
c-Myc Targets HDAC3 to Suppress NKG2DL Expression and Innate Immune Response in N-Type SCLC through Histone Deacetylation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and Transfection
2.3. MYC siRNA Transfection
2.4. Inhibitor Incubation
2.5. Cell Proliferation Analysis
2.6. Cell Migration Analysis
2.7. Western Blotting
2.8. Immunofluorescence Analysis
2.9. Flow Cytometry Analysis
2.10. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR) Analysis
2.11. Co-Immunoprecipitation (Co-IP) Assay
2.12. Chromatin Immunoprecipitation (ChIP)-qPCR Assay
2.13. Cytotoxicity Assay
2.14. Analysis of Differential Gene Expression from Cancer Cell Line Encyclopedia (CCLE) Dataset
2.15. Kaplan-Meier Survival Analysis
2.16. Statistical Analysis
3. Results
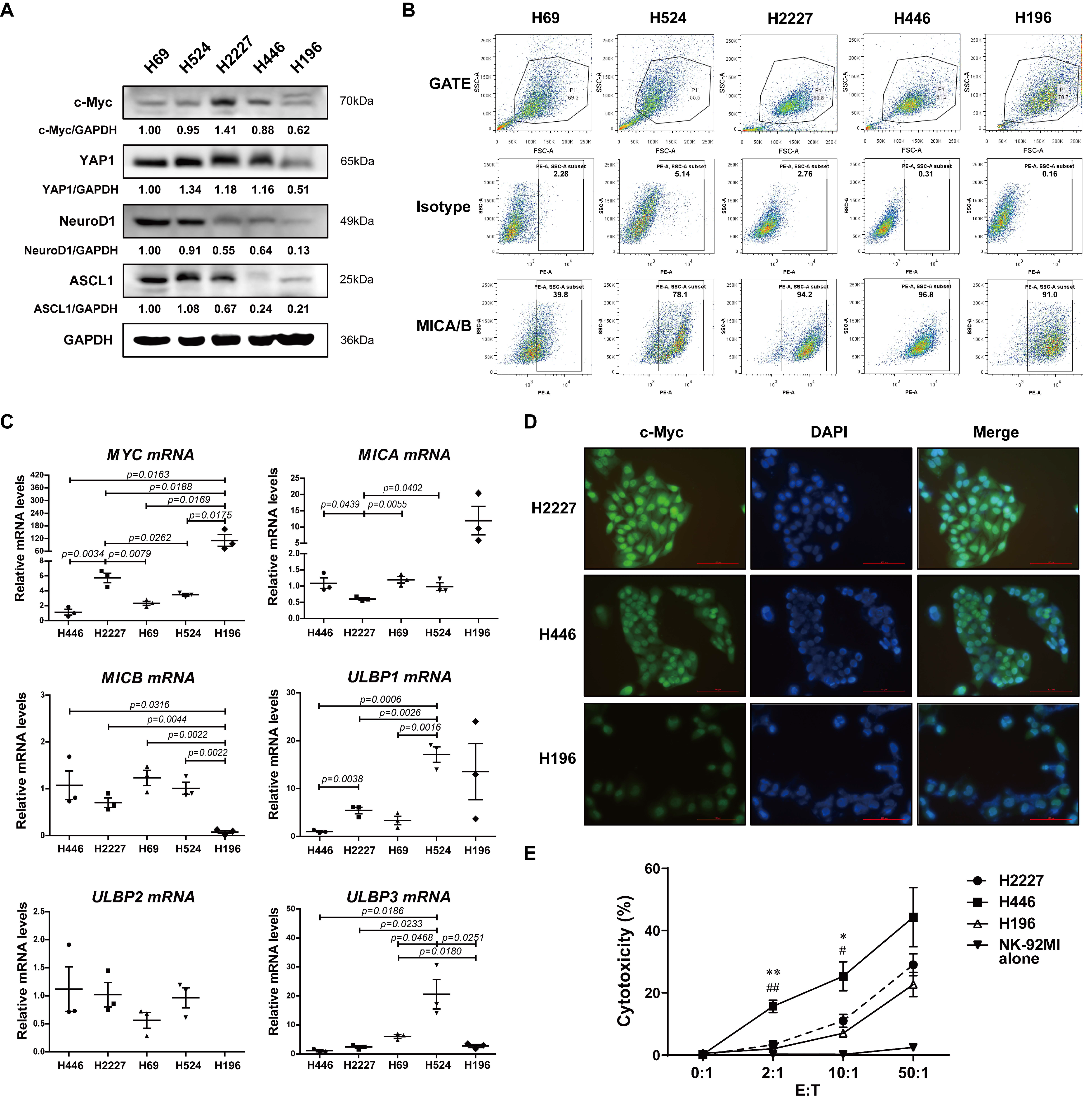
3.1. c-Myc, NKG2DL Expression and Susceptibility to NK Cell Killing of SCLC Cell Lines
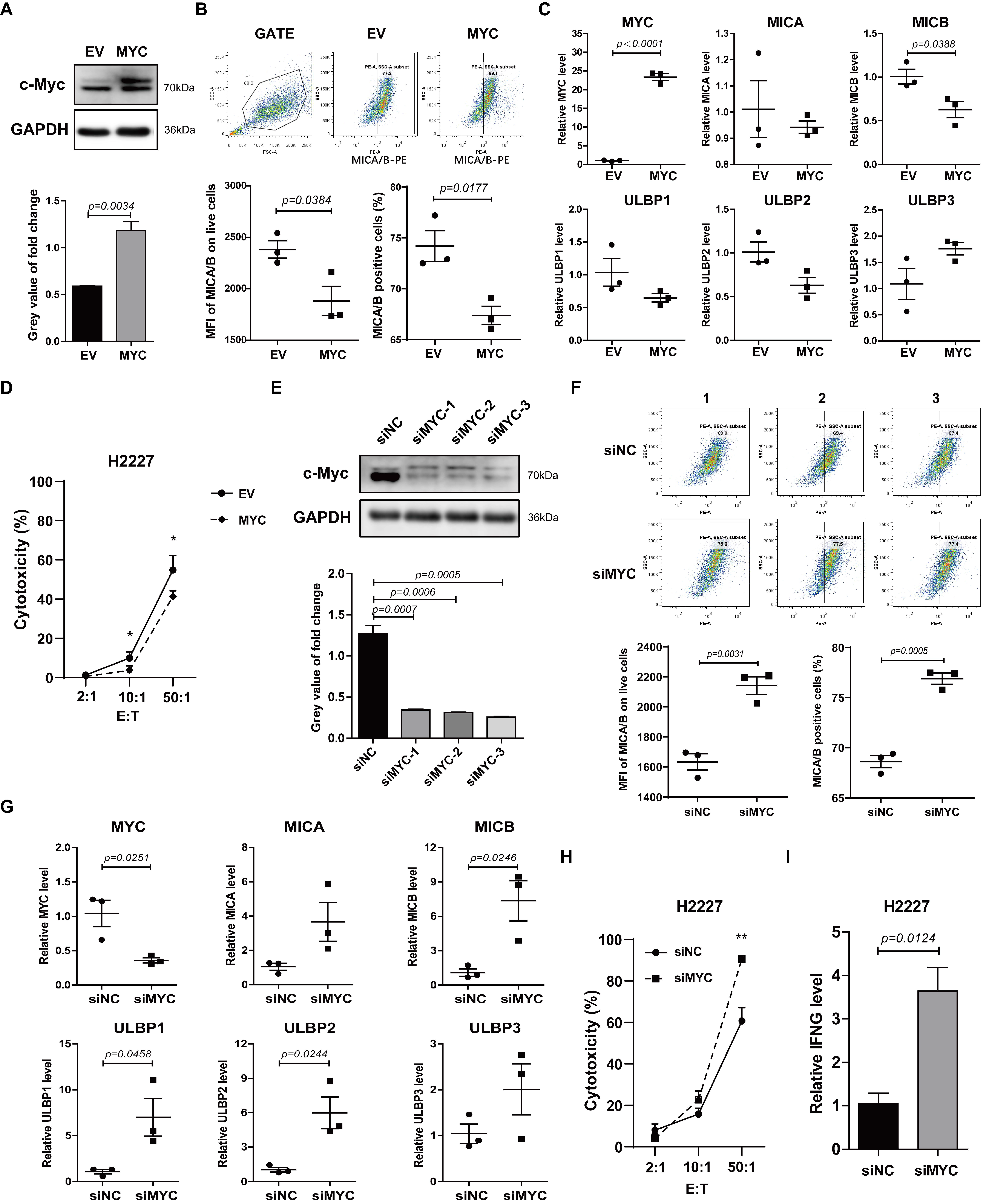
3.2. c-Myc Inhibited Expression of NKG2DL and Susceptibility to NK Cell Killing in SCLC-N Cells
3.3. HDACs Involved in the Modulation of NKG2DL Expression in SCLC-N Cells
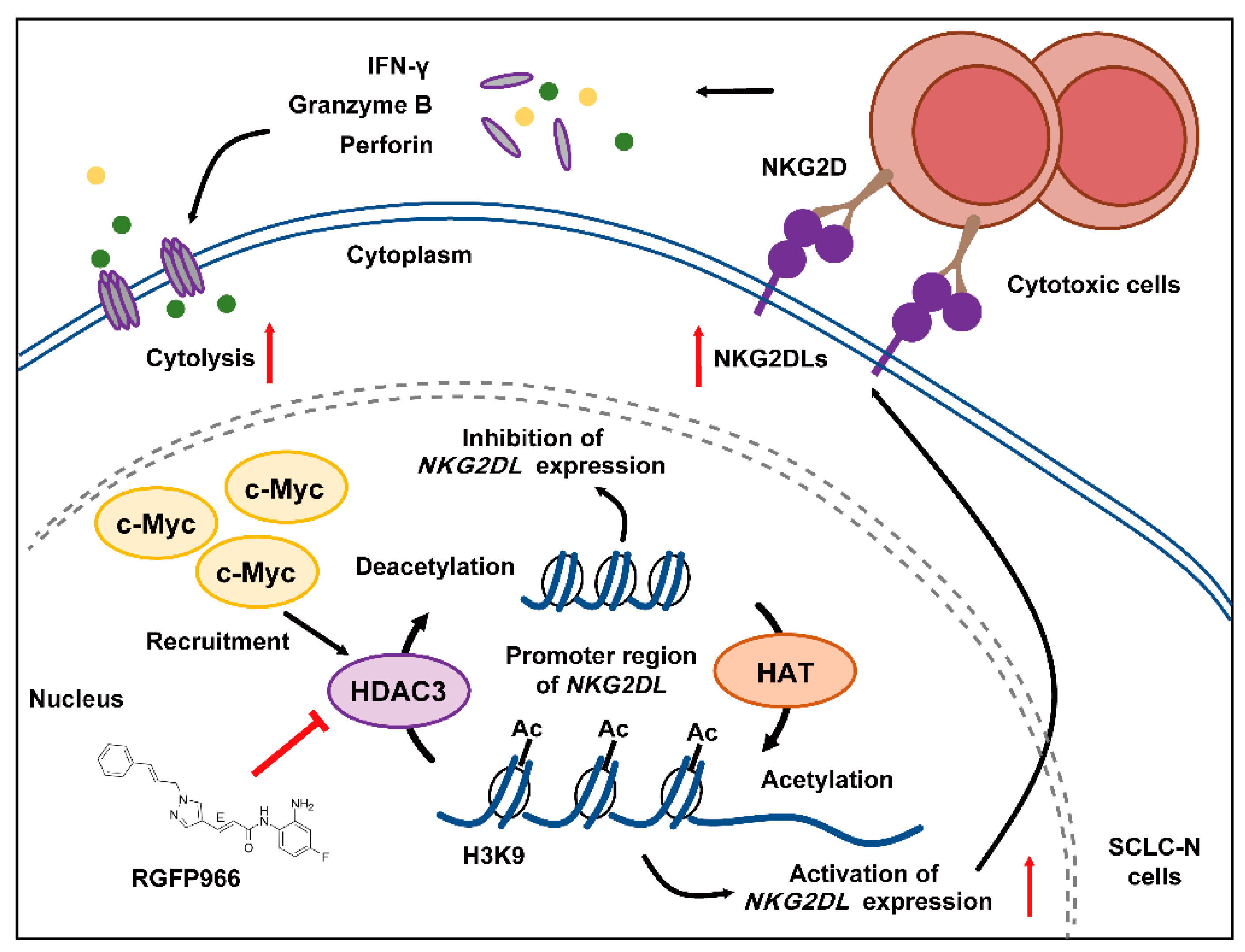
3.4. c-Myc Regulated NKG2DL Expression on SCLC-N Cells through HDAC3
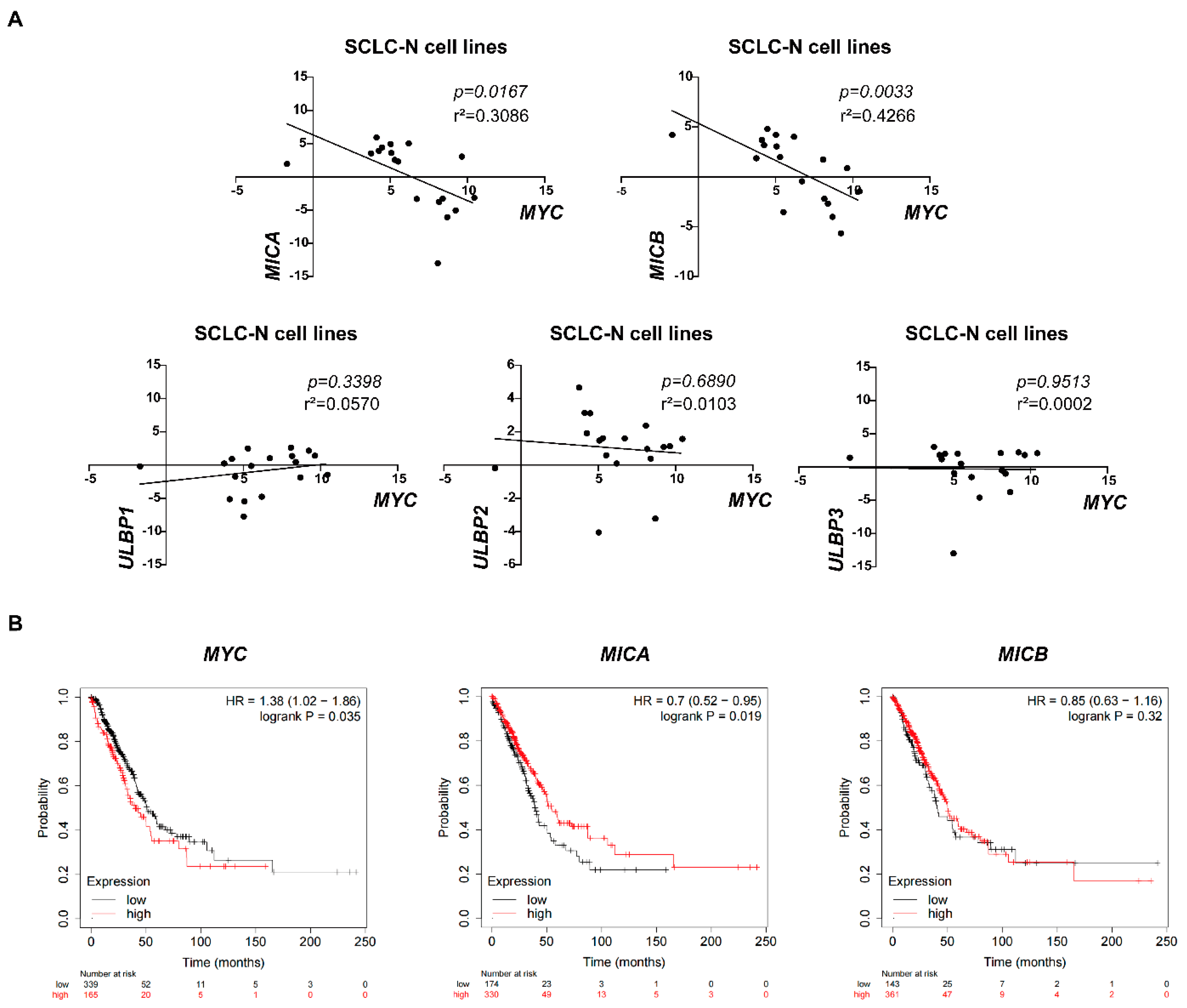
3.5. Correlation between MYC and NKG2DL Expression in SCLC Cells and Lung Cancer Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nicholson, A.G.; Chansky, K.; Crowley, J.; Beyruti, R.; Kubota, K.; Turrisi, A.; Eberhardt, W.E.E.; van Meerbeeck, J.; Rami-Porta, R. The International Association for the Study of Lung Cancer Lung Cancer Staging Project: Proposals for the Revision of the Clinical and Pathologic Staging of Small Cell Lung Cancer in the Forthcoming Eighth Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016, 11, 300–311. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; di Maio, M.; Chiodini, P.; Rudd, R.M.; Okamoto, H.; Skarlos, D.V.; Früh, M.; Qian, W.; Tamura, T.; Samantas, E.; et al. Carboplatin- or cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: The COCIS meta-analysis of individual patient data. J. Clin. Oncol. 2012, 30, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.C.; Piha-Paul, S.A.; Lopez-Martin, J.; Schellens, J.H.M.; Kao, S.; Miller, W.H., Jr.; Delord, J.P.; Gao, B.; Planchard, D.; Gottfried, M.; et al. Pembrolizumab After Two or More Lines of Previous Therapy in Patients with Recurrent or Metastatic SCLC: Results From the KEYNOTE-028 and KEYNOTE-158 Studies. J. Thorac. Oncol. 2020, 15, 618–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Hamilton, G.; Rath, B. Immunotherapy for small cell lung cancer: Mechanisms of resistance. Expert Opin. Biol. Ther. 2019, 19, 423–432. [Google Scholar] [CrossRef]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021. [Google Scholar] [CrossRef]
- Busch, S.E.; Hanke, M.L.; Kargl, J.; Metz, H.E.; MacPherson, D.; Houghton, A.M. Lung Cancer Subtypes Generate Unique Immune Responses. J. Immunol. 2016, 197, 4493–4503. [Google Scholar] [CrossRef] [Green Version]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar] [CrossRef] [Green Version]
- Cong, J.; Wang, X.; Zheng, X.; Wang, D.; Fu, B.; Sun, R.; Tian, Z.; Wei, H. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab. 2018, 28, 243–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, S.A.; Hess, J.B.; Souza-Fonseca-Guimaraes, F.; Cursons, J.; Kersbergen, A.; Dong, X.; Rautela, J.; Hyslop, S.R.; Ritchie, M.E.; Davis, M.J.; et al. Harnessing Natural Killer Immunity in Metastatic SCLC. J. Thorac. Oncol. 2020, 15, 1507–1521. [Google Scholar] [CrossRef]
- Lowry, L.E.; Zehring, W.A. Potentiation of Natural Killer Cells for Cancer Immunotherapy: A Review of Literature. Front. Immunol. 2017, 8, 1061. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verneris, M.R.; Karimi, M.; Baker, J.; Jayaswal, A.; Negrin, R.S. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood 2004, 103, 3065–3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Yang, L.; Li, X.; Lu, W.; Lu, F.; Wang, S.; Wang, Y.; Hua, L.; Cui, C.; Dong, B.; et al. Rae1 drives NKG2D binding-dependent tumor development in mice by activating mTOR and STAT3 pathways in tumor cells. Cancer Sci. 2020, 111, 2234–2247. [Google Scholar] [CrossRef]
- Zhu, M.; Huang, Y.; Bender, M.E.; Girard, L.; Kollipara, R.; Eglenen-Polat, B.; Naito, Y.; Savage, T.K.; Huffman, K.E.; Koyama, S.; et al. Evasion of Innate Immunity Contributes to Small Cell Lung Cancer Progression and Metastasis. Cancer Res. 2021, 81, 1813–1826. [Google Scholar] [CrossRef]
- Brägelmann, J.; Böhm, S.; Guthrie, M.R.; Mollaoglu, G.; Oliver, T.G.; Sos, M.L. Family matters: How MYC family oncogenes impact small cell lung cancer. Cell Cycle 2017, 16, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Dammert, M.A.; Bragelmann, J.; Olsen, R.R.; Bohm, S.; Monhasery, N.; Whitney, C.P.; Chalishazar, M.D.; Tumbrink, H.L.; Guthrie, M.R.; Klein, S.; et al. MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat. Commun. 2019, 10, 3485. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef]
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Soltero, D.; et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020, 38, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Quintanal-Villalonga, Á.; Gao, V.R.; Xie, Y.; Allaj, V.; Chaudhary, O.; Masilionis, I.; Egger, J.; Chow, A.; Walle, T.; et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell 2021. [Google Scholar] [CrossRef]
- Liu, M.; Meng, Y.; Zhang, L.; Han, Z.; Feng, X. High-efficient generation of natural killer cells from peripheral blood with preferable cell vitality and enhanced cytotoxicity by combination of IL-2, IL-15 and IL-18. Biochem. Biophys. Res. Commun. 2021, 534, 149–156. [Google Scholar] [CrossRef]
- Kloss, S.; Oberschmidt, O.; Morgan, M.; Dahlke, J.; Arseniev, L.; Huppert, V.; Granzin, M.; Gardlowski, T.; Matthies, N.; Soltenborn, S.; et al. Optimization of Human NK Cell Manufacturing: Fully Automated Separation, Improved Ex Vivo Expansion Using IL-21 with Autologous Feeder Cells, and Generation of Anti-CD123-CAR-Expressing Effector Cells. Hum. Gene Ther. 2017, 28, 897–913. [Google Scholar] [CrossRef]
- Wang, J.; Elahi, A.; Ajidahun, A.; Clark, W.; Hernandez, J.; Achille, A.; Hao, J.-h.; Seto, E.; Shibata, D. The interplay between histone deacetylases and c-Myc in the transcriptional suppression of HPP1 in colon cancer. Cancer Biol. Ther. 2014, 15, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieszczad, K.M.; Bechay, K.; Rusche, J.R.; Jacques, V.; Kudugunti, S.; Miao, W.; Weinberger, N.M.; McGaugh, J.L.; Wood, M.A. Histone Deacetylase Inhibition via RGFP966 Releases the Brakes on Sensory Cortical Plasticity and the Specificity of Memory Formation. J. Neurosci. 2015, 35, 13124–13132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.J.; Manjegowda, M.C.; Kain, J.; Anandh, S.; Bochkis, I.M. Hdac3, Setdb1, and Kap1 mark H3K9me3/H3K14ac bivalent regions in young and aged liver. Aging Cell 2020, 19, e13092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimoto, K.; Niida, H.; Uchida, C.; Ohhata, T.; Kitagawa, K.; Motegi, A.; Suda, T.; Kitagawa, M. HDAC3 Is Required for XPC Recruitment and Nucleotide Excision Repair of DNA Damage Induced by UV Irradiation. Mol. Cancer Res. 2020, 18, 1367–1378. [Google Scholar] [CrossRef]
- Yuliana, A.; Jheng, H.-F.; Kawarasaki, S.; Nomura, W.; Takahashi, H.; Ara, T.; Kawada, T.; Goto, T. β-adrenergic Receptor Stimulation Revealed a Novel Regulatory Pathway via Suppressing Histone Deacetylase 3 to Induce Uncoupling Protein 1 Expression in Mice Beige Adipocyte. Int. J. Mol. Sci. 2018, 19, 2436. [Google Scholar] [CrossRef] [Green Version]
- Dora, D.; Rivard, C.; Yu, H.; Bunn, P.; Suda, K.; Ren, S.; Lueke Pickard, S.; Laszlo, V.; Harko, T.; Megyesfalvi, Z.; et al. Neuroendocrine subtypes of small cell lung cancer differ in terms of immune microenvironment and checkpoint molecule distribution. Mol. Oncol. 2020, 14, 1947–1965. [Google Scholar] [CrossRef]
- Textor, S.; Fiegler, N.; Arnold, A.; Porgador, A.; Hofmann, T.G.; Cerwenka, A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 2011, 71, 5998–6009. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Lakshmikanth, T.; Garofalo, C.; Enge, M.; Spinnler, C.; Anichini, A.; Szekely, L.; Kärre, K.; Carbone, E.; Selivanova, G. Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle 2011, 10, 3346–3358. [Google Scholar] [CrossRef] [Green Version]
- Soriani, A.; Borrelli, C.; Ricci, B.; Molfetta, R.; Zingoni, A.; Fionda, C.; Carnevale, S.; Abruzzese, M.P.; Petrucci, M.T.; Ricciardi, M.R.; et al. p38 MAPK differentially controls NK activating ligands at transcriptional and post-transcriptional level on multiple myeloma cells. Oncoimmunology 2017, 6, e1264564. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Sun, B.; Xu, K.; He, Y.; Zhang, T.; Hall, S.R.R.; Tan, S.T.; Schmid, R.A.; Peng, R.W.; Hu, G.; et al. Pharmaco-transcriptomic correlation analysis reveals novel responsive signatures to HDAC inhibitors and identifies Dasatinib as a synergistic interactor in small-cell lung cancer. eBioMedicine 2021, 69, 103457. [Google Scholar] [CrossRef]
- Tlemsani, C.; Pongor, L.; Elloumi, F.; Girard, L.; Huffman, K.E.; Roper, N.; Varma, S.; Luna, A.; Rajapakse, V.N.; Sebastian, R.; et al. SCLC-CellMiner: A Resource for Small Cell Lung Cancer Cell Line Genomics and Pharmacology Based on Genomic Signatures. Cell Rep. 2020, 33, 108296. [Google Scholar] [CrossRef] [PubMed]
- Owonikoko, T.K.; Dwivedi, B.; Chen, Z.; Zhang, C.; Barwick, B.; Ernani, V.; Zhang, G.; Gilbert-Ross, M.; Carlisle, J.; Khuri, F.R.; et al. YAP1 Expression in Small Cell Lung Cancer Defines a Distinct Subtype with T-cell Inflamed Phenotype. J. Thorac. Oncol. 2020, 16, 464–476. [Google Scholar] [CrossRef]
- Masso-Valles, D.; Beaulieu, M.E.; Soucek, L. MYC, MYCL, and MYCN as therapeutic targets in lung cancer. Expert Opin. Ther. Targets 2020, 24, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, X.; Lin, J.; Lwin, T.; Wright, G.; Moscinski, L.C.; Dalton, W.S.; Seto, E.; Wright, K.; Sotomayor, E.; et al. Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene 2012, 31, 3002–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, R.; Gong, J.-N.; Chen, M.-T.; Song, L.; Shen, C.; Zhang, X.-H.; Yin, X.-L.; Ning, H.-M.; Liu, B.; Wang, F.; et al. c-Myc suppresses miR-451⊣YWTAZ/AKT axis via recruiting HDAC3 in acute myeloid leukemia. Oncotarget 2016, 7, 77430–77443. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.-Y.; Roe, J.-S.; Seol, J.-H.; Kim, H.-R.; Cho, E.-J.; Youn, H.-D. pVHL-mediated transcriptional repression of c-Myc by recruitment of histone deacetylases. Mol. Cells 2012, 33, 195–201. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, P.; Sun, X.; Li, H.; Liu, Y.; Cui, Y.; Tian, L.; Cheng, Y. c-Myc Targets HDAC3 to Suppress NKG2DL Expression and Innate Immune Response in N-Type SCLC through Histone Deacetylation. Cancers 2022, 14, 457. https://doi.org/10.3390/cancers14030457
Zhao P, Sun X, Li H, Liu Y, Cui Y, Tian L, Cheng Y. c-Myc Targets HDAC3 to Suppress NKG2DL Expression and Innate Immune Response in N-Type SCLC through Histone Deacetylation. Cancers. 2022; 14(3):457. https://doi.org/10.3390/cancers14030457
Chicago/Turabian StyleZhao, Peiyan, Xiaodan Sun, Hui Li, Yan Liu, Yanan Cui, Lin Tian, and Ying Cheng. 2022. "c-Myc Targets HDAC3 to Suppress NKG2DL Expression and Innate Immune Response in N-Type SCLC through Histone Deacetylation" Cancers 14, no. 3: 457. https://doi.org/10.3390/cancers14030457
APA StyleZhao, P., Sun, X., Li, H., Liu, Y., Cui, Y., Tian, L., & Cheng, Y. (2022). c-Myc Targets HDAC3 to Suppress NKG2DL Expression and Innate Immune Response in N-Type SCLC through Histone Deacetylation. Cancers, 14(3), 457. https://doi.org/10.3390/cancers14030457