
Factors and Mechanisms That Influence Chromatin-Mediated Enhancer–Promoter Interactions and Transcriptional Regulation


Abstract
:Simple Summary
Abstract
1. Introduction
2. Histone Modifications Involved in E–P Interactions

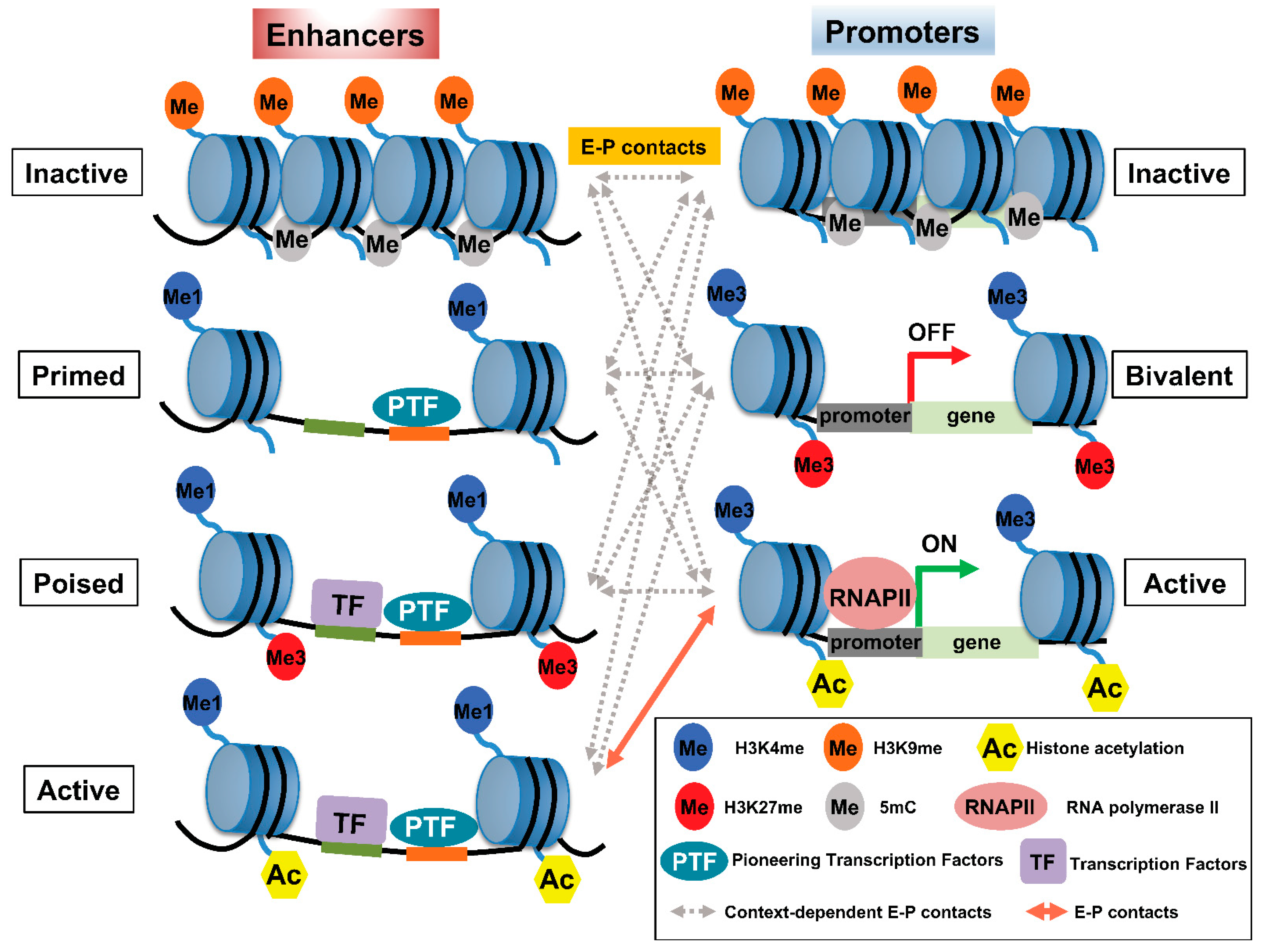{kind=link}
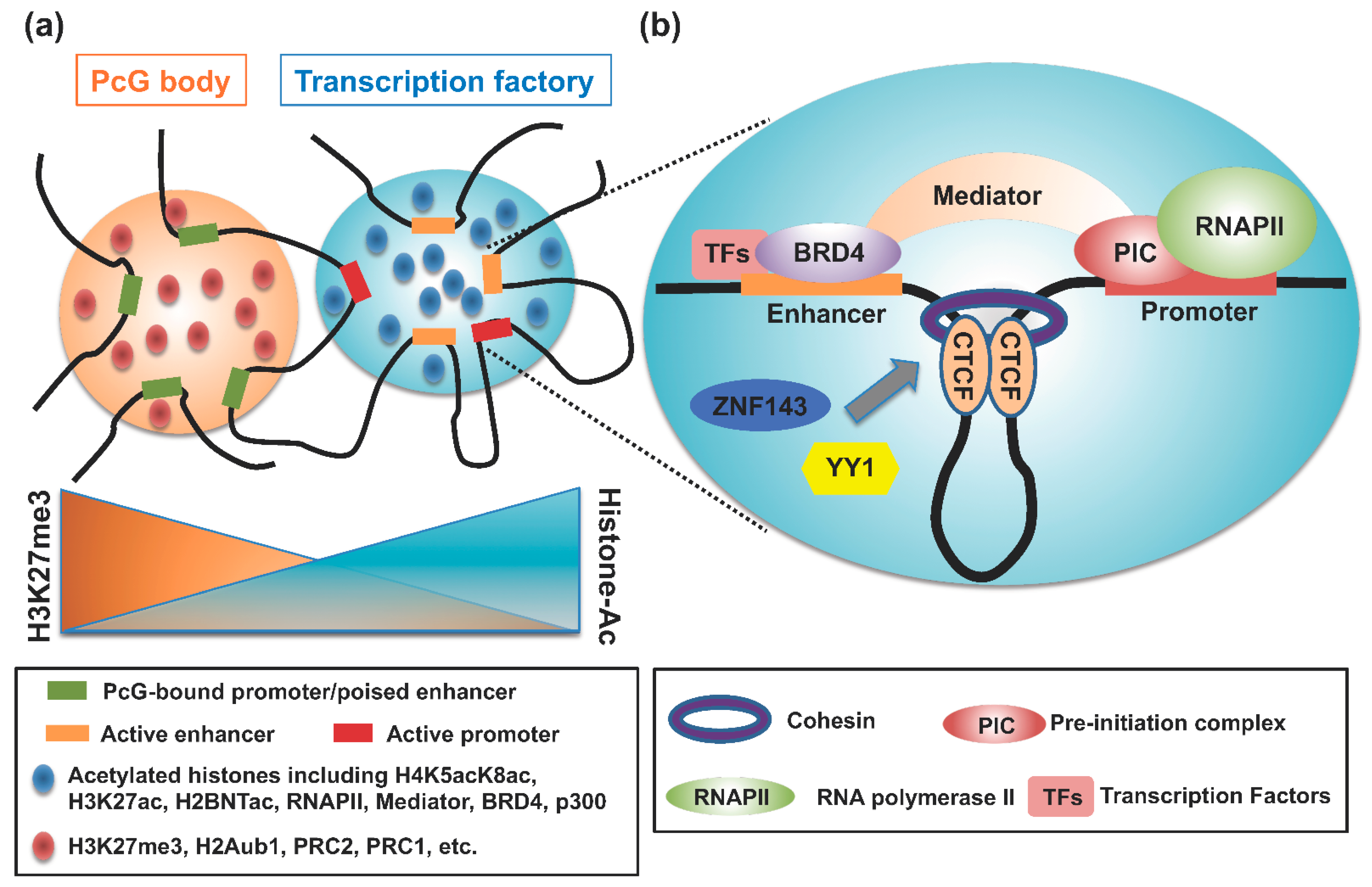{kind=link}
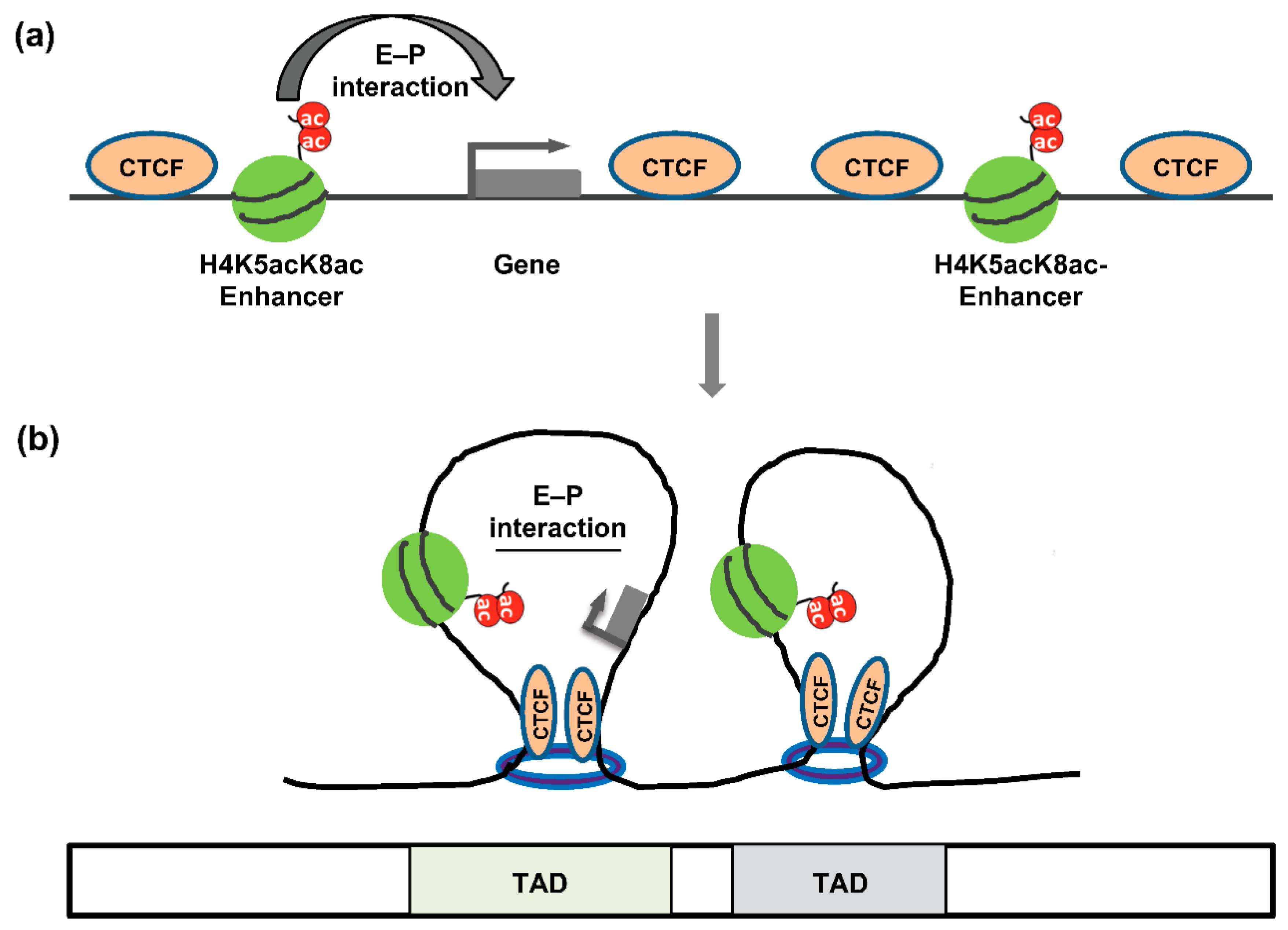{kind=link}
| Histone Modification | Putative Role in E–P Interactions and Transcription | Most Enriched Region | Reference |
|---|---|---|---|
| H3K4me3 | Activation | Promoters, bivalent promoters | [19] |
| H3K4me1 | Activation | Enhancers | [14] |
| H3K27ac | Activation | Enhancers, promoters | [11,12,13,14] |
| H4K5acK8ac | Activation | Enhancers, promoters | [15] |
| H4K16ac | Activation | Enhancers | [18] |
| H2BNTac (K5, K12, K16, K20) | Activation | Enhancers | [16] |
| H3K27me3 | Repression | Bivalent Promoters, poised enhancers | [30,31,32,33,34] |
3. Factors Regulating E–P Interactions
4. TFs and Pioneer TFs Involved in E–P Communication
5. The Architectural Proteins CTCF and Cohesin
6. BRD4 and the Mediator Complex
7. Polycomb Group (PcG) Complexes and LIM Domain Binding Proteins
8. E–P Interactions in LLPS
9. E–P Interactions Associated with Chromatin and Diseases
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Merkenschlager, M.; Nora, E.P. CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation. Annu. Rev. Genom. Hum. Genet. 2016, 17, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Ren, B. The Three-Dimensional Organization of Mammalian Genomes. Annu. Rev. Cell Dev. Biol. 2017, 33, 265–289. [Google Scholar] [CrossRef] [PubMed]
- Pombo, A.; Dillon, N. Three-dimensional genome architecture: Players and mechanisms. Nat. Rev. Mol. Cell Biol. 2015, 16, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Workman, J.L.; Kingston, R.E. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 1998, 67, 545–579. [Google Scholar] [CrossRef] [Green Version]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640. [Google Scholar] [CrossRef]
- Trojer, P.; Reinberg, D. Facultative heterochromatin: Is there a distinctive molecular signature? Mol. Cell 2007, 28, 1–13. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Shendure, J. Mechanisms of Interplay between Transcription Factors and the 3D Genome. Mol. Cell 2019, 76, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handoko, L.; Kaczkowski, B.; Hon, C.C.; Lizio, M.; Wakamori, M.; Matsuda, T.; Ito, T.; Jeyamohan, P.; Sato, Y.; Sakamoto, K.; et al. JQ1 affects BRD2-dependent and independent transcription regulation without disrupting H4-hyperacetylated chromatin states. Epigenetics 2018, 13, 410–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, T.; Higashijima, Y.; Kilic, S.; Liebner, T.; Walter, J.; Choudhary, C. A unique H2B acetylation signature marks active enhancers and predicts their target genes. bioRxiv 2022. [Google Scholar] [CrossRef]
- Pradeepa, M.M.; Grimes, G.R.; Kumar, Y.; Olley, G.; Taylor, G.C.; Schneider, R.; Bickmore, W.A. Histone H3 globular domain acetylation identifies a new class of enhancers. Nat. Genet. 2016, 48, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.C.; Eskeland, R.; Hekimoglu-Balkan, B.; Pradeepa, M.M.; Bickmore, W.A. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res. 2013, 23, 2053–2065. [Google Scholar] [CrossRef] [Green Version]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Goudarzi, A.; Shiota, H.; Rousseaux, S.; Khochbin, S. Genome-scale acetylation-dependent histone eviction during spermatogenesis. J. Mol. Biol. 2014, 426, 3342–3349. [Google Scholar] [CrossRef]
- Shiota, H.; Barral, S.; Buchou, T.; Tan, M.; Coute, Y.; Charbonnier, G.; Reynoird, N.; Boussouar, F.; Gerard, M.; Zhu, M.; et al. Nut Directs p300-Dependent, Genome-Wide H4 Hyperacetylation in Male Germ Cells. Cell Rep. 2018, 24, 3477–3487.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alekseyenko, A.A.; Walsh, E.M.; Wang, X.; Grayson, A.R.; Hsi, P.T.; Kharchenko, P.V.; Kuroda, M.I.; French, C.A. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Genes Dev. 2015, 29, 1507–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baud, M.G.J.; Lin-Shiao, E.; Cardote, T.; Tallant, C.; Pschibul, A.; Chan, K.H.; Zengerle, M.; Garcia, J.R.; Kwan, T.T.; Ferguson, F.M.; et al. Chemical biology. A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes. Science 2014, 346, 638–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [Green Version]
- Devaiah, B.N.; Case-Borden, C.; Gegonne, A.; Hsu, C.H.; Chen, Q.; Meerzaman, D.; Dey, A.; Ozato, K.; Singer, D.S. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016, 23, 540–548. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Li, Q.; Helfer, C.M.; Jiao, J.; You, J. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J. Biol. Chem. 2012, 287, 10738–10752. [Google Scholar] [CrossRef] [Green Version]
- Eagen, K.P.; French, C.A. Supercharging BRD4 with NUT in carcinoma. Oncogene 2021, 40, 1396–1408. [Google Scholar] [CrossRef]
- Rosencrance, C.D.; Ammouri, H.N.; Yu, Q.; Ge, T.; Rendleman, E.J.; Marshall, S.A.; Eagen, K.P. Chromatin Hyperacetylation Impacts Chromosome Folding by Forming a Nuclear Subcompartment. Mol. Cell 2020, 78, 112–126.e12. [Google Scholar] [CrossRef]
- Sharif, J.; Endo, T.A.; Ito, S.; Ohara, O.; Koseki, H. Embracing change to remain the same: Conservation of polycomb functions despite divergence of binding motifs among species. Curr. Opin. Cell Biol. 2013, 25, 305–313. [Google Scholar] [CrossRef]
- Voigt, P.; Tee, W.W.; Reinberg, D. A double take on bivalent promoters. Genes Dev. 2013, 27, 1318–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stutzer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngan, C.Y.; Wong, C.H.; Tjong, H.; Wang, W.; Goldfeder, R.L.; Choi, C.; He, H.; Gong, L.; Lin, J.; Urban, B.; et al. Chromatin interaction analyses elucidate the roles of PRC2-bound silencers in mouse development. Nat. Genet. 2020, 52, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Zeitlinger, J.; Stark, A.; Kellis, M.; Hong, J.W.; Nechaev, S.; Adelman, K.; Levine, M.; Young, R.A. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat. Genet. 2007, 39, 1512–1516. [Google Scholar] [CrossRef]
- Spivakov, M.; Fisher, A.G. Epigenetic signatures of stem-cell identity. Nat. Rev. Genet. 2007, 8, 263–271. [Google Scholar] [CrossRef]
- Hafner, A.; Boettiger, A. The spatial organization of transcriptional control. Nat. Rev. Genet. 2022. [Google Scholar] [CrossRef]
- Robson, M.I.; Ringel, A.R.; Mundlos, S. Regulatory Landscaping: How Enhancer-Promoter Communication Is Sculpted in 3D. Mol. Cell 2019, 74, 1110–1122. [Google Scholar] [CrossRef]
- Chong, S.; Dugast-Darzacq, C.; Liu, Z.; Dong, P.; Dailey, G.M.; Cattoglio, C.; Heckert, A.; Banala, S.; Lavis, L.; Darzacq, X.; et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 2018, 361, eaar2555. [Google Scholar] [CrossRef] [Green Version]
- Zabidi, M.A.; Arnold, C.D.; Schernhuber, K.; Pagani, M.; Rath, M.; Frank, O.; Stark, A. Enhancer-core-promoter specificity separates developmental and housekeeping gene regulation. Nature 2015, 518, 556–559. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, Y.; Loh, Y.P.; Tng, J.Q.; Lim, M.C.; Cao, Z.; Raju, A.; Lieberman Aiden, E.; Li, S.; Manikandan, L.; et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 2021, 12, 719. [Google Scholar] [CrossRef]
- Sungalee, S.; Liu, Y.; Lambuta, R.A.; Katanayeva, N.; Donaldson Collier, M.; Tavernari, D.; Roulland, S.; Ciriello, G.; Oricchio, E. Histone acetylation dynamics modulates chromatin conformation and allele-specific interactions at oncogenic loci. Nat. Genet. 2021, 53, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Larson, E.D.; Marsh, A.J.; Harrison, M.M. Pioneering the developmental frontier. Mol. Cell 2021, 81, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanborn, A.L.; Rao, S.S.; Huang, S.C.; Durand, N.C.; Huntley, M.H.; Jewett, A.I.; Bochkov, I.D.; Chinnappan, D.; Cutkosky, A.; Li, J.; et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. USA 2015, 112, E6456–E6465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, M.; Bickmore, W.; Furlong, E.E.M.; Narlikar, G. Chromatin topology, condensates and gene regulation: Shifting paradigms or just a phase? Development 2019, 146, dev182766. [Google Scholar] [CrossRef] [Green Version]
- Sabari, B.R.; Dall’Agnese, A.; Young, R.A. Biomolecular Condensates in the Nucleus. Trends Biochem. Sci. 2020, 45, 961–977. [Google Scholar] [CrossRef]
- Long, H.K.; Prescott, S.L.; Wysocka, J. Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 2016, 167, 1170–1187. [Google Scholar] [CrossRef] [Green Version]
- Iwafuchi-Doi, M.; Donahue, G.; Kakumanu, A.; Watts, J.A.; Mahony, S.; Pugh, B.F.; Lee, D.; Kaestner, K.H.; Zaret, K.S. The Pioneer Transcription Factor FoxA Maintains an Accessible Nucleosome Configuration at Enhancers for Tissue-Specific Gene Activation. Mol. Cell 2016, 62, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Dodonova, S.O.; Zhu, F.; Dienemann, C.; Taipale, J.; Cramer, P. Nucleosome-bound SOX2 and SOX11 structures elucidate pioneer factor function. Nature 2020, 580, 669–672. [Google Scholar] [CrossRef]
- Kumasaka, N.; Knights, A.J.; Gaffney, D.J. High-resolution genetic mapping of putative causal interactions between regions of open chromatin. Nat. Genet. 2019, 51, 128–137. [Google Scholar] [CrossRef]
- Deng, W.; Lee, J.; Wang, H.; Miller, J.; Reik, A.; Gregory, P.D.; Dean, A.; Blobel, G.A. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 2012, 149, 1233–1244. [Google Scholar] [CrossRef] [Green Version]
- Zaret, K.S. Pioneer Transcription Factors Initiating Gene Network Changes. Annu. Rev. Genet. 2020, 54, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Donaghey, J.; Thakurela, S.; Charlton, J.; Chen, J.S.; Smith, Z.D.; Gu, H.; Pop, R.; Clement, K.; Stamenova, E.K.; Karnik, R.; et al. Genetic determinants and epigenetic effects of pioneer-factor occupancy. Nat. Genet. 2018, 50, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 2008, 132, 958–970. [Google Scholar] [CrossRef] [Green Version]
- Mayran, A.; Khetchoumian, K.; Hariri, F.; Pastinen, T.; Gauthier, Y.; Balsalobre, A.; Drouin, J. Pioneer factor Pax7 deploys a stable enhancer repertoire for specification of cell fate. Nat. Genet. 2018, 50, 259–269. [Google Scholar] [CrossRef]
- Reddington, J.P.; Garfield, D.A.; Sigalova, O.M.; Karabacak Calviello, A.; Marco-Ferreres, R.; Girardot, C.; Viales, R.R.; Degner, J.F.; Ohler, U.; Furlong, E.E.M. Lineage-Resolved Enhancer and Promoter Usage during a Time Course of Embryogenesis. Dev. Cell 2020, 55, 648–664.e9. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Akopyan, G.; Garban, H.; Bonavida, B. Transcription factor YY1: Structure, function, and therapeutic implications in cancer biology. Oncogene 2006, 25, 1125–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017, 171, 1573–1588.e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beagan, J.A.; Duong, M.T.; Titus, K.R.; Zhou, L.; Cao, Z.; Ma, J.; Lachanski, C.V.; Gillis, D.R.; Phillips-Cremins, J.E. YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res. 2017, 27, 1139–1152. [Google Scholar] [CrossRef] [Green Version]
- Khachigian, L.M. The Yin and Yang of YY1 in tumor growth and suppression. Int. J. Cancer 2018, 143, 460–465. [Google Scholar] [CrossRef]
- Bailey, S.D.; Zhang, X.; Desai, K.; Aid, M.; Corradin, O.; Cowper-Sal Lari, R.; Akhtar-Zaidi, B.; Scacheri, P.C.; Haibe-Kains, B.; Lupien, M. ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nat. Commun. 2015, 2, 6186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yu, M.; Tirado-Magallanes, R.; Li, B.; Kong, L.; Guo, M.; Tan, Z.H.; Lee, S.; Chai, L.; Numata, A.; et al. ZNF143 mediates CTCF-bound promoter-enhancer loops required for murine hematopoietic stem and progenitor cell function. Nat. Commun. 2021, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Ortabozkoyun, H.; Huang, P.Y.; Cho, H.; Narendra, V.; LeRoy, G.; Gonzalez-Buendia, E.; Skok, J.A.; Tsirigos, A.; Mazzoni, E.O.; Reinberg, D. CRISPR and biochemical screens identify MAZ as a cofactor in CTCF-mediated insulation at Hox clusters. Nat. Genet. 2022, 54, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Parelho, V.; Hadjur, S.; Spivakov, M.; Leleu, M.; Sauer, S.; Gregson, H.C.; Jarmuz, A.; Canzonetta, C.; Webster, Z.; Nesterova, T.; et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 2008, 132, 422–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, E.D.; Reiss, D.J.; Welcsh, P.L.; Disteche, C.M.; Filippova, G.N.; Baliga, N.S.; Aebersold, R.; Ranish, J.A.; Krumm, A. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. USA 2008, 105, 8309–8314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losada, A.; Hirano, M.; Hirano, T. Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes Dev. 1998, 12, 1986–1997. [Google Scholar] [CrossRef] [Green Version]
- Yusufzai, T.M.; Tagami, H.; Nakatani, Y.; Felsenfeld, G. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol. Cell 2004, 13, 291–298. [Google Scholar] [CrossRef]
- Hou, C.; Zhao, H.; Tanimoto, K.; Dean, A. CTCF-dependent enhancer-blocking by alternative chromatin loop formation. Proc. Natl. Acad. Sci. USA 2008, 105, 20398–20403. [Google Scholar] [CrossRef] [Green Version]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Xu, Q.; Canzio, D.; Shou, J.; Li, J.; Gorkin, D.U.; Jung, I.; Wu, H.; Zhai, Y.; Tang, Y.; et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 2015, 162, 900–910. [Google Scholar] [CrossRef]
- Nanni, L.; Ceri, S.; Logie, C. Spatial patterns of CTCF sites define the anatomy of TADs and their boundaries. Genome Biol. 2020, 21, 197. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Dadon, D.B.; Powell, B.E.; Fan, Z.P.; Borges-Rivera, D.; Shachar, S.; Weintraub, A.S.; Hnisz, D.; Pegoraro, G.; Lee, T.I.; et al. 3D Chromosome Regulatory Landscape of Human Pluripotent Cells. Cell Stem Cell 2016, 18, 262–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Y.; Mariani, L.; Barozzi, I.; Schulz, E.G.; Bluthgen, N.; Stadler, M.; Tiana, G.; Giorgetti, L. Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes. Genome Res. 2017, 27, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelleher, R.J., 3rd; Flanagan, P.M.; Kornberg, R.D. A novel mediator between activator proteins and the RNA polymerase II transcription apparatus. Cell 1990, 61, 1209–1215. [Google Scholar] [CrossRef]
- Di Micco, R.; Fontanals-Cirera, B.; Low, V.; Ntziachristos, P.; Yuen, S.K.; Lovell, C.D.; Dolgalev, I.; Yonekubo, Y.; Zhang, G.; Rusinova, E.; et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014, 9, 234–247. [Google Scholar] [CrossRef] [Green Version]
- El Khattabi, L.; Zhao, H.; Kalchschmidt, J.; Young, N.; Jung, S.; Van Blerkom, P.; Kieffer-Kwon, P.; Kieffer-Kwon, K.R.; Park, S.; Wang, X.; et al. A Pliable Mediator Acts as a Functional Rather Than an Architectural Bridge between Promoters and Enhancers. Cell 2019, 178, 1145–1158.e20. [Google Scholar] [CrossRef]
- Lai, F.; Orom, U.A.; Cesaroni, M.; Beringer, M.; Taatjes, D.J.; Blobel, G.A.; Shiekhattar, R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 2013, 494, 497–501. [Google Scholar] [CrossRef] [Green Version]
- Soutourina, J. Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 2018, 19, 262–274. [Google Scholar] [CrossRef]
- Crump, N.T.; Ballabio, E.; Godfrey, L.; Thorne, R.; Repapi, E.; Kerry, J.; Tapia, M.; Hua, P.; Lagerholm, C.; Filippakopoulos, P.; et al. BET inhibition disrupts transcription but retains enhancer-promoter contact. Nat. Commun. 2021, 12, 223. [Google Scholar] [CrossRef]
- Kim, J.J.; Kingston, R.E. Context-specific Polycomb mechanisms in development. Nat. Rev. Genet. 2022, 23, 680–695. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Kingston, R.E. Mechanisms of polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Muller, J.; Hart, C.M.; Francis, N.J.; Vargas, M.L.; Sengupta, A.; Wild, B.; Miller, E.L.; O’Connor, M.B.; Kingston, R.E.; Simon, J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 2002, 111, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Isono, K.; Endo, T.A.; Ku, M.; Yamada, D.; Suzuki, R.; Sharif, J.; Ishikura, T.; Toyoda, T.; Bernstein, B.E.; Koseki, H. SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev. Cell 2013, 26, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Robinson, A.K.; Leal, B.Z.; Chadwell, L.V.; Wang, R.; Ilangovan, U.; Kaur, Y.; Junco, S.E.; Schirf, V.; Osmulski, P.A.; Gaczynska, M.; et al. The growth-suppressive function of the polycomb group protein polyhomeotic is mediated by polymerization of its sterile alpha motif (SAM) domain. J. Biol. Chem. 2012, 287, 8702–8713. [Google Scholar] [CrossRef] [Green Version]
- Seif, E.; Kang, J.J.; Sasseville, C.; Senkovich, O.; Kaltashov, A.; Boulier, E.L.; Kapur, I.; Kim, C.A.; Francis, N.J. Phase separation by the polyhomeotic sterile alpha motif compartmentalizes Polycomb Group proteins and enhances their activity. Nat. Commun. 2020, 11, 5609. [Google Scholar] [CrossRef]
- Kondo, T.; Isono, K.; Kondo, K.; Endo, T.A.; Itohara, S.; Vidal, M.; Koseki, H. Polycomb potentiates meis2 activation in midbrain by mediating interaction of the promoter with a tissue-specific enhancer. Dev. Cell 2014, 28, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Ogiyama, Y.; Schuettengruber, B.; Papadopoulos, G.L.; Chang, J.M.; Cavalli, G. Polycomb-Dependent Chromatin Looping Contributes to Gene Silencing during Drosophila Development. Mol. Cell 2018, 71, 73–88.e5. [Google Scholar] [CrossRef]
- Kondo, T.; Ito, S.; Koseki, H. Polycomb in Transcriptional Phase Transition of Developmental Genes. Trends Biochem. Sci. 2016, 41, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Krivega, I.; Dean, A. Chromatin looping as a target for altering erythroid gene expression. Ann. N. Y. Acad. Sci. 2016, 1368, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breen, J.J.; Agulnick, A.D.; Westphal, H.; Dawid, I.B. Interactions between LIM domains and the LIM domain-binding protein Ldb1. J. Biol. Chem. 1998, 273, 4712–4717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, A.J.; Jeffries, C.M.; Trewhella, J.; Matthews, J.M. LIM domain binding proteins 1 and 2 have different oligomeric states. J. Mol. Biol. 2010, 399, 133–144. [Google Scholar] [CrossRef]
- Krivega, I.; Dale, R.K.; Dean, A. Role of LDB1 in the transition from chromatin looping to transcription activation. Genes Dev. 2014, 28, 1278–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855.e16. [Google Scholar] [CrossRef] [Green Version]
- Sanulli, S.; Trnka, M.J.; Dharmarajan, V.; Tibble, R.W.; Pascal, B.D.; Burlingame, A.L.; Griffin, P.R.; Gross, J.D.; Narlikar, G.J. HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 2019, 575, 390–394. [Google Scholar] [CrossRef]
- Zhang, Y.; Kutateladze, T.G. Liquid-liquid phase separation is an intrinsic physicochemical property of chromatin. Nat. Struct. Mol. Biol. 2019, 26, 1085–1086. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [Green Version]
- Shrinivas, K.; Sabari, B.R.; Coffey, E.L.; Klein, I.A.; Boija, A.; Zamudio, A.V.; Schuijers, J.; Hannett, N.M.; Sharp, P.A.; Young, R.A.; et al. Enhancer Features that Drive Formation of Transcriptional Condensates. Mol. Cell 2019, 75, 549–561.e7. [Google Scholar] [CrossRef]
- Cook, P.R. A model for all genomes: The role of transcription factories. J. Mol. Biol. 2010, 395, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.K.; Spille, J.H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, B.A.; Doolittle, L.K.; Schneider, M.W.G.; Jensen, L.E.; Gamarra, N.; Henry, L.; Gerlich, D.W.; Redding, S.; Rosen, M.K. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 2019, 179, 470–484.e21. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, K.; Cai, W.; Liu, X.; Zhang, Y.; Orkin, S.H.; Xu, J.; Yuan, G.C. Dissecting super-enhancer hierarchy based on chromatin interactions. Nat. Commun. 2018, 9, 943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, B.; Levine, M.S. Enhancer-promoter communication: Hubs or loops? Curr. Opin. Genet. Dev. 2021, 67, 5–9. [Google Scholar] [CrossRef]
- Agelopoulos, M.; Foutadakis, S.; Thanos, D. The Causes and Consequences of Spatial Organization of the Genome in Regulation of Gene Expression. Front. Immunol. 2021, 12, 682397. [Google Scholar] [CrossRef]
- Deng, W.; Rupon, J.W.; Krivega, I.; Breda, L.; Motta, I.; Jahn, K.S.; Reik, A.; Gregory, P.D.; Rivella, S.; Dean, A.; et al. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell 2014, 158, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Levo, M.; Barinov, L.; Fujioka, M.; Jaynes, J.B.; Gregor, T. Dynamic interplay between enhancer-promoter topology and gene activity. Nat Genet 2018, 50, 1296–1303. [Google Scholar] [CrossRef]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Barakat, T.S.; Halbritter, F.; Zhang, M.; Rendeiro, A.F.; Perenthaler, E.; Bock, C.; Chambers, I. Functional Dissection of the Enhancer Repertoire in Human Embryonic Stem Cells. Cell Stem Cell 2018, 23, 276–288.e8. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Z.; Dong, Q.; Xiong, J.; Zhu, B. Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol. 2020, 21, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankar, A.; Mohammad, F.; Sundaramurthy, A.K.; Wang, H.; Lerdrup, M.; Tatar, T.; Helin, K. Histone editing elucidates the functional roles of H3K27 methylation and acetylation in mammals. Nat. Genet. 2022, 54, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Donaldson-Collier, M.C.; Sungalee, S.; Zufferey, M.; Tavernari, D.; Katanayeva, N.; Battistello, E.; Mina, M.; Douglass, K.M.; Rey, T.; Raynaud, F.; et al. EZH2 oncogenic mutations drive epigenetic, transcriptional, and structural changes within chromatin domains. Nat. Genet. 2019, 51, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Henikoff, S. Modulation of a transcription factor counteracts heterochromatic gene silencing in Drosophila. Cell 2001, 104, 839–847. [Google Scholar] [CrossRef]
- Carpenter, A.E.; Memedula, S.; Plutz, M.J.; Belmont, A.S. Common effects of acidic activators on large-scale chromatin structure and transcription. Mol. Cell Biol. 2005, 25, 958–968. [Google Scholar] [CrossRef] [Green Version]
- Therizols, P.; Illingworth, R.S.; Courilleau, C.; Boyle, S.; Wood, A.J.; Bickmore, W.A. Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science 2014, 346, 1238–1242. [Google Scholar] [CrossRef]
- Boettiger, A.N.; Bintu, B.; Moffitt, J.R.; Wang, S.; Beliveau, B.J.; Fudenberg, G.; Imakaev, M.; Mirny, L.A.; Wu, C.T.; Zhuang, X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 2016, 529, 418–422. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.; Feodorova, Y.; Naumova, N.; Imakaev, M.; Lajoie, B.R.; Leonhardt, H.; Joffe, B.; Dekker, J.; Fudenberg, G.; Solovei, I.; et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 2019, 570, 395–399. [Google Scholar] [CrossRef]
- Gassler, J.; Brandao, H.B.; Imakaev, M.; Flyamer, I.M.; Ladstatter, S.; Bickmore, W.A.; Peters, J.M.; Mirny, L.A.; Tachibana, K. A mechanism of cohesin-dependent loop extrusion organizes zygotic genome architecture. EMBO J. 2017, 36, 3600–3618. [Google Scholar] [CrossRef]
- Nora, E.P.; Goloborodko, A.; Valton, A.L.; Gibcus, J.H.; Uebersohn, A.; Abdennur, N.; Dekker, J.; Mirny, L.A.; Bruneau, B.G. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 2017, 169, 930–944.e22. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suva, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelham-Webb, B.; Polyzos, A.; Wojenski, L.; Kloetgen, A.; Li, J.; Di Giammartino, D.C.; Sakellaropoulos, T.; Tsirigos, A.; Core, L.; Apostolou, E. H3K27ac bookmarking promotes rapid post-mitotic activation of the pluripotent stem cell program without impacting 3D chromatin reorganization. Mol. Cell 2021, 81, 1732–1748.e8. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, B.; Tavares-Cadete, F.; Young, A.N.; Sugar, R.; Schoenfelder, S.; Ferreira, L.; Wingett, S.W.; Andrews, S.; Grey, W.; Ewels, P.A.; et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015, 47, 598–606. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Furlan-Magaril, M.; Mifsud, B.; Tavares-Cadete, F.; Sugar, R.; Javierre, B.M.; Nagano, T.; Katsman, Y.; Sakthidevi, M.; Wingett, S.W.; et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 2015, 25, 582–597. [Google Scholar] [CrossRef]



| Complex | Subunit | Domain | Potential Function |
|---|---|---|---|
| PRC1 | RING1A/RING1B | RING finger | H2AK119 ubiquitination |
| PCGF1-6 | RING finger | Enhancer of RING1A/RING1B activity | |
| PHC1-3 | SAM | Oligomerization | |
| CBX2,-4,-6,-7,-8 | Chromodomain | H3K27me3 binding | |
| PRC2 | EZH1/EZH2 | SET | H3K27 methylation |
| EED | WD40 | H3K27me3 binding | |
| SUZ12 | Zinc finger | PRC2 stability | |
| RBBP4/RBBP7 | WD40 | Nucleosome binding |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, S.; Das, N.D.; Umehara, T.; Koseki, H. Factors and Mechanisms That Influence Chromatin-Mediated Enhancer–Promoter Interactions and Transcriptional Regulation. Cancers 2022, 14, 5404. https://doi.org/10.3390/cancers14215404
Ito S, Das ND, Umehara T, Koseki H. Factors and Mechanisms That Influence Chromatin-Mediated Enhancer–Promoter Interactions and Transcriptional Regulation. Cancers. 2022; 14(21):5404. https://doi.org/10.3390/cancers14215404
Chicago/Turabian StyleIto, Shinsuke, Nando Dulal Das, Takashi Umehara, and Haruhiko Koseki. 2022. "Factors and Mechanisms That Influence Chromatin-Mediated Enhancer–Promoter Interactions and Transcriptional Regulation" Cancers 14, no. 21: 5404. https://doi.org/10.3390/cancers14215404
APA StyleIto, S., Das, N. D., Umehara, T., & Koseki, H. (2022). Factors and Mechanisms That Influence Chromatin-Mediated Enhancer–Promoter Interactions and Transcriptional Regulation. Cancers, 14(21), 5404. https://doi.org/10.3390/cancers14215404