
Novel BH4-BCL-2 Domain Antagonists Induce BCL-2-Mediated Apoptosis in Triple-Negative Breast Cancer
 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Compounds
2.2. Covalent Docking
2.3. Cell Culture
2.4. Surface Plasmon Resonance
2.5. ESI Mass Spectrometry
2.6. Immunoblotting
2.7. BCL2 Gene Expression Analysis by qPCR
2.8. MTT Cell Viability Assay
2.9. BrdU Cell Proliferation Assay
2.10. Annexin V/Propidium Iodide Assay
2.11. Cellular BH4 Profiling Assay
2.12. Cytochrome C Release Assays
2.13. Clonogenic Colony Formation Assay
2.14. Soft Agar Colony Formation Assay
2.15. Wound Healing Assay
2.16. Transwell Cell Migration Assay
2.17. Transwell Cell Invasion Assay
2.18. Angiogenic Tube Formation Assay
2.19. Animals and Treatment Groups
2.20. Immunohistochemistry of Treatment Groups—Animal Tissues
2.21. Plasma Stability and Hemolytic Assays
2.22. Statistical Analysis
3. Results
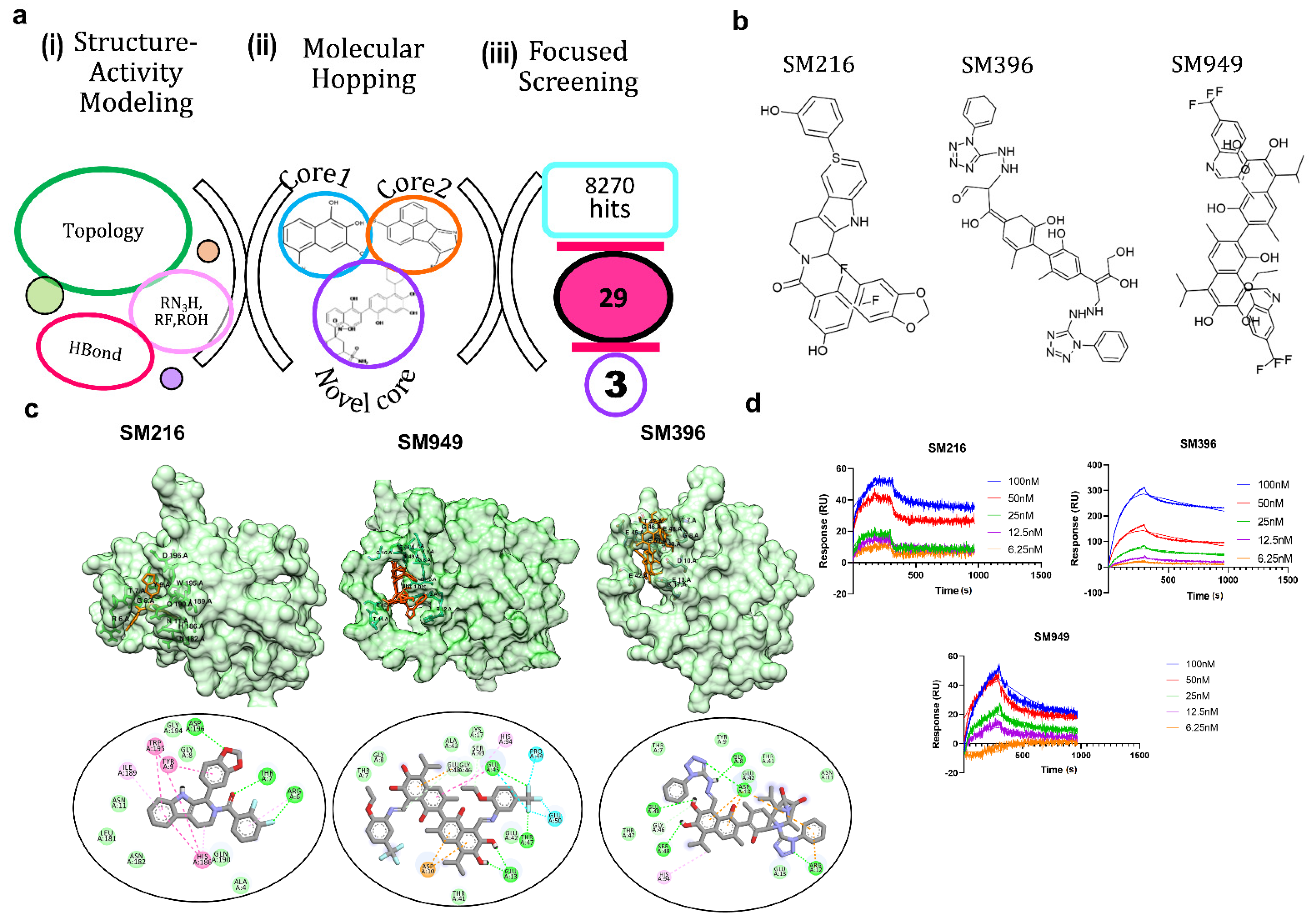
3.1. SM216, SM396, and SM949 Are the Top Three Lead Molecules from Molecular Remodeling and Binding Analysis
3.2. SM396 Binds Covalently to the BH4 Domain of BCL-2
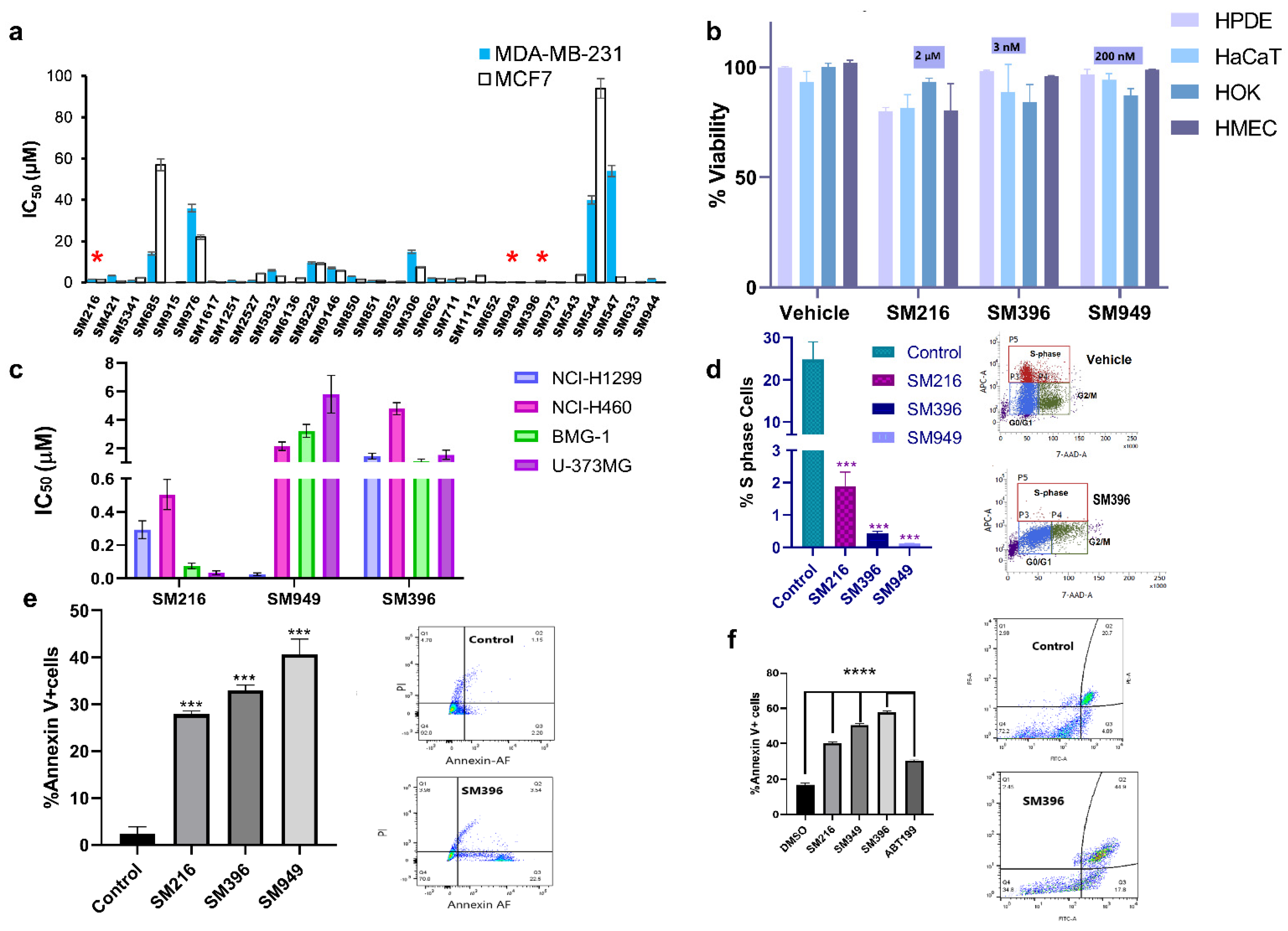
3.3. BCL-2 Profiling Shows Heterogenous Expressions in Different Cancer Cell Lines
3.4. SM216, SM396, and SM949 Explicitly Kill Cancer Cells, and Not the Normal Cells
3.5. SM216, SM396, and SM949 Induce Membrane Depolarization and Cytochrome C Release
3.6. SM216, SM396, and SM949 Attenuate the Transforming Capabilities of Breast Cancer Cells
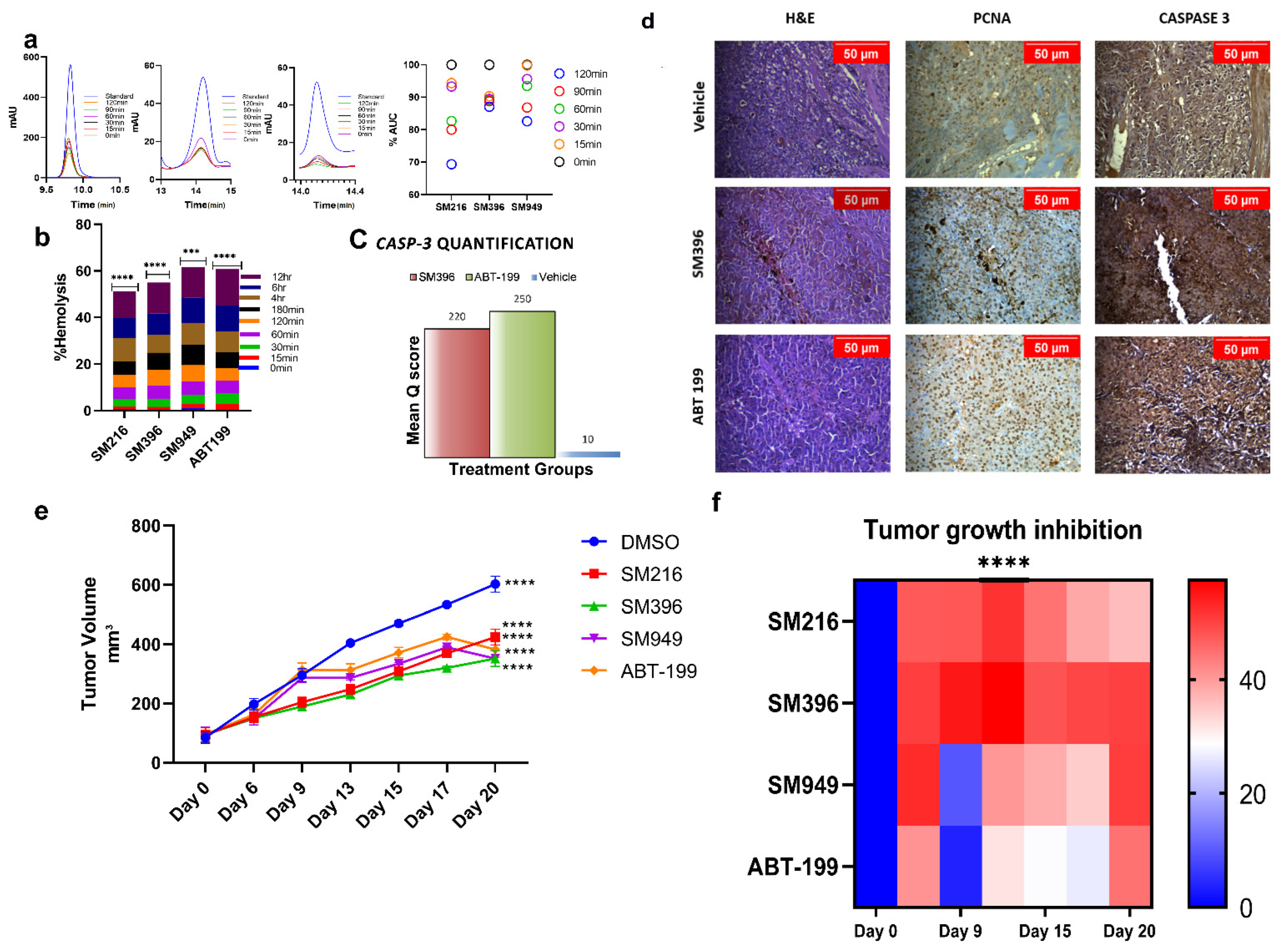
3.7. SM216, SM396, and SM949 Are Highly Tolerated by Erythrocytes at Efficacious Doses
3.8. SM216, SM396, and SM949 Showed Anti-Tumor Activity on Human Breast Cancer Xenografts on Athymic Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Gross, A. BCL-2 family proteins as regulators of mitochondria metabolism. Biochim. Biophys. Acta 2016, 1857, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nougarède, A.; Rimokh, R.; Gillet, G. BH4-mimetics and -antagonists: An emerging class of Bcl-2 protein modulators for cancer therapy. Oncotarget 2018, 9, 35291–35292. [Google Scholar] [CrossRef] [PubMed]
- Lavik, A.R.; Zhong, F.; Chang, M.J.; Greenberg, E.; Choudhary, Y.; Smith, M.R.; McColl, K.S.; Pink, J.; Reu, F.J.; Matsuyama, S.; et al. A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget 2015, 6, 27388–27402. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wild, C.; Ding, Y.; Ye, N.; Chen, H.; Wold, E.A.; Zhou, J. BH4 domain of Bcl-2 as a novel target for cancer therapy. Drug Discov. Today 2016, 21, 989–996. [Google Scholar] [CrossRef] [Green Version]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Rody, A.; Karn, T.; Liedtke, C.; Pusztai, L.; Ruckhaeberle, E.; Hanker, L.; Gaetje, R.; Solbach, C.; Ahr, A.; Metzler, D.; et al. A clinically relevant gene signature in triple negative and basal like breast cancer. Breast Cancer Res. 2011, 13, R97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; McBride, D.J.; Lin, M.L.; Varela, I.; Pleasance, E.D.; Simpson, J.T. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009, 462, 1005–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Eiermann, W.; Bergh, J.; Cardoso, F.; Conte, P.; Crown, J.; Curtin, N.J.; Gligorov, J.; Gusterson, B.; Joensuu, H.; Linderholm, B.K.; et al. Triple negative breast cancer: Proposals for a pragmatic definition and implications for patient management and trial design. Breast 2012, 21, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. 2013, 19, 5533–5540. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Pietenpol, J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef]
- Abdel-Fatah, T.M.; Perry, C.; Dickinson, P.; Ball, G.; Moseley, P.; Madhusudan, S.; Ellis, I.O.; Chan, S.Y. Bcl2 is an independent prognostic marker of triple negative breast cancer (TNBC) and predicts response to anthracycline combination (ATC) chemotherapy (CT) in adjuvant and neoadjuvant settings. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 2801–2807. [Google Scholar] [CrossRef]
- Dawson, S.J.; Makretsov, N.; Blows, F.M.; Driver, K.E.; Provenzano, E.; Le Quesne, J.; Baglietto, L.; Severi, G.; Giles, G.G.; McLean, C.A. BCL2 in breast cancer: A favourable prognostic marker across molecular subtypes and independent of adjuvant therapy received. Br. J. Cancer 2010, 103, 668–675. [Google Scholar] [CrossRef]
- Gandhi, L.; Camidge, D.R.; Ribeiro de Oliveira, M.; Bonomi, P.; Gandara, D.; Khaira, D.; Hann, C.L.; McKeegan, E.M.; Litvinovich, E.; Hemken, P.M.; et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J. Clin. Oncol. 2011, 29, 909–916. [Google Scholar] [CrossRef]
- Parrondo, R.; de Las Pozas, A.; Reiner, T.; Perez-stable, C. ABT-737, a small molecule Bcl-2/Bcl-xL antagonist, increases antimitotic-mediated apoptosis in human prostate cancer cells. PeerJ 2013, 1, e144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199 a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, F.; Merino, D.; Lee, L.; Breslin, K.; Pal, B.; Ritchie, M.E.; Smyth, G.K.; Christie, M.; Phillipson, L.J.; Burns, C.J.; et al. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell 2013, 24, 120–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fresquet, V.; Rieger, M.; Carolis, C.; García-Barchino, M.J.; Martinez-Climent, J.A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 2014, 123, 4111–4119. [Google Scholar] [CrossRef] [Green Version]
- Tahir, S.K.; Smith, M.L.; Hessler, P.; Rapp, L.R.; Idler, K.B.; Park, C.H.; Leverson, J.D.; Lam, L.T. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer 2017, 17, 399. [Google Scholar] [CrossRef] [Green Version]
- Blombery, P.; Anderson, M.A.; Gong, J.-N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Kanakaveti, V.; Sakthivel, R.; Rayala, S.K.; Gromiha, M.M. Importance of functional groups in predicting the activity of small molecule inhibitors for Bcl-2 and Bcl-xL. Chem. Biol. Drug Des. 2017, 90, 308–316. [Google Scholar] [CrossRef]
- Kanakaveti, V.; Sakthivel, R.; Rayala, S.K.; Gromiha, M.M. Forging new scaffolds from old: Combining Scaffold hopping and Hierarchical virtual screening for identifying novel Bcl-2 inhibitors. Curr. Top Med. Chem. 2019, 19, 1162–1172. [Google Scholar] [CrossRef]
- Birkinshaw, R.W.; Gong, J.N.; Luo, C.S.; Lio, D.; White, C.A.; Anderson, M.A.; Blombery, P.; Lessene, G.; Majewski, I.J.; Thijssen, R.; et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat. Commun. 2019, 10, 2385. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, G.; Forli, S.; Goodsell, D.S.; Olson, A.J. Covalent docking using autodock: Two-point attractor and flexible side chain methods. Protein Sci. 2016, 25, 295–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Xu, Q.W.; Wang, R.; Uszkoreit, J.; Griss, J.; Sanchez, A.; Reisinger, F.; Csordas, A.; Ternent, T.; del Toro, N.; et al. PRIDE Inspector Toolsuite: Moving towards a universal visualization tool for proteomics data standard formats and quality assessment of ProteomeXchange datasets. Mol. Cell Proteom. 2016, 15, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Fraser, C.; Ryan, J.; Sarosiek, K. BH3 Profiling: A Functional Assay to Measure Apoptotic Priming and Dependencies. Methods Mol. Biol. 2019, 1877, 61–76. [Google Scholar]
- Ryan, J.; Letai, A. BH3 profiling in whole cells by fluorimeter or FACS. Methods 2013, 61, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Karl, E.; Warner, K.; Zeitlin, B.; Kaneko, T.; Wurtzel, L.; Jin, T.; Chang, J.; Wang, S.; Wang, C.Y.; Strieter, R.M.; et al. Bcl-2 acts in a proangiogenic signaling pathway through nuclear factor-kappaB and CXC chemokines. Cancer Res. 2005, 65, 5063–5069. [Google Scholar] [CrossRef] [Green Version]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.M.; Cook, R.S. Bcl-2 family proteins in breast development and cancer: Could Mcl-1 targeting overcome therapeutic resistance? Oncotarget 2015, 6, 3519–3530. [Google Scholar] [CrossRef] [Green Version]
- Cang, S.; Iragavarapu, C.; Savooji, J.; Song, Y.; Liu, D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 2015, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogler, M. Targeting BCL2-Proteins for the Treatment of Solid Tumours. Adv. Med. 2014, 2014, 943648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, V.; Kell, D.L.; Van de Rijn, M.; Warnke, R.A. Bcl-2 immunoreactivity in breast carcinoma correlates with hormone receptor positivity. Am. J. Pathol. 1994, 145, 535–540. [Google Scholar] [PubMed]
- Oakes, S.R.; Vaillant, F.; Lim, E.; Lee, L.; Breslin, K.; Feleppa, F.; Deb, S.; Ritchie, M.E.; Takano, E.; Ward, T.; et al. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc. Natl. Acad. Sci. USA 2012, 109, 2766–2771. [Google Scholar] [CrossRef] [Green Version]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Inao, T.; Iida, Y.; Moritani, T.; Okimoto, T.; Tanino, R.; Kotani, H.; Harada, M. Bcl-2 inhibition sensitizes triple-negative human breast cancer cells to doxorubicin. Oncotarget 2018, 9, 25545–25556. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lead Compounds | Predicted IC50 (µM) | Experimental IC50 (µM) | Binding Energy (kcal mol−1) |
|---|---|---|---|
| SM216 | 1.2 | 1.602 | −9 |
| SM421 | 2.9 | 3.38 | −5 |
| SM5341 | 1.4 | 1.183 | −4.4 |
| SM685 | 13 | 14 | −5.2 |
| SM915 | 0.00039 | 0.0004 | −4.7 |
| SM976 | 32 | 36 | −5.8 |
| SM1617 | 0.65 | 0.74 | −3.7 |
| SM1251 | 1.6 | 1.15 | −4.9 |
| SM2527 | 2.1 | 1.16 | −4.6 |
| SM5832 | 6.1 | 5.923 | −3.4 |
| SM6136 | 13 | 0.178 | −3.9 |
| SM8228 | 41 | 9.49 | −4.9 |
| SM9146 | 10 | 7.09 | −3.2 |
| SM850 | 4 | 3.002 | −4.5 |
| SM851 | 1.7 | 0.88 | −5.6 |
| SM852 | 0.47 | 0.423 | −5.9 |
| SM306 | 6 | 14.9 | −6.5 |
| SM662 | 2.1 | 2.054 | −6.8 |
| SM711 | 1.8 | 1.58 | −4.3 |
| SM1112 | 0.567 | 0.654 | −4 |
| SM652 | 0.324 | 0.373 | −5.9 |
| SM949 | 0.18 | 0.172 | −8.6 |
| SM396 | 0.0016 | 0.00273 | −9.3 |
| SM973 | 0.23 | 0.201 | −8.9 |
| SM543 | 0.0016 | 0.00182 | −7.6 |
| SM544 | 31 | 40 | −8.3 |
| SM547 | 56 | 54 | −5.7 |
| SM633 | 0.013 | 0.006 | −4.6 |
| SM944 | 1.8 | 1.795 | −10.8 |
| Compound | KD (nM) |
|---|---|
| SM216 | 3.92 |
| SM396 | 6.37 |
| SM949 | 78 |
| ABT-199 | 116 |
| Groups | Tumor Volume (mm3) | Tumor Volume (mm3) | Tumor Volume (mm3) | Tumor Volume (mm3) | Tumor Volume (mm3) | Tumor Volume (mm3) | Tumor Volume (mm3) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 0 | Day 6 | Day 9 | Day 13 | Day 15 | Day 17 | Day 20 | ||||||||
| Mean | SEM | Mean | SEM | SEM | SEM | Mean | SEM | Mean | SEM | Mean | SEM | Mean | SEM | |
| DMSO | 84.84 | 19.16 | 197.77 | 19.33 | 295.63 | 21.97 | 404.38 | 3.78 | 470.33 | 7.94 | 532.7 | 6.09 | 602.01 | 5.85 |
| SM216 | 94.01 | 23.93 | 153.57 | 17.17 | 204.5 | 16.69 | 248.27 | 16.37 | 308.59 | 12.10 | 369.88 | 13.0 | 424.26 | 11.41 |
| SM396 | 94.04 | 24.32 | 150.29 | 12.83 | 189.48 | 11.53 | 230.27 | 11.32 | 294.6 | 3.09 | 320.51 | 5.32 | 351.81 | 4.68 |
| SM949 | 95.6 | 25.45 | 149.12 | 21.54 | 286.11 | 15.57 | 286.11 | 6.63 | 334.58 | 3.68 | 389.36 | 3.57 | 351.73 | 88.06 |
| ABT-199 | 95.35 | 23.42 | 162.15 | 20.46 | 313.94 | 23.32 | 313.03 | 20.43 | 371.94 | 17.45 | 424.83 | 9.64 | 382.6 | 95.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanakaveti, V.; Ramasamy, S.; Kanumuri, R.; Balasubramanian, V.; Saravanan, R.; Ezhil, I.; Pitani, R.; Venkatraman, G.; Rayala, S.K.; Gromiha, M.M. Novel BH4-BCL-2 Domain Antagonists Induce BCL-2-Mediated Apoptosis in Triple-Negative Breast Cancer. Cancers 2022, 14, 5241. https://doi.org/10.3390/cancers14215241
Kanakaveti V, Ramasamy S, Kanumuri R, Balasubramanian V, Saravanan R, Ezhil I, Pitani R, Venkatraman G, Rayala SK, Gromiha MM. Novel BH4-BCL-2 Domain Antagonists Induce BCL-2-Mediated Apoptosis in Triple-Negative Breast Cancer. Cancers. 2022; 14(21):5241. https://doi.org/10.3390/cancers14215241
Chicago/Turabian StyleKanakaveti, Vishnupriya, Sakthivel Ramasamy, Rahul Kanumuri, Vaishnavi Balasubramanian, Roshni Saravanan, Inemai Ezhil, Ravishankar Pitani, Ganesh Venkatraman, Suresh Kumar Rayala, and M. Michael Gromiha. 2022. "Novel BH4-BCL-2 Domain Antagonists Induce BCL-2-Mediated Apoptosis in Triple-Negative Breast Cancer" Cancers 14, no. 21: 5241. https://doi.org/10.3390/cancers14215241