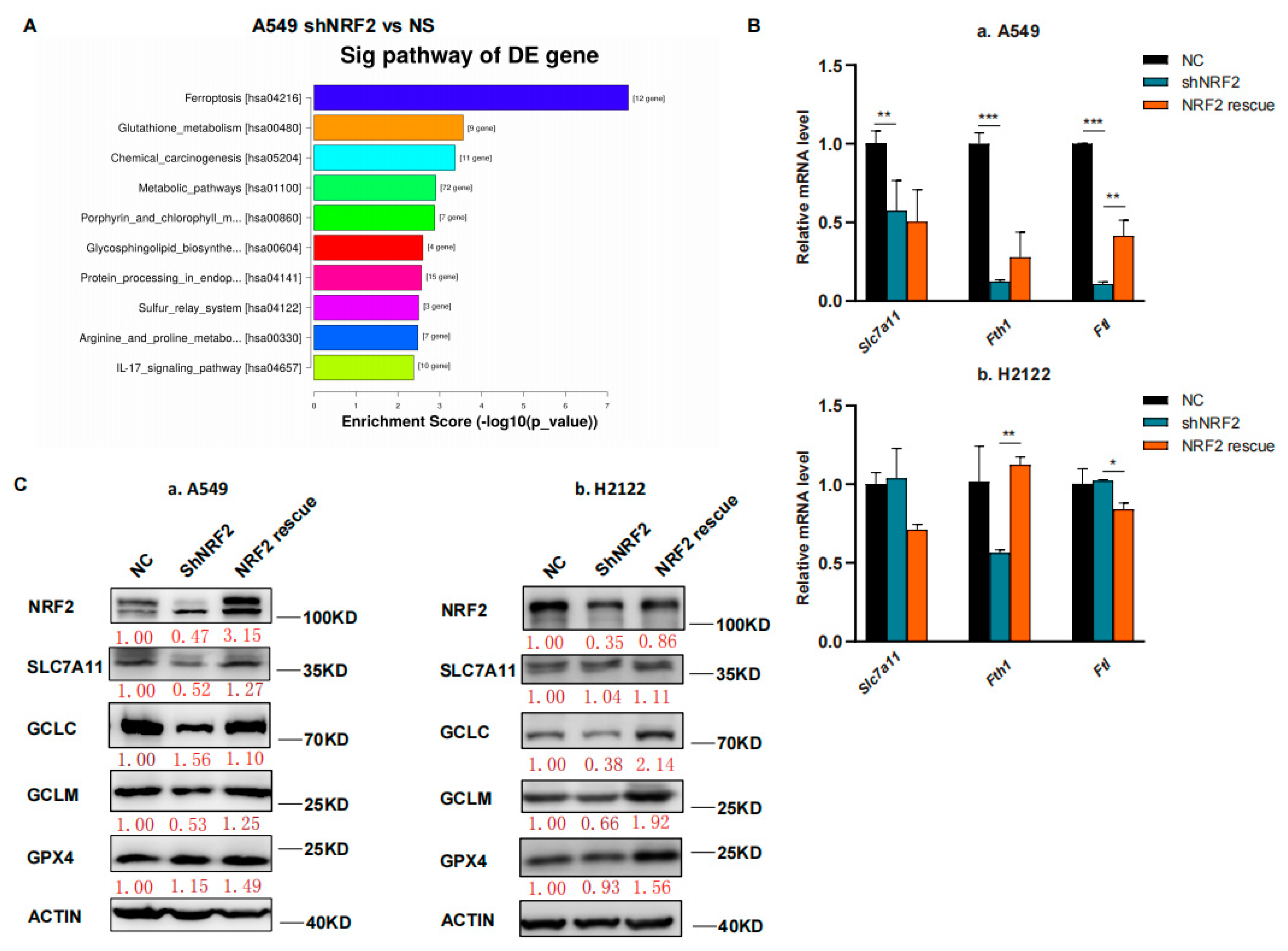
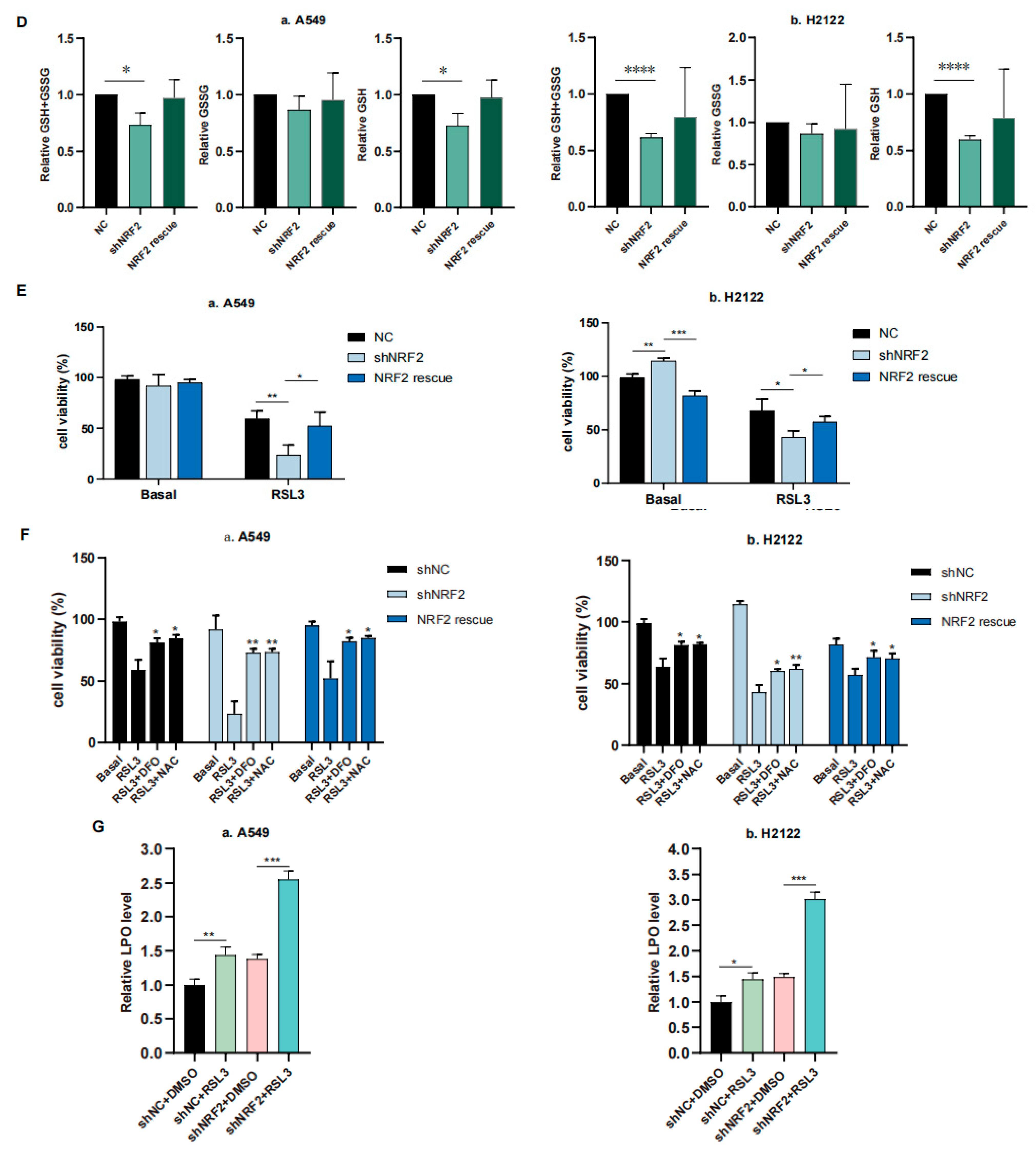
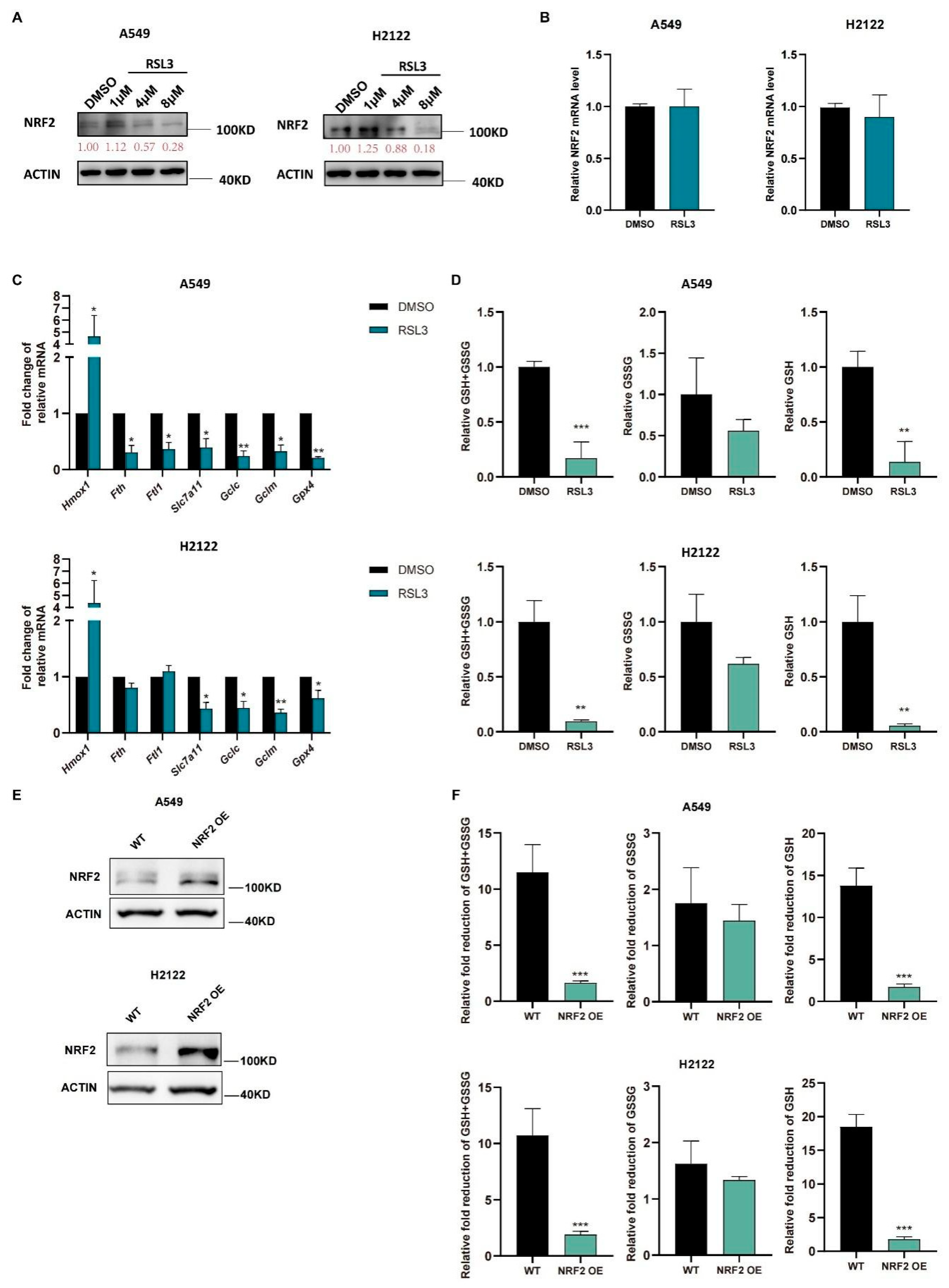
3.1. RSL3 Induction of KLK LUAD Cell Ferroptosis Sensitized by the NRF2-GSH Inhibition
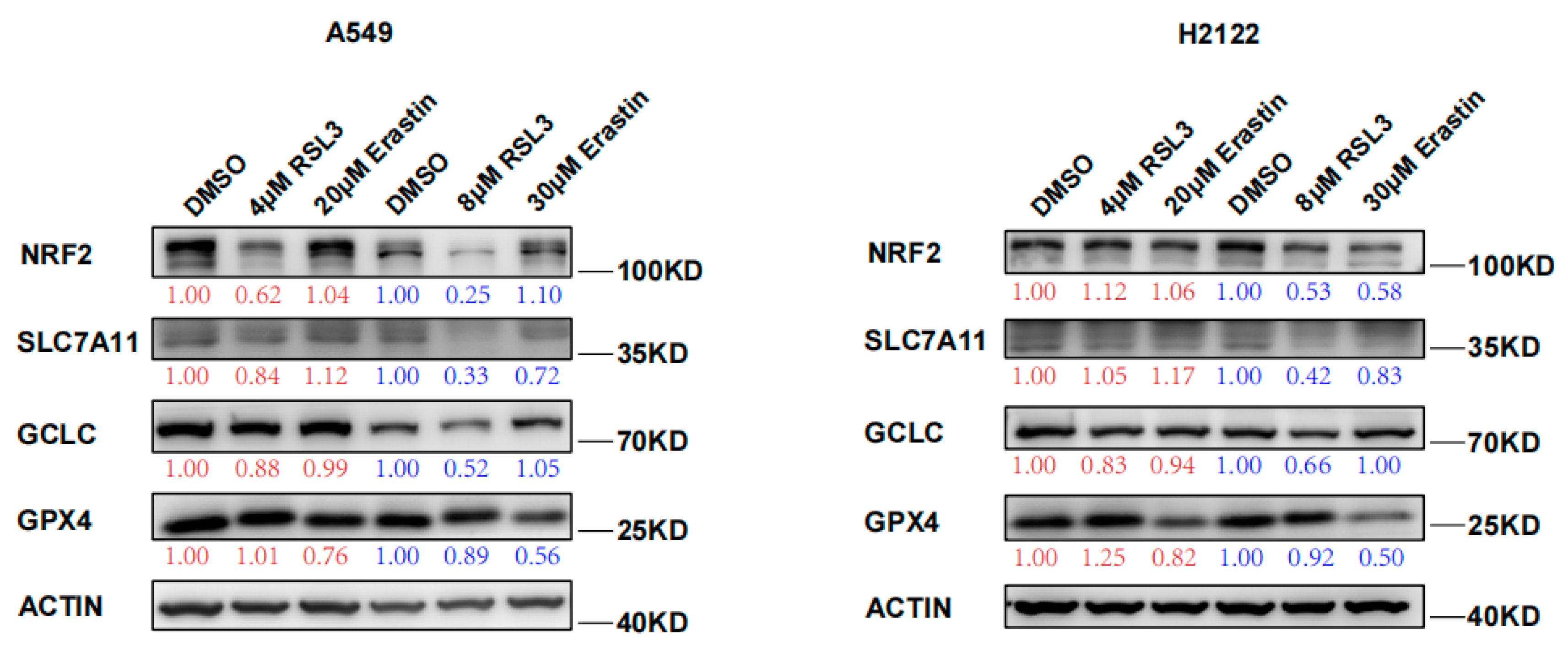
3.2. RSL3 Reduction of NRF2 Expression in KLK LUAD Cells during Tumor Cell Ferroptosis
3.3. RSL3 Inhibition of the NRF2-GSH Axis during Ferroptosis in KLK LUAD Cells In Vitro and In Vivo
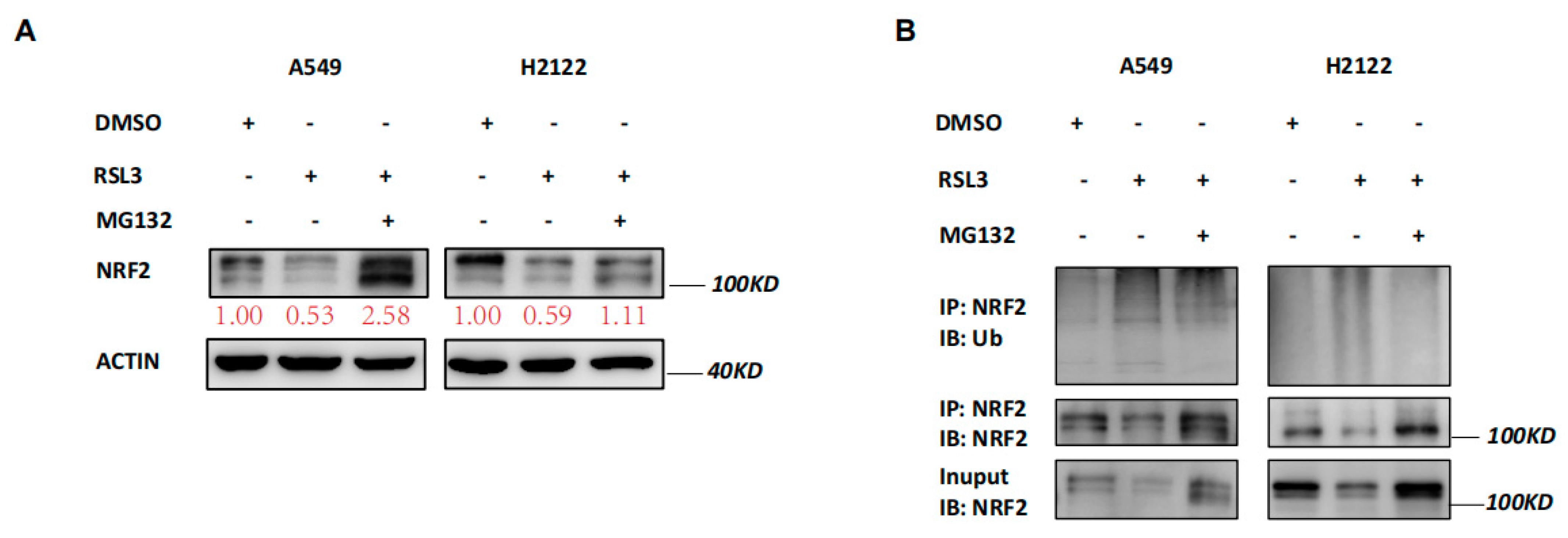
3.4. RSL3 Reduction of NRF2 Expression by Promoting Its Ubiquination in KLK LUAD Cells
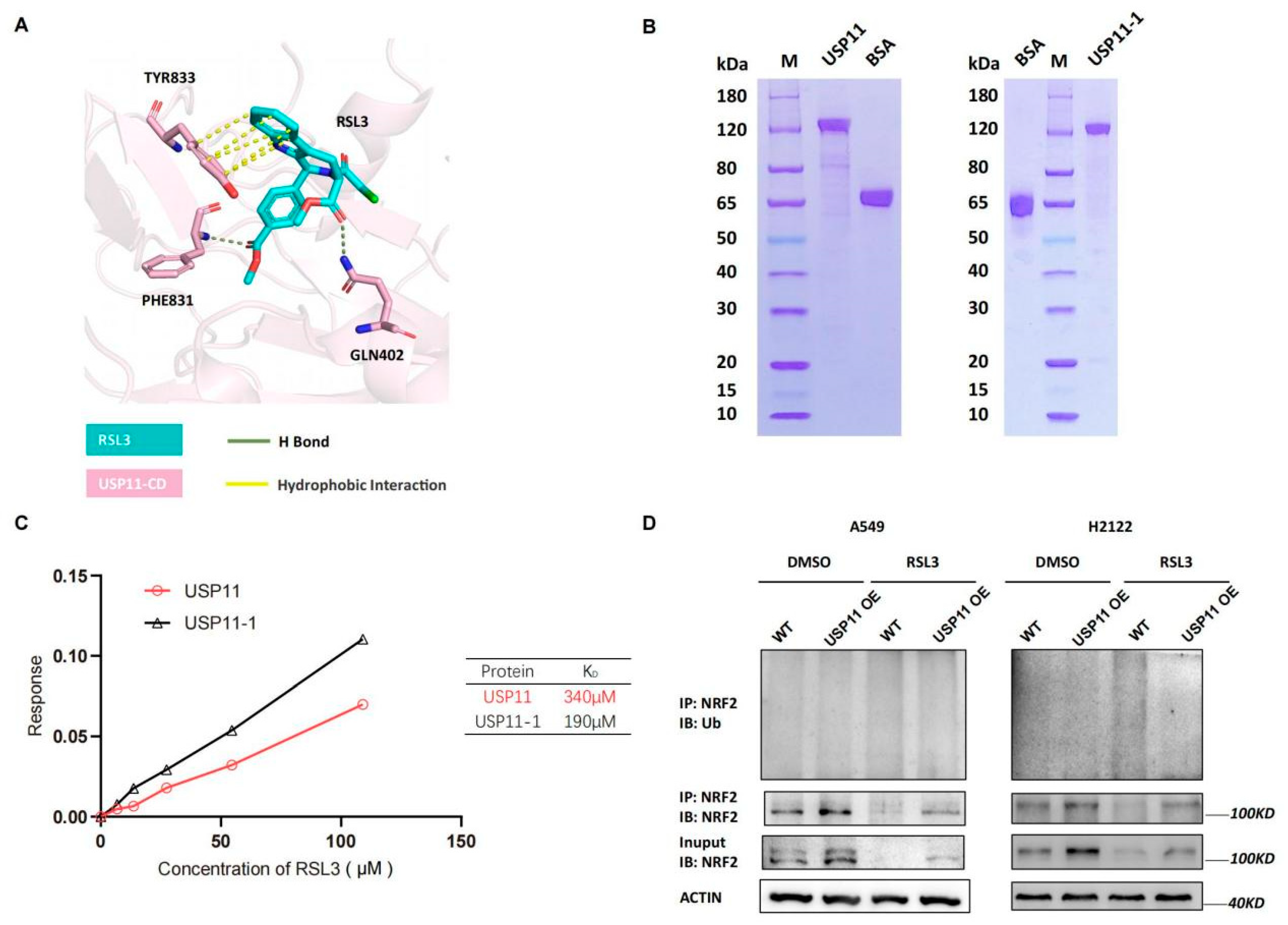
3.5. RSL3 Directly Targeting USP11 in Promotion of NRF2 Ubiquination in KLK LUAD Cells
4. Discussion
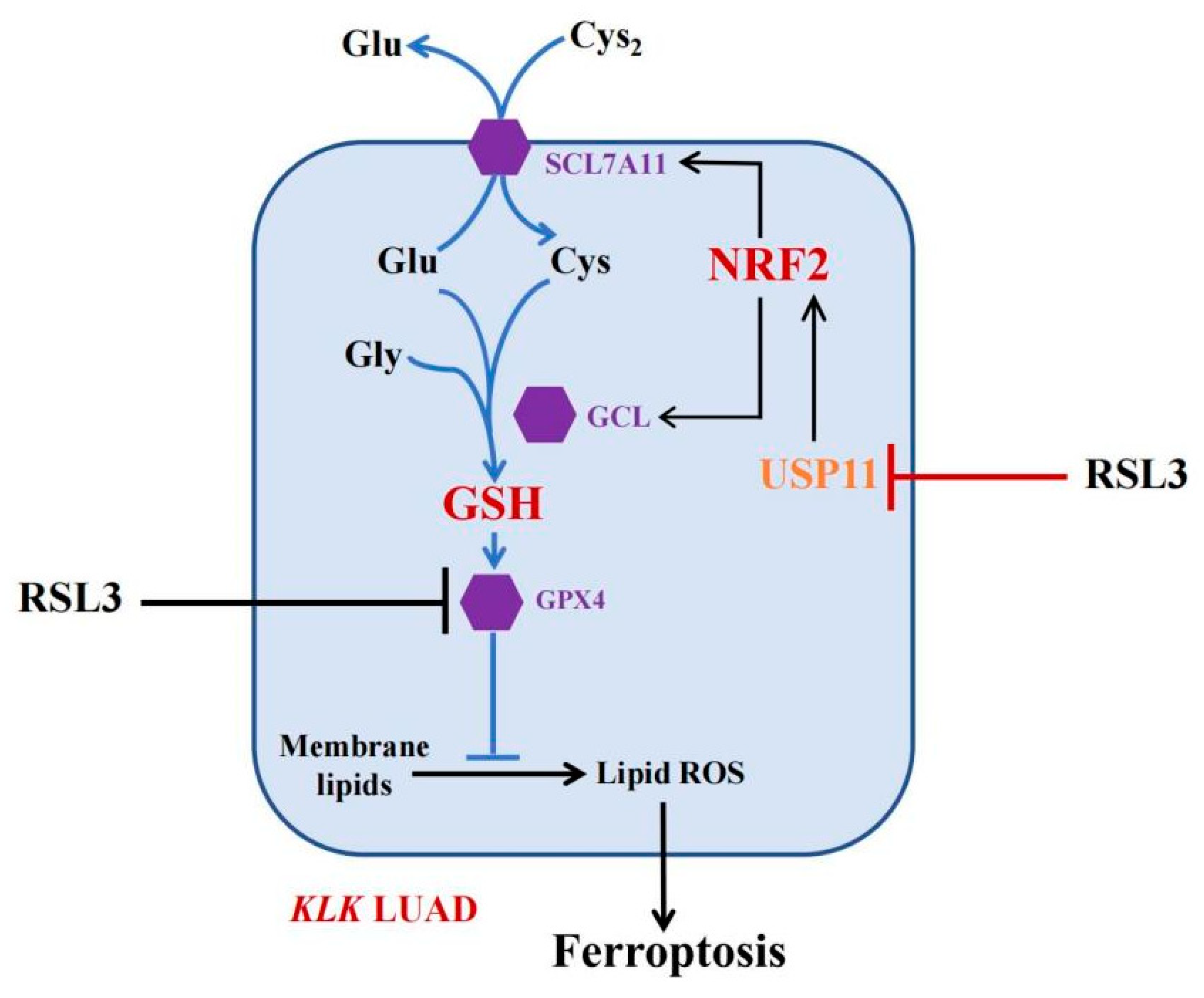
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, B.A.; Woo, M.S.; Getz, G.; Perner, S.; Ding, L.; Beroukhim, R.; Lin, W.M.; Province, M.A.; Kraja, A.; Johnson, L.A.; et al. Characterizing the cancer genome in lung adenocarcinoma. Nature 2007, 450, 893–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [Green Version]
- Calles, A.; Sholl, L.M.; Rodig, S.J.; Pelton, A.K.; Hornick, J.L.; Butaney, M.; Lydon, C.; Dahlberg, S.E.; Oxnard, G.R.; Jackman, D.M.; et al. Immunohistochemical Loss of LKB1 Is a Biomarker for More Aggressive Biology in KRAS-Mutant Lung Adenocarcinoma. Clin. Cancer Res. 2015, 21, 2851–2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Huang, Q.; Xia, J.; Cheng, S.; Pei, D.; Zhang, X.; Shu, X. The E3 Ligase MIB1 Promotes Proteasomal Degradation of NRF2 and Sensitizes Lung Cancer Cells to Ferroptosis. Mol. Cancer Res. 2022, 20, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [Green Version]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes. Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Meng, C.; Zhan, J.; Chen, D.; Shao, G.; Zhang, H.; Gu, W.; Luo, J. The deubiquitinase USP11 regulates cell proliferation and ferroptotic cell death via stabilization of NRF2 USP11 deubiquitinates and stabilizes NRF2. Oncogene 2021, 40, 1706–1720. [Google Scholar] [CrossRef]
- Snyder, N.A.; Silva, G.M. Deubiquitinating enzymes (DUBs): Regulation, homeostasis, and oxidative stress response. J. Biol. Chem. 2021, 297, 101077. [Google Scholar] [CrossRef]
- Xu, J.; Guo, H.; Xing, Z.; Zhang, W.; He, J.; Cheng, J.; Cai, R. Mild Oxidative Stress Reduces NRF2 SUMOylation to Promote Kras/Lkb1/Keap1 Mutant Lung Adenocarcinoma Cell Migration and Invasion. Oxid. Med. Cell. Longev. 2020, 2020, 6240125. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Szankasi, P.; Roberts, C.J.; Murray, A.W.; Friend, S.H. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997, 278, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Li, K.; Lv, J.; Feng, J.; Chen, J.; Wu, H.; Cheng, F.; Jiang, W.; Wang, J.; Pei, H.; et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Investig. 2020, 130, 1752–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olayanju, A.; Copple, I.M.; Bryan, H.K.; Edge, G.T.; Sison, R.L.; Wong, M.W.; Lai, Z.Q.; Lin, Z.X.; Dunn, K.; Sanderson, C.M.; et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic. Biol. Med. 2015, 78, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Peddibhotla, S.; Fontaine, P.; Leung, C.K.; Maloney, P.; Hershberger, P.M.; Wang, Y.; Bousquet, M.S.; Luesch, H.; Mangravita-Novo, A.; Pinkerton, A.B.; et al. Discovery of ML358, a Selective Small Molecule Inhibitor of the SKN-1 Pathway Involved in Drug Detoxification and Resistance in Nematodes. ACS Chem. Biol. 2015, 10, 1871–1879. [Google Scholar] [CrossRef]
- Duong, H.Q.; Yi, Y.W.; Kang, H.J.; Hong, Y.B.; Tang, W.; Wang, A.; Seong, Y.S.; Bae, I. Inhibition of NRF2 by PIK-75 augments sensitivity of pancreatic cancer cells to gemcitabine. Int. J. Oncol. 2014, 44, 959–969. [Google Scholar] [CrossRef] [Green Version]
- Seiler, A.; Schneider, M.; Forster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Radmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Love, K.R.; Catic, A.; Schlieker, C.; Ploegh, H.L. Mechanisms, biology and inhibitors of deubiquitinating enzymes. Nat. Chem. Biol. 2007, 3, 697–705. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayir, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef] [Green Version]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Li, W.; Liu, X.; Cheng, X.; Zhang, W.; Gong, C.; Gao, C.; Peng, H.; Yang, B.; Tang, S.; Tao, H. Effect of Malt-PEG-Abz@RSL3 micelles on HepG2 cells based on NADPH depletion and GPX4 inhibition in ferroptosis. J. Drug. Target 2022, 30, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Jiang, L.; Tavana, O.; Gu, W. The Deubiquitylase OTUB1 Mediates Ferroptosis via Stabilization of SLC7A11. Cancer Res. 2019, 79, 1913–1924. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, X.; Yang, Q.; Chen, J.; Huang, Q.; Yao, L.; Yan, D.; Wu, J.; Zhang, P.; Tang, D.; et al. Broad Spectrum Deubiquitinase Inhibition Induces Both Apoptosis and Ferroptosis in Cancer Cells. Front. Oncol. 2020, 10, 949. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Kroemer, G.; Tang, D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021, 28, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Luo, M.; Zhang, K.; Zhang, J.; Gao, T.; Connell, D.O.; Yao, F.; Mu, C.; Cai, B.; Shang, Y.; et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 2020, 11, 433. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies, Chemicals and Reagents | SOURCE | Identifier |
|---|---|---|
| Anti-NRF2 antibody | Abcam | ab62352 |
| Anti-GCLC antibody | Abcam | ab190685 |
| Anti-GCLM antibody | Abcam | ab126704 |
| Anti-GPX4 antibody | Abcam | ab125066 |
| Anti-SLC7A11 antibody | Cell Signaling Technology (Danvers, MA, USA) | 12691S |
| Anti-USP11 antibody | Abcam (Cambridge, UK) | ab109232 |
| Anti-Ubiquitin antibody | Cell Signaling Technology | 3936S |
| Anti-β-actin antibody | Abcam | ab8226 |
| RSL3 | SELLECK (Houston, TX, USA) | S8155 |
| Erastin | SELLECK | S7242 |
| DMSO | Sigma-Aldrich | D4540 |
| TRIzol | TIANGEN (Kusatsu City, Japan) | DP424 |
| Fast King gDNA Dispelling RT SuperMix kit | TIANGEN | KR118 |
| TB Green Premix | TAKARA | RR420Q |
| ECL substrate | Tanon (Shanghai, China) | 180–506 |
| CCK-8 | Dojindo (Rockville, MD, USA) | CK04 |
| GSH and GSSG Assay Kit | Beyotime (Shanghai, China) | S0053 |
| BODIPY™ 665/676 | Invitrogen | B3932 |
| FerroOrange | Dojindo | F374 |
| Corn oil | Abcam | S6701 |
| MDA Assay Kit | Beyotime | S0131S |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, W.; Li, X.; Xu, J.; Wang, Y.; Xing, Z.; Hu, S.; Fan, Q.; Lu, S.; Cheng, J.; Gu, J.; et al. The RSL3 Induction of KLK Lung Adenocarcinoma Cell Ferroptosis by Inhibition of USP11 Activity and the NRF2-GSH Axis. Cancers 2022, 14, 5233. https://doi.org/10.3390/cancers14215233
Zhang W, Li X, Xu J, Wang Y, Xing Z, Hu S, Fan Q, Lu S, Cheng J, Gu J, et al. The RSL3 Induction of KLK Lung Adenocarcinoma Cell Ferroptosis by Inhibition of USP11 Activity and the NRF2-GSH Axis. Cancers. 2022; 14(21):5233. https://doi.org/10.3390/cancers14215233
Chicago/Turabian StyleZhang, Wenlong, Xiaohe Li, Jiaqian Xu, Ying Wang, Zhengcao Xing, Shuming Hu, Qiuju Fan, Shaoyong Lu, Jinke Cheng, Jianmin Gu, and et al. 2022. "The RSL3 Induction of KLK Lung Adenocarcinoma Cell Ferroptosis by Inhibition of USP11 Activity and the NRF2-GSH Axis" Cancers 14, no. 21: 5233. https://doi.org/10.3390/cancers14215233
APA StyleZhang, W., Li, X., Xu, J., Wang, Y., Xing, Z., Hu, S., Fan, Q., Lu, S., Cheng, J., Gu, J., & Cai, R. (2022). The RSL3 Induction of KLK Lung Adenocarcinoma Cell Ferroptosis by Inhibition of USP11 Activity and the NRF2-GSH Axis. Cancers, 14(21), 5233. https://doi.org/10.3390/cancers14215233