
A Novel apaQTL-SNP for the Modification of Non-Small-Cell Lung Cancer Susceptibility across Histological Subtypes

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
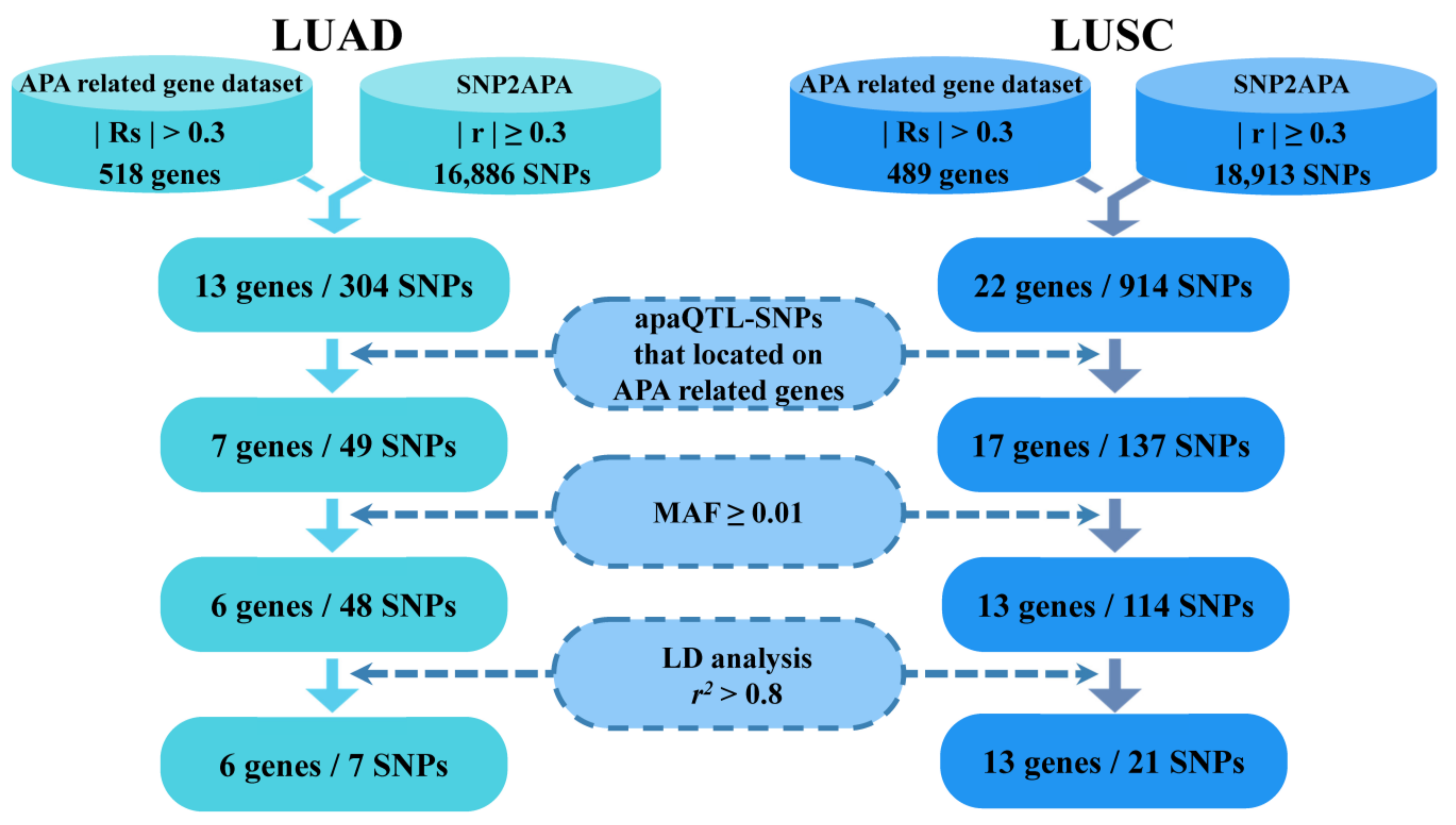
2.2. Identification of APA-Related Genes Associated with LUAD and LUSC
2.3. Selection of Respective apaQTL-SNPs in APA-Related LUAD and LUSC Genes
2.4. Genotyping of FLCCA GWAS in Phase I
2.5. Genotyping of Candidate apaQTL-SNPs in Phase II
2.6. 3′-Rapid Amplification of cDNA Ends (3′RACE) Experiments
2.7. Vectors Construction and qRT-PCR Assay
2.8. Statistical Analysis
3. Results
3.1. Characteristics of the Study Subjects
3.2. Identification of APA-Related Genes Associated with LUAD and LUSC
3.3. Screening of Candidate apaQTL-SNPs
3.4. Association between Candidate apaQTL-SNPs and NSCLC Risk in Phase I
3.5. Evaluation of the Association between apaQTL-SNP (rs10138506) and LUAD Risk in Phase II
3.6. Evaluation of the Association between apaQTL-SNPs (rs1130698 and rs1130719) and LUSC Risk in Phase II
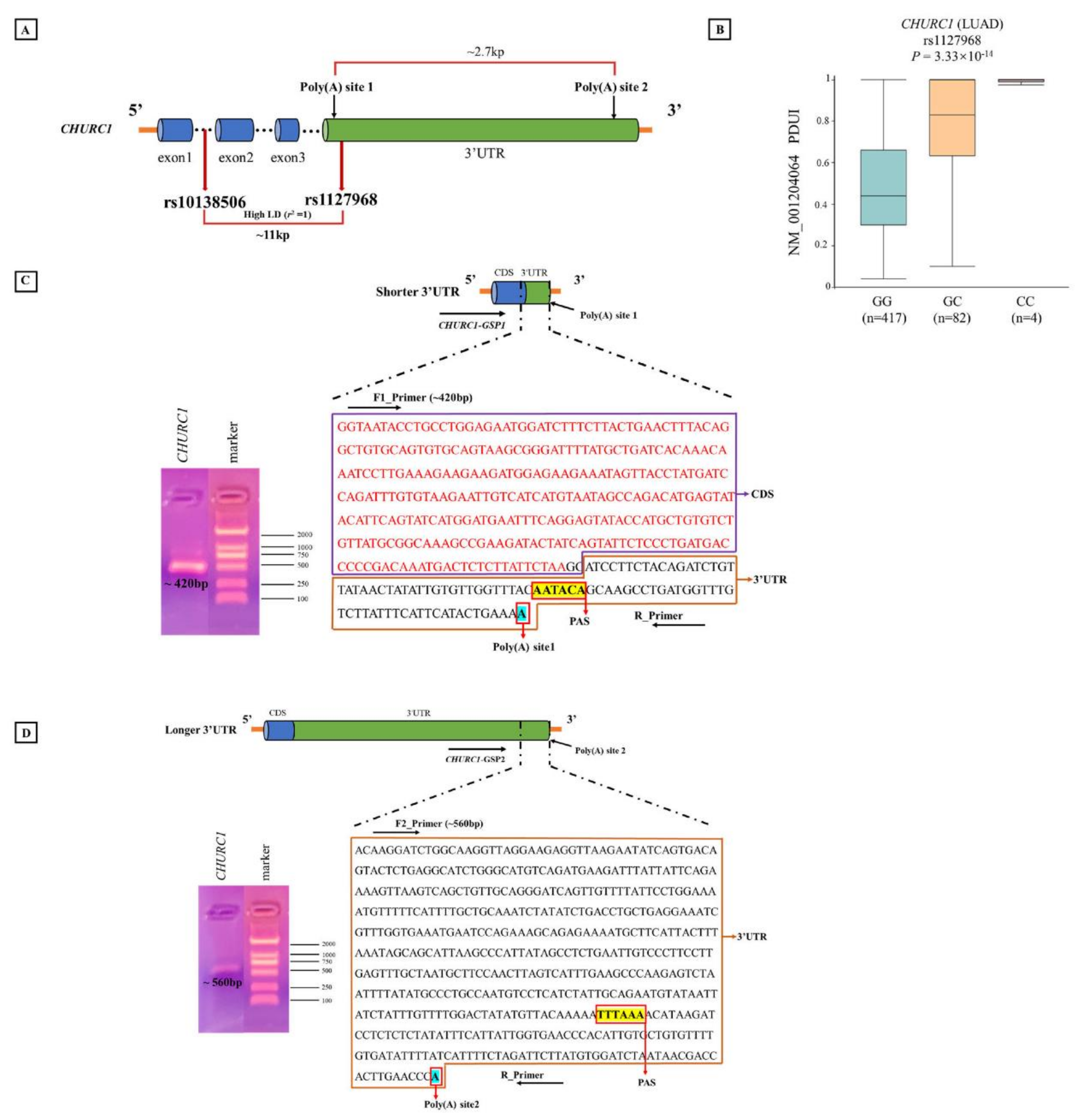
3.7. APA Analysis for rs10138506
3.8. CHURC1 Expression
3.9. Survival Analysis
3.10. Analysis of Poly(A) Sites of CHURC1 Based on the 3′RACE Experiment
3.11. The Expression of CHURC1 Isoforms under Different Genotypes of apaQTL-SNP rs10138506
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, W.; Zheng, R.; Zhang, S.; Ji, J.S.; Zou, X.; Xia, C.; Sun, K.; Yang, Z.; Li, H.; et al. Changing cancer survival in China during 2003–15: A pooled analysis of 17 population-based cancer registries. Lancet Glob. Health 2018, 6, e555–e567. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Ran, X.; An, L.; Zheng, R.; Zhang, S.; Ji, J.S.; Zhang, Y.; Chen, W.; Wei, W.; He, J.; et al. Disparities in stage at diagnosis for five common cancers in China: A multicentre, hospital-based, observational study. Lancet Public Health 2021, 6, e877–e887. [Google Scholar] [CrossRef]
- St Claire, S.; Gouda, H.; Schotte, K.; Fayokun, R.; Fu, D.; Varghese, C.; Prasad, V.M. Lung health, tobacco, and related products: Gaps, challenges, new threats, and suggested research. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 318, L1004–L1007. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Lv, J.; Zhu, M.; Wang, Y.; Qin, N.; Ma, H.; He, Y.-Q.; Zhang, R.; Tan, W.; Fan, J.; et al. Identification of risk loci and a polygenic risk score for lung cancer: A large-scale prospective cohort study in Chinese populations. Lancet Respir. Med. 2019, 7, 881–891. [Google Scholar] [CrossRef]
- Yang, H.D.; Nam, S.W. Pathogenic diversity of RNA variants and RNA variation-associated factors in cancer development. Exp. Mol. Med. 2020, 52, 582–593. [Google Scholar] [CrossRef]
- Shao, L.; Zuo, X.; Yang, Y.; Zhang, Y.; Yang, N.; Shen, B.; Wang, J.; Wang, X.; Li, R.; Jin, G.; et al. The inherited variations of a p53-responsive enhancer in 13q12.12 confer lung cancer risk by attenuating TNFRSF19 expression. Genome Biol. 2019, 20, 103. [Google Scholar] [CrossRef]
- Ishigaki, K.; Akiyama, M.; Kanai, M.; Takahashi, A.; Kawakami, E.; Sugishita, H.; Sakaue, S.; Matoba, N.; Low, S.-K.; Okada, Y.; et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 2020, 52, 669–679. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Mitra, M.; Lee, H.N.; Coller, H.A. Splicing Busts a Move: Isoform Switching Regulates Migration. Trends Cell Biol. 2020, 30, 74–85. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef]
- Xiang, Y.; Ye, Y.; Lou, Y.; Yang, Y.; Cai, C.; Zhang, Z.; Mills, T.; Chen, N.-Y.; Kim, Y.; Ozguc, F.M.; et al. Comprehensive Characterization of Alternative Polyadenylation in Human Cancer. J. Natl. Cancer Inst. 2018, 110, 379–389. [Google Scholar] [CrossRef]
- Mittleman, B.E.; Pott, S.; Warland, S.; Zeng, T.; Mu, Z.; Kaur, M.; Gilad, Y.; Li, Y. Alternative polyadenylation mediates genetic regulation of gene expression. eLife 2020, 9, e57492. [Google Scholar] [CrossRef]
- Mariella, E.; Marotta, F.; Grassi, E.; Gilotto, S.; Provero, P. The Length of the Expressed 3′ UTR Is an Intermediate Molecular Phenotype Linking Genetic Variants to Complex Diseases. Front. Genet. 2019, 10, 714. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Donehower, L.A.; Cooper, T.A.; Neilson, J.R.; Wheeler, D.A.; Wagner, E.J.; Li, W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat. Commun. 2014, 5, 5274. [Google Scholar] [CrossRef] [Green Version]
- Yuan, F.; Hankey, W.; Wagner, E.J.; Li, W.; Wang, Q. Alternative polyadenylation of mRNA and its role in cancer. Genes Dis. 2021, 8, 61–72. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, X.; Lei, W.; Liang, J.; Xu, Y.; Liu, H.; Ma, S. Genome-wide profiling reveals alternative polyadenylation of mRNA in human non-small cell lung cancer. J. Transl. Med. 2019, 17, 257. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Qiu, Q.; Zhou, Q.; Ding, J.; Lu, Y.; Liu, P. Alternative polyadenylation: Methods, mechanism, function, and role in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 51. [Google Scholar] [CrossRef] [PubMed]
- Shulman, E.D.; Elkon, R. Systematic identification of functional SNPs interrupting 3′UTR polyadenylation signals. PLoS Genet. 2020, 16, e1008977. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Huang, K.-L.; Gao, Y.; Cui, Y.; Wang, G.; Elrod, N.D.; Li, Y.; Chen, Y.E.; Ji, P.; Peng, F.; et al. An atlas of alternative polyadenylation quantitative trait loci contributing to complex trait and disease heritability. Nat. Genet. 2021, 53, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Q.; Miao, Y.-R.; Yang, J.; Yang, W.; Yu, F.; Wang, D.; Guo, A.-Y.; Gong, J. SNP2APA: A database for evaluating effects of genetic variants on alternative polyadenylation in human cancers. Nucleic Acids Res. 2020, 48, D226–D232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Q.; Hsiung, C.A.; Matsuo, K.; Hong, Y.-C.; Seow, A.; Wang, Z.; Hosgood, H.D., 3rd; Chen, K.; Wang, J.-C.; Chatterjee, N.; et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat. Genet. 2012, 44, 1330–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Mao, L.; Lu, X.; Yuan, W.; Chen, Y.; Jiang, L.; Ding, L.; Sang, L.; Xu, Z.; Tian, T.; et al. Functional Variant in 3′UTR of FAM13A Is Potentially Associated with Susceptibility and Survival of Lung Squamous Carcinoma. DNA Cell Biol. 2019, 38, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, P.H.; Trubetskoy, V.; Nurhussein-Patterson, A.; Hall, J.P.; Nath, A.; Huo, D.; Fleming, G.F.; Ingle, J.N.; Abramson, V.G.; Morrow, P.K.; et al. Clinical evaluation of germline polymorphisms associated with capecitabine toxicity in breast cancer: TBCRC-015. Breast Cancer Res. Treat. 2020, 181, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Chen, Z.; Bao, J.; Long, Q.; Shu, X.-O.; Zheng, W.; Guo, X. Genetic variations of DNA bindings of FOXA1 and co-factors in breast cancer susceptibility. Nat. Commun. 2021, 12, 5318. [Google Scholar] [CrossRef]
- Strub, M.D.; Gao, L.; Tan, K.; McCray, P.B., Jr. Analysis of multiple gene co-expression networks to discover interactions favoring CFTR biogenesis and ΔF508-CFTR rescue. BMC Med. Genom. 2021, 14, 258. [Google Scholar] [CrossRef]
- Das, T.K.; Sangodkar, J.; Negre, N.; Narla, G.; Cagan, R.L. Sin3a acts through a multi-gene module to regulate invasion in Drosophila and human tumors. Oncogene 2013, 32, 3184–3197. [Google Scholar] [CrossRef]
- Xu, J.-Y.; Zhang, C.; Wang, X.; Zhai, L.; Ma, Y.; Mao, Y.; Qian, K.; Sun, C.; Liu, Z.; Jiang, S.; et al. Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell 2020, 182, 245–261.e17. [Google Scholar] [CrossRef]
- Buccitelli, C.; Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Jemal, A.; Ward, E.M.; Johnson, C.J.; Cronin, K.A.; Ma, J.; Ryerson, A.B.; Mariotto, A.; Lake, A.J.; Wilson, R.; Sherman, R.L.; et al. Annual Report to the Nation on the Status of Cancer, 1975–2014, Featuring Survival. J. Natl. Cancer Inst. 2017, 109, djx030. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-C.; Tsai, S.-W.; Shie, R.-H.; Zeng, C.; Yang, H.-Y. Indoor Air Pollution Increases the Risk of Lung Cancer. Int. J. Environ. Res. Public Health 2022, 19, 1164. [Google Scholar] [CrossRef]
- Turner, M.C.; Andersen, Z.J.; Baccarelli, A.; Diver, W.R.; Gapstur, S.M.; Pope, C.A., 3rd; Prada, D.; Samet, J.; Thurston, G.; Cohen, A. Outdoor air pollution and cancer: An overview of the current evidence and public health recommendations. CA Cancer J. Clin. 2020, 70, 460–479. [Google Scholar] [CrossRef]
- Qin, N.; Li, Y.; Wang, C.; Zhu, M.; Dai, J.; Hong, T.; Albanes, D.; Lam, S.; Tardon, A.; Chen, C.; et al. Comprehensive functional annotation of susceptibility variants identifies genetic heterogeneity between lung adenocarcinoma and squamous cell carcinoma. Front. Med. 2021, 15, 275–291. [Google Scholar] [CrossRef]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Möller, S.; Unger, R.H.; et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Begik, O.; Oyken, M.; Alican, T.C.; Can, T.; Erson-Bensan, A.E. Alternative Polyadenylation Patterns for Novel Gene Discovery and Classification in Cancer. Neoplasia 2017, 19, 574–582. [Google Scholar] [CrossRef]
- Kim, K.; Chadalapaka, G.; Lee, S.-O.; Yamada, D.; Sastre-Garau, X.; Defossez, P.-A.; Park, Y.-Y.; Lee, J.-S.; Safe, S. Identification of oncogenic microRNA-17-92/ZBTB4/specificity protein axis in breast cancer. Oncogene 2012, 31, 1034–1044. [Google Scholar] [CrossRef] [Green Version]
- Mayr, C. What Are 3′ UTRs Doing? Cold Spring Harb. Perspect. Biol. 2019, 11, a034728. [Google Scholar] [CrossRef]
- Mayr, C. Evolution and Biological Roles of Alternative 3′UTRs. Trends Cell Biol. 2016, 26, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.-H.; Zhang, Z.-T.; Wei, H.-X.; Ning, L.; Ai, J.-S.; Li, W.-H.; Zhang, H.; Wang, S.-Q. LncRNA ST7-AS1, by regulating miR-181b-5p/KPNA4 axis, promotes the malignancy of lung adenocarcinoma. Cancer Cell Int. 2020, 20, 568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jiang, H.; Zhang, H.; Liu, J.; Hu, X.; Chen, L. miR-4262, low level of which predicts poor prognosis, targets proto-oncogene CD163 to suppress cell proliferation and invasion in gastric cancer. OncoTargets Ther. 2019, 12, 599–607. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Screening (FLCCA GWAS) | Validation (Taqman) | ||||
|---|---|---|---|---|---|---|
| Case | Control | p | Case | Control | p | |
| (N = 4107) | (N = 3710) | (N = 779) | (N = 667) | |||
| Age, N (100%) | <0.001 | 0.358 | ||||
| ≤60 | 2028 (49.38) | 2013 (54.26) | 294 (37.74) | 268 (40.18) | ||
| >60 | 2079 (50.62) | 1697 (45.74) | 485 (62.26) | 399 (59.82) | ||
| Gender, N (100%) | 0.781 | |||||
| Male | 0 | 0 | 511 (65.60) | 443 (66.42) | ||
| Female | 4107 (100) | 3710 (100) | 268 (34.40) | 224 (33.58) | ||
| Smoking status, N (100%) | <0.001 | |||||
| Ever | 0 | 0 | 421 (54.04) | 297 (44.53) | ||
| Never | 4107 (100) | 3710 (100) | 358 (45.96) | 370 (55.47) | ||
| Histology type, N (100%) | ||||||
| Squamous cell carcinoma | 654 (15.92) | 203 (26.06) | ||||
| Adenocarcinoma | 3453 (84.08) | 576 (73.94) | ||||
| No. | Histology Type | SNP | Location | Gene | apaQTL-p Value | MAF (CHB) | MAF (CHS) | MAF (JPT) |
|---|---|---|---|---|---|---|---|---|
| 1 | LUAD | rs1130663 | chr11:837582 | CD151 | 8.11 × 10−92 | 0.1359 | 0.1048 | 0.0817 |
| 2 | LUAD | rs4899152 | chr14:65387116 | CHURC1 | 1.75 × 10−95 | 0.1456 | 0.1333 | 0.1298 |
| 3 | LUAD | rs10138506 | chr14:65388243 | CHURC1 | 3.33 × 10−14 | 0.0534 | 0.0333 | 0.0529 |
| 4 | LUAD | rs3785 | chr15:23005202 | NIPA2 | 6.09 × 10−15 | 0.3058 | 0.4190 | 0.3654 |
| 5 | LUAD | rs8069673 | chr17:30661250 | C17orf75 | 2.02 × 10−14 | 0.0340 | 0.0667 | 0.1587 |
| 6 | LUAD | rs4802607 | chr19:49959364 | ALDH16A1 | 1.14 × 10−13 | 0.3883 | 0.4381 | 0.3029 |
| 7 | LUAD | rs6151429 | chr22:51063477 | ARSA | 1.90 × 10−41 | 0.0291 | 0.0238 | 0.0144 |
| 8 | LUSC | rs17116169 | chr5:153837482 | SAP30L | 6.23 × 10−12 | 0.0534 | 0.0524 | 0.0288 |
| 9 | LUSC | rs2268314 | chr7:44695725 | OGDH | 1.34 × 10−13 | 0.4320 | 0.4571 | 0.4571 |
| 10 | LUSC | rs10999323 | chr10:72174390 | EIF4EBP2 | 4.04 × 10−11 | 0.3398 | 0.2714 | 0.4471 |
| 11 | LUSC | rs1130698 | chr11:838542 | CD151 | 6.51 × 10−80 | 0.1019 | 0.0762 | 0..0529 |
| 12 | LUSC | rs1130719 | chr11:838760 | CD151 | 2.02 × 10−80 | 0.1408 | 0.1048 | 0.0817 |
| 13 | LUSC | rs7302556 | chr12:66532242 | TMBIM4 | 4.97 × 10−31 | 0.0194 | 0.0333 | 0.0192 |
| 14 | LUSC | rs1615416 | chr12:66549321 | TMBIM4 | 2.23 × 10−19 | 0.1117 | 0.1095 | 0.0673 |
| 15 | LUSC | rs11176067 | chr12:66557162 | TMBIM4 | 3.38 × 10−107 | 0.3641 | 0.3667 | 0.3462 |
| 16 | LUSC | rs1185888 | chr12:66560879 | TMBIM4 | 9.61 × 10−103 | 0.1650 | 0.2048 | 0.2019 |
| 17 | LUSC | rs169562 | chr13:32998362 | N4BP2L2 | 3.54 × 10−16 | 0.2718 | 0.3524 | 0.3558 |
| 18 | LUSC | rs798272 | chr13:33061719 | N4BP2L2 | 5.48 × 10−13 | 0.4466 | 0.4429 | 0.4471 |
| 19 | LUSC | rs45604 | chr13:33099347 | N4BP2L2 | 3.89 × 10−13 | 0.3301 | 0.3905 | 0.4087 |
| 20 | LUSC | rs10138534 | chr14:65387989 | CHURC1 | 4.33 × 10−134 | 0.1456 | 0.1333 | 0.1298 |
| 21 | LUSC | rs72726301 | chr14:65395668 | CHURC1 | 1.52 × 10−36 | 0.0922 | 0.1000 | 0.0769 |
| 22 | LUSC | rs1064108 | chr14:65400265 | CHURC1 | 3.48 × 10−61 | 0.2670 | 0.2667 | 0.3654 |
| 23 | LUSC | rs7224742 | chr17:30657058 | C17orf75 | 1.00 × 10−15 | 0.0340 | 0.0667 | 0.1587 |
| 24 | LUSC | rs73572386 | chr20:3849736 | MAVS | 1.25 × 10−12 | 0.2864 | 0.3048 | 0.2837 |
| 25 | LUSC | rs8141941 | chr22:19166263 | SLC25A1 | 4.32 × 10−15 | 0.2573 | 0.2762 | 0.1779 |
| 26 | LUSC | rs2481 | chr22:36677400 | MYH9 | 2.63 × 10−15 | 0.3786 | 0.3571 | 0.3462 |
| 27 | LUSC | rs7073 | chr22:43266363 | PACSIN2 | 8.75 × 10−12 | 0.0728 | 0.0810 | 0.0433 |
| 28 | LUSC | rs6151429 | chr22:51063477 | ARSA | 7.04 × 10−35 | 0.0291 | 0.0238 | 0.0144 |
| SNPs | Histology Type | Gene | Alleles | Cases | Controls | MAF (Cases) | MAF (Controls) | OR (95% CI) a | p a |
|---|---|---|---|---|---|---|---|---|---|
| rs10138506 | LUAD | CHURC1 | A > G | 3048/390/15 | 3324/380/6 | 0.061 | 0.053 | 1.16(1.01–1.33) | 0.034 |
| LUSC | CHURC1 | A > G | 596/55/3 | 3324/380/6 | 0.047 | 0.053 | 0.87(0.66–1.15) | 0.325 | |
| rs1130698 | LUAD | CD151 | T > C | 2729/675/49 | 2915/742/53 | 0.112 | 0.114 | 0.98(0.88–1.08) | 0.687 |
| LUSC | CD151 | T > C | 493/149/12 | 2915/742/53 | 0.132 | 0.114 | 1.19(1.00–1.41) | 0.049 | |
| rs1130719 | LUAD | CD151 | A > T | 2560/824/69 | 2751/882/77 | 0.139 | 0.140 | 1.00(0.91–1.10) | 0.990 |
| LUSC | CD151 | A > T | 455/175/24 | 2751/882/77 | 0.170 | 0.140 | 1.26(1.08–1.48) | 0.002 |
| Histology Type | SNPs | Phase | Genotypes | Cases, N (100%) | Controls, N (100%) | Adjusted OR (95% CI) a | p a |
|---|---|---|---|---|---|---|---|
| LUAD | rs10138506 | Screening | AA | 3048 (88.27) | 3324 (89.60) | 1 (ref) | -- |
| CHURC1 | AG | 390 (11.30) | 380 (10.24) | 1.12 (0.97–1.30) | 0.133 | ||
| GG | 15 (0.43) | 6 (0.16) | 2.72 (1.05–7.02) | 0.038 | |||
| Dominant model | 2.69 (1.04–6.94) | 0.041 | |||||
| Recessive model | 1.14 (0.99–1.32) | 0.071 | |||||
| Additive model | 1.16 (1.01–1.33) | 0.034 | |||||
| Validation | AA | 489 (84.90) | 591 (89.01) | 1 (ref) | -- | ||
| AG | 86 (14.93) | 72 (10.84) | 1.45 (1.03–2.03) | 0.033 | |||
| GG | 1 (0.17) | 1 (0.15) | 1.06 (0.07–17.00) | 0.969 | |||
| Dominant model | 1.44 (1.03–2.02) | 0.034 | |||||
| Additive model | 1.42 (1.02–1.98) | 0.038 | |||||
| LUSC | rs1130698 | Screening | TT | 493 (75.38) | 2915 (78.57) | 1 (ref) | -- |
| CD151 | TC | 149 (22.78) | 742 (20.00) | 1.19 (0.98–1.44) | 0.087 | ||
| CC | 12 (1.84) | 53 (1.43) | 1.39 (0.75–2.56) | 0.295 | |||
| Dominant model | 1.34 (0.73–2.46) | 0.351 | |||||
| Recessive model | 1.20 (0.99–1.45) | 0.060 | |||||
| Additive model | 1.19 (1.00–1.41) | 0.049 | |||||
| Validation | TT | 153 (75.37) | 535 (80.21) | 1 (ref) | -- | ||
| TC | 47 (23.15) | 126 (18.89) | 1.36 (0.91–2.02) | 0.138 | |||
| CC | 3 (1.48) | 6 (0.90) | 1.46 (0.33–6.48) | 0.615 | |||
| Dominant model | 1.36 (0.92–2.02) | 0.125 | |||||
| Additive model | 1.32 (0.92–1.90) | 0.130 | |||||
| LUSC | rs1130719 | Screening | AA | 455 (69.57) | 2751 (74.15) | 1 (ref) | -- |
| CD151 | AT | 175 (26.76) | 882 (23.77) | 1.20 (1.00–1.45) | 0.052 | ||
| TT | 24 (3.67) | 77 (2.08) | 1.86 (1.18–2.95) | 0.008 | |||
| Dominant model | 1.78 (1.13–2.81) | 0.014 | |||||
| Recessive model | 1.25 (1.05–1.50) | 0.012 | |||||
| Additive model | 1.26 (1.08–1.48) | 0.002 | |||||
| Validation | AA | 147 (72.41) | 482 (73.59) | 1 (ref) | -- | ||
| AT | 49 (24.14) | 154 (23.51) | 1.17 (0.79–1.73) | 0.428 | |||
| TT | 7 (3.45) | 19 (2.90) | 1.28 (0.50–3.26) | 0.611 | |||
| Dominant model | 1.18 (0.81–1.72) | 0.378 | |||||
| Additive model | 1.15 (0.84–1.58) | 0.372 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, A.; Xu, H.; Mao, L.; Xu, B.; Fu, X.; Cheng, J.; Zhao, R.; Cheng, Z.; Liu, X.; Xu, J.; et al. A Novel apaQTL-SNP for the Modification of Non-Small-Cell Lung Cancer Susceptibility across Histological Subtypes. Cancers 2022, 14, 5309. https://doi.org/10.3390/cancers14215309
Qiu A, Xu H, Mao L, Xu B, Fu X, Cheng J, Zhao R, Cheng Z, Liu X, Xu J, et al. A Novel apaQTL-SNP for the Modification of Non-Small-Cell Lung Cancer Susceptibility across Histological Subtypes. Cancers. 2022; 14(21):5309. https://doi.org/10.3390/cancers14215309
Chicago/Turabian StyleQiu, Anni, Huiwen Xu, Liping Mao, Buyun Xu, Xiaoyu Fu, Jingwen Cheng, Rongrong Zhao, Zhounan Cheng, Xiaoxuan Liu, Jingsheng Xu, and et al. 2022. "A Novel apaQTL-SNP for the Modification of Non-Small-Cell Lung Cancer Susceptibility across Histological Subtypes" Cancers 14, no. 21: 5309. https://doi.org/10.3390/cancers14215309
APA StyleQiu, A., Xu, H., Mao, L., Xu, B., Fu, X., Cheng, J., Zhao, R., Cheng, Z., Liu, X., Xu, J., Zhou, Y., Dong, Y., Tian, T., Tian, G., & Chu, M. (2022). A Novel apaQTL-SNP for the Modification of Non-Small-Cell Lung Cancer Susceptibility across Histological Subtypes. Cancers, 14(21), 5309. https://doi.org/10.3390/cancers14215309