Simple Summary
Natural killer group 2, member D ligand (NKG2DL) is the most relevant ligand of NK cells to perform immune surveillance and is rarely expressed in most small cell lung cancer (SCLC) with the unclear mechanism. This study aimed to investigate the mechanisms underlying the NKG2DL deficiency in C-MYC (MYC)-amplificated N-type SCLC (SCLC-N) with less immune infiltrate. Our data showed that c-Myc was the suppressor of NKG2DL in SCLC-N. Further, c-Myc suppressed the transcription of NKG2DL by recruiting HDAC3 to deacetylate H3K9ac at the promoter of MICA and MICB in SCLC-N and inhibited the cytotoxicity of NK cells. The above findings revealed the role of c-Myc/HDAC3 axis in the regulation of NKG2DL expression, supplying a new perception for comprehending the mechanism of SCLC-N immune escape, which was poorly understood and providing the therapeutic targets that SCLC-N may benefit from.
Abstract
SCLC is an aggressive malignancy with a very poor prognosis and limited effective therapeutic options. Despite the high tumor mutational burden, responses to immunotherapy are rare in SCLC patients, which may be due to the lack of immune surveillance. Here, we aimed to examine the role and mechanism of oncogene MYC in the regulation of NKG2DL, the most relevant NK-activating ligand in SCLC-N. Western Blotting, Immunofluorescence, flow cytometry, quantitative real-time PCR (qRT-PCR), Co-Immunoprecipitation (Co-IP), chromatin immunoprecipitation (ChIP), and Cytotoxicity assay were used on H2227 cells, H446 cells, and other SCLC cell lines, and we found that c-Myc negatively regulated NKG2DL expression in SCLC-N cells. Mechanistically, c-Myc recruited HDAC3 to deacetylate H3K9ac at the promoter regions of MICA and MICB, suppressing the MICA/B expression of SCLC-N cells and the cytotoxicity of NK cells. Treatment with selective HDAC3 inhibitor up-regulated the expression of NKG2DL on SCLC-N cells and increased the cytotoxicity of NK cells. Furthermore, analysis of the CCLE and Kaplan-Meier plotter data performed the negative correlation between MYC and NKG2DL in SCLC-N cells and the correlation with the prognosis of lung cancer patients. Collectively, the results provided the new insight into the role and mechanism of c-Myc/HDAC3 axis in NKG2DL expression and innate immune escape of SCLC-N, suggesting the potential target for SCLC-N immunotherapy.
1. Introduction
Small cell lung cancer (SCLC), an extremely malignant subtype of lung cancer, is featured by rapid growth and tendency to metastasize, with a dismal prognosis and high relapse rate. Among all patients with SCLC, about two-thirds are diagnosed with advanced-stage disease that is generally accompanied by distant metastasis and has a five-year survival rate of less than 5% [1]. SCLC is sensitive to chemoradiotherapy, but most patients experience disease relapse and drug resistance within six months [2]. In the immunotherapy era, despite the addition of immunotherapy to frontline chemotherapy, the absolute improvements in progression-free survival (PFS) and overall survival (OS) are modest and the effective rate of immune checkpoint inhibitor monotherapy is only 10–20% [3,4,5,6]. Therefore, there is an urgent need to identify new targets or treatment modalities to improve the effectiveness of immunotherapy for SCLC. Abundant clinical trials of immunotherapy that do not differentiate among SCLC patients are underway, but the results of the experiments are disappointing.
In SCLC, responses to immune checkpoint blockade (ICB) are rare, which may be related with the low infiltration by immune cells, especially cytotoxic lymphocytes [7,8]. SCLC has an immune cold tumor microenvironment (TME) with few infiltrated cytotoxic lymphocytes and one-fifth total immune cells compared with NSCLC [9]. Antitumor activity of the immune system largely depends on cytotoxic cells, T cells, and NK cells. While T cells depend on specific antigens and act as an important component of adaptive immune response, NK cells are part of innate immunity and recognize tumors by germline-encoded patterns [10]. NK cells are critical in preventing lung tumor growth, as depletion of NK cells were shown to facilitate lung cancer initiation and metastasis in experimental models [11,12]. They are activated by natural killer group 2, member D ligand (NKG2DL) present on the surface of tumor cells and attack tumor cells by secreting cytotoxic proteins, such as perforin and granzymes [13,14]. In humans, NKG2DL can be classified into two subsets, MICA/B and ULBP. While mice only have orthologs of human RAET1 genes, including Rae1α/β/γ/δ/ε, MULT1, and H60a/b/c [15]. NKG2D is expressed on cytotoxic T cells, and NKG2DL can stimulate cytotoxicity of T cells [16]. Our previous research found that NKG2DL could recruit the cytotoxicity immune cells into the tumor nest [17]. However, according to reports, SCLC cell lines and patient-derived SCLC showed a significantly lower level of total NKG2DL compared with NSCLC cell lines [18], suggesting that NKG2DL might be the key molecule that modulates the immunosuppressive TME of SCLC.
SCLC has been treated as a single disease without patient stratification and exhibits genetic loss of both tumor suppressors RB1 and TP53, along with mutually exclusive expression of MYC paralog (MYC, MYCN, MYCL) [19,20,21,22,23]. Nowadays, next-generation gene sequencing-based gene expression analysis of human tumors and cell lines revealed that SCLC comprised four distinct subtypes based on the expression of ASCL1(SCLC-A), NeuroD1(SCLC-N), YAP1(SCLC-Y), and POU2F3(SCLC-P) [24]. The research on SCLC typing is endless, and the typing methods are not completely consistent. The researchers analyzed the sequencing results of patients enrolled in the IMpower133 study and named the ASCL1−NeuroD1−POU2F3− tumor tissues as subtype-I (SCLC-I) with high expression of inflammatory genes and increased number of immune cells including T cells, NK cells, and macrophages [8]. Survival analysis showed that SCLC-I tumors derived greater benefit from ICB. According to the reports that MYC drove the SCLC-N and SCLC-Y subtype of SCLC in a temporal evolution by reprograming neuroendocrine fate [25], SCLC-N cell lines highly expressed c-Myc with low level of NKG2DL and showed less immune infiltrate than other types [26], and SCLC-Y cell lines expressed low level of c-Myc with high level of NKG2DL [8], we speculated that MYC may contribute to the construction of SCLC-N immune microenvironment and the effect of immunotherapy through modulating NKG2DL.
Here, the study aimed to identify the c-Myc-dependent mechanism of suppressing NKG2DL expression and innate immune response in SCLC-N and found that c-Myc targeted HDAC3 to deacetylate H3K9ac at NKG2DL promoter and inhibited the transcription of NKG2DL in SCLC-N cells. The study might suggest the inhibition of HDAC3 to be the selectively beneficial therapeutic approaches for MYC-amplificated SCLC-N.
2. Materials and Methods
2.1. Cell Culture
Human SCLC cell lines NCI-H2227 (H2227), NCI-H446 (H446), NCI-H69 (H69), NCI-H524 (H524), and NCI-H196 (H196) (American Type Culture Collection, Manassas, VA, USA) were maintained at 37 °C in RPMI-1640 medium or Dulbecco’s Modified Eagle Media supplemented with 10% (v/v) FBS and antibiotics (100 IU/mL of penicillin and streptomycin). Human malignant non-Hodgkins Lymphoma cell line NK-92MI were maintained at 37 °C in NK-92MI special complete medium (Procell, Wuhan, China). All the cell lines were cultured in a 5% CO2 in air humidified incubator.
2.2. Plasmids and Transfection
For the overexpression of c-Myc, full-length MYC encoding sequence (CDS) (Gene ID: 4609) was subcloned into CMV-MCS-IRES-EGFP-SV40-Neomycin vector (MYC-OE, GENECHEM, Shanghai, China). The empty vector was used as a negative control. All plasmids were isolated using TIANprep Mini Plasmid Kit (DP103, TIANGEN, Beijing, China) and transfected using Lipofectamine 3000 Reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacture’s protocol. After 48 or 72 h, the cells were collected for the following studies.
2.3. MYC siRNA Transfection
Endogenous c-Myc expression in H446 cells and H2227 cells was reduced using transient siRNA transfection. Around 24 h post-seeding when reaching 50% cell density, the cells were transfected with either 40 nM of MYC siRNAs (siMYC#1, GAGGAUAUCUGGAAGAAAUTT; siMYC#2, GCUUGUACCUGCAGGAUCUTT; siMYC#3, GGAAGAAAUCGAUGUUGUUTT) or 40 nM of Negative Control siRNA (NC-siRNA, UUCUCCGAACGUGUCACGUTT) (GeneParma, Shanghai, China). The transfection was performed for 4 h using a dilution of 1:50 in Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) in serum-free DMEM followed by a 48 or 72 h incubation in FBS containing media.
2.4. Inhibitor Incubation
H2227, H446, and H196 cells were co-cultured with Entinostat (HY-12163, MCE, Monmouth Junction, NJ, USA) or 10058F4 (SC6650, Beyotime, Shanghai, China) for 48 h and collected for the following studies. The H2227 and H446 cells transfected with MYC-OE plasmid for 48 h were incubated with RGFP966 (HY-13909, MCE, Monmouth Junction, NJ, USA). After 48 h, the cells were collected for flow cytometry analysis.
2.5. Cell Proliferation Analysis
SCLC cells (5 × 103) were seeded into 96-well plates treated with different concentrations of Entinostat (1 μM, 2 μM, 4 μM, 8 μM, 16 μM, and 32 μM). After 48 h, the numbers of live cells were detected by Cell Counting Kit-8 (CCK8) (C0038, Beyotime, Shanghai, China). Absorbance at 450 nm was measured. Cell viability = (ODsample − ODspontaneous)/(ODcontrol − ODspontaneous) × 100%.
2.6. Cell Migration Analysis
SCLC cells (5 × 104) were seeded into the chamber of BD BioCoat Matrigel coated plates (354480, BD, Franklin Lakes, NJ, USA). After 24 h, migrated cells were recorded after staining with crystal violet.
2.7. Western Blotting
Western blotting was conducted as described previously [17]. The parental and 72 h-transfected cells were lysed in RIPA buffer containing 1 mM phenylmethylsulphonyl fluoride. The supernatant was collected after the centrifuge at 15,000× g for 15 min at 4 °C and quantified using Enhanced BCA Protein Assay Kit (P0010, Beyotime, Shanghai, China). Equal amounts of protein were fractionated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), blocked with skimmed milk and then incubated overnight at 4 °C with different primary antibodies in buffer containing 5% skimmed milk. Membranes were washed with TBS containing 0.05% Tween-20 three times, incubated with a secondary antibody for 1 h at room temperature, and then washed again three times. The blotting was examined using chemiluminescence (P0018AM, Beyotime, Shanghai, China) and analyzed with Image J software (version 1.8.0). Antibodies against GAPDH (AP0066, bioworld, MN, USA), c-Myc (WL01781, Wanleibio, Shenyang, China; 10828-1-AP, Proteintech, Chicago, IL, USA), NeuroD1 (4373S, CST, Boston, MA, USA), YAP1 (14074S, CST, Boston, MA, USA), ASCL1 (ab74065, Abcam, Cambridge, UK), HDAC1 (WL01297, Wanleibio, Shenyang, China), HDAC2 (WL03149, Wanleibio, Shenyang, China), HDAC3 (WL02946, Wanleibio, Shenyang, China), and horseradish peroxidase-conjugated goat anti-rabbit IgG (RM3002, Ray Antibody Biotech, Beijing, China) were used.
2.8. Immunofluorescence Analysis
The cells were seeded in 24-well plates. After overnight incubation, the cells were washed with PBS and fixed with 4% paraformaldehyde for 20 min. After being washed in PBS, the cells were permeabilized with 0.1% Triton X-100 for 10 min and blocked with 5% bovine serum albumin. After 1 h incubation the cells were stained with rabbit anti-human c-Myc (10828-1-AP, Proteintech, Chicago, IL, USA, 1:500) at 4 °C overnight, followed by being incubated with fluorescein (FITC)-conjugated goat anti-rabbit IgG (SA00003-2, Proteintech, Chicago, IL, USA, 1:100) for 1 h at room temperature. After being washed, the cells were counterstained with antifade mounting medium with DAPI (P0131, Beyotime, Shanghai, China). The images of the cells were taken under the fluorescence microscope.
2.9. Flow Cytometry Analysis
The cultured cells were harvested, washed twice with PBS containing 2% FBS, and stained with PE-labelled mouse anti-human MICA/MICB (320906) or PE-labelled mouse IgG2a κ isotype ctrl antibody (400212, BioLegend, San Diego, CA, USA) for 30 min on ice in the dark followed by being washed twice with PBS containing 2% FBS. All stained cells were analyzed by FACSCanto II (BD, Franklin Lakes, NJ, USA). Live cells were carefully gated by forward and side scatter. Data were analyzed using FlowJo software (version 10).
2.10. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR) Analysis
Total RNA was extracted using TRIzol Reagent (15596026, Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions and reverse transcribed using First Strand cDNA Synthesis Kit (TOYOBO, Shanghai, China). Analyses of qRT-PCR were performed using SYBR qPCR Mix (QPS-201, TOYOBO, Shanghai, China) on a system (Cobas z480, Roche, Basel, Switzerland). The primers used for analysis were shown in Supplementary Table S1. The fold change of target mRNA expression was calculated based on the threshold cycle (Ct). ACTB was used as an internal control. Relative mRNA expression levels were analyzed using the 2−ΔΔCt method.
2.11. Co-Immunoprecipitation (Co-IP) Assay
The lysates of the cultured H446 and H2227 cells were harvested and subjected to c-Myc immunoprecipitation using anti-c-Myc antibody (4 μg, 10828-1-AP, Proteintech, Chicago, IL, USA). Antibody-protein complexes were captured using 20 μL protein A + G sepharose beads (P2012, Beyotime, Shanghai, China). Immunoprecipitates were then analyzed by Western Blotting. The HRP conjugated light-chain specific mouse anti-rabbit IgG antibody (93702, CST, Boston, MA, USA) was used as secondary antibody, and rabbit IgG (3900S, CST, Boston, MA, USA) was used as a negative control.
2.12. Chromatin Immunoprecipitation (ChIP)-qPCR Assay
The predicted promoter sequence of MICA and MICB promoters were searched by UCSC Genome Browser (http://genome.ucsc.edu/; accessed on 4 February 2021) and Cistrome Data Browser (http://cistrome.org/db/#/; accessed on 4 February 2021). ChIP assay kit (P2078, Beyotime, Shanghai, China) was used for ChIP assay following the manufacturer’s instructions. Briefly, cells were fixed with 1% formaldehyde and quenched with 0.125 M glycine. Next, the cells were sonicated using an ultrasonic cell disruptor in the lysis buffer. DNA was immunoprecipitated with either control IgG (B900610, Proteintech, Chicago, IL, USA), H3K9ac (AF5611, Beyotime, Shanghai, China), H3K14ac (AF5614, Beyotime, Shanghai, China), H3K27ac (AF5620, Beyotime, Shanghai, China), HDAC3 (10255-1-AP, Proteintech, Chicago, IL, USA), or c-Myc (10828-1-AP, Proteintech, Chicago, IL, USA) primary antibody. RNA and protein were digested using RNase A (ST576, Beyotime, Shanghai, China) and Protein K (ST533, Beyotime, Shanghai, China), respectively. DNA were purified using DNA Purification Kit (D0033, Beyotime, Shanghai, China) followed by qPCR analysis. The qPCR primers are listed in Supplementary Table S2.
2.13. Cytotoxicity Assay
H2227, H196, or H446 cells (T) were seeded into 96-well plates (5 × 103 cells per well) and cultured overnight. Next, the SCLC cell lines were co-cultured with NK-92MI cells (E) at different E/T ratio (2:1, 10:1, 50:1). After 4 h, the lysate of SCLC cells was tested using LDH Cytotoxicity Assay Kit (C0017, Beyotime, Shanghai, China). Absorbance at 490 nm was measured using a microplate reader (CLARIOstar, BMG LABTECH, Offenburg, Germany). Percent cell death was calculated as (ODsample − ODspontaneous)/(ODMax − ODspontaneous) × 100%.
2.14. Analysis of Differential Gene Expression from Cancer Cell Line Encyclopedia (CCLE) Dataset
Gene expression data of the SCLC cell lines were downloaded from CCLE (https://sites.broadinstitute.org/ccle; accessed on 9 March 2021). The correlation between MYC expression and NKG2DL expression was evaluated by Linear Regression test.
2.15. Kaplan-Meier Survival Analysis
To investigate the association between MYC, MICA, or MICB and the survival of patients, we downloaded the information related to the survival time of patients with lung cancer from Kaplan-Meier plotter (http://kmplot.com/analysis/; accessed on 8 January 2022).
2.16. Statistical Analysis
All experiments were performed in triplicate. Data were analyzed using GraphPad Prism software (version 8.0), and the results are presented as the mean ± SD. Comparisons between groups were conducted using analysis of unpaired t tests. Correlations between MYC and NKG2DL in SCLC cell lines were analyzed by Spearman’s rank test. p value < 0.05 was considered statistically significant.
3. Results
3.1. c-Myc, NKG2DL Expression and Susceptibility to NK Cell Killing of SCLC Cell Lines
To explore the expression levels of c-Myc and NKG2DL of SCLC cell lines, we firstly detected the expression of c-Myc, key transcription regulators of SCLC-N and SCLC-Y (ASCL1, NeuroD1, and YAP1) [24] and major NKG2DL (MICA, MICB, ULBP1-3) [18] in H2227, H446, H69, H524, and H196 cells by Western Blotting, flow cytometry, and qRT-PCR. Consistent with the previous reports, H2227 and H446 cells were MYC-amplificated SCLC-N cells [25], both of which expressed c-Myc and neuroendocrine markers (Figure 1A and File S1). While the expression levels of MICA/B were exactly different: H446 and H196 cells expressed the highest level of MICA/B and H69 expressed the lowest level of MICA/B; H2227 and H524 cells expressed equivalent levels of MICA/B (Figure 1B). The qRT-PCR results showed that the expression level of MYC was higher in H2227 cells than in H446 cells, while the expression level of MICA was lower in H2227 cells than in H446 cells (Figure 1C). Further, the immunofluorescence assay and cytotoxicity assay were used to verify the c-Myc expression and susceptibility to NK cell killing of H2227, H446, and H196 cells. We found that compared to H446 and H196 cells, H2227 expressed a higher level of c-Myc and obtained a lower susceptibility to the killing by NK-92MI cells (Figure 1D,E). The above results indicated that c-Myc may be negatively correlated with NKG2DL expression and susceptibility to NK cell killing in SCLC-N cells.
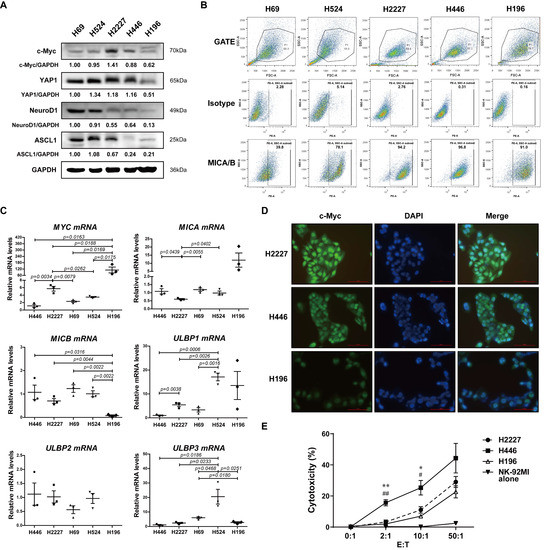
Figure 1.
The expression of c-Myc and NKG2DL, and the susceptibility to NK-92MI cell killing in SCLC cell lines. (A) Western blotting spots of c-Myc, YAP1, NeuroD1, ASCL1, and GAPDH in H69, H524, H2227, H446, and H196 cells. (B) Flow cytometry analysis of MICA/B expression levels on the surface of the above cells. (C) qRT-PCR analysis of MYC, MICA, MICB, and ULBP1-3 mRNA expression levels of the above cells. (D) Immunofluorescence staining of c-Myc in H2227, H446, and H196 cells (40×). Scale bar = 100 μm. (E) Cytotoxicity assay of the susceptibility of H2227, H446, and H196 cells to NK-92MI cell killing, respectively. Data are represented as mean ± SD (n = 3). *, #, p < 0.05; **, ##, p < 0.01. * p: H446 group vs. H2227 group; # p: H446 group vs. H196 group.
3.2. c-Myc Inhibited Expression of NKG2DL and Susceptibility to NK Cell Killing in SCLC-N Cells
To verify whether c-Myc was involved in regulating the expression of NKG2DL in SCLC-N cells, we constructed the overexpression plasmid containing the CDS of human MYC gene and named it MYC-OE. H2227 and H446 cells were transfected with MYC-OE and the overexpression of c-Myc in H2227 cells was determined by western blotting (Figure 2A and Figure S1A). Further, flow cytometry analysis showed that the Median Fluorescence Intensity (MFI) of MICA/B and the percentage of MICA/B+ cells were lower in MYC-OE-transfected H2227 cells than in empty vector (EV)-transfected cells (Figure 2B). The results of qRT-PCR also showed that as MYC was up-regulated, the expression level of MICB in H2227 cells was down-regulated (Figure 2C). The cytotoxicity assay revealed that the susceptibility of H2227 cells to the killing by NK-92MI cells was decreased after MYC-OE transfection (Figure 2D).
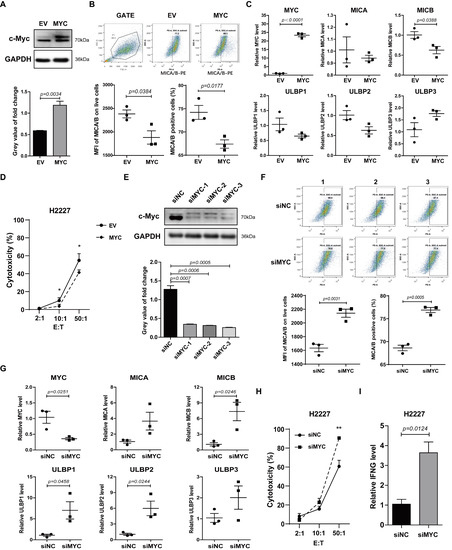
Figure 2.
c-Myc was a negative regulator of NKG2DL in H2227 cells. H2227 cells were transfected with MYC-OE or MYC siRNAs (40 pmol/mL). (A,E) Western blotting analysis of the c-Myc expression levels in the transfected H2227 cells. (B,F) Flow cytometry analysis of MICA/B expression on the transfected H2227 cells. (C,G) qRT-PCR analysis of MYC, MICA, MICB, and ULBP1-3 mRNA expression levels in H2227 cells after transfected with MYC-OE or MYC siRNA-3, respectively. (D,H) LDH analysis of the susceptibility of transfected H2227 cells to NK-92MI cell killing. (I) qRT-PCR analysis of IFNG mRNA expression levels in the transfected H2227 cells. Data are represented as mean ± SD (n = 3). *, p < 0.05; **, p < 0.01.
Next, we transfected the human MYC siRNA (siMYC) into H2227 cells to silence c-Myc. As a result, siMYC transfection silenced the expression of c-Myc (Figure 2E), up-regulated the expression of MICA/B protein (Figure 2F), and MICB, ULBP1, and ULBP2 mRNA (Figure 2G), enhanced the susceptibility to the killing by NK-92MI cells (Figure 2H) and the expression levels of IFNG in NK-92MI cells (Figure 2I). Similar results were obtained from H446 cells, in which c-Myc expression was up-regulated and silenced, respectively (Figures S1B and S2). The incubation of 10058F4 (the inhibitor of c-Myc) with SCLC cells obviously caused the co-cultured NK cells to aggregate into larger tumor spheres (Figure S3) which represented higher killing ability [27,28]. These data indicated a potential role for c-Myc in suppressing the expression of NKG2DL and anti-tumor immune response of NK cells in SCLC-N.
3.3. HDACs Involved in the Modulation of NKG2DL Expression in SCLC-N Cells
To explore the mechanism of c-Myc inhibiting MICA/B in SCLC, we used ChIP-qPCR assay to perform whether c-Myc directly binds to the promoter region of MICA or MICB. The data showed that c-Myc may not directly bind to the promoter of MICA or MICB in H2227 or H446 cells (Figure 3A). Recently, NKG2DL on SCLC-A cells was reported to be modulated by HDAC [18], which could interact with c-Myc to regulate gene expression [29]. Therefore, we speculated that c-Myc might regulate the expression of NKG2DL through HDACs in SCLC-N. Here, we found that the inhibitor of class I HDACs, the highly-expressed HDACs in the locally advanced, dedifferentiated, and strongly proliferating tumors [30], Entinostat, could inhibit the expression of HDAC1, HDAC2, and HDAC3, the three most studied class Ⅰ HDACs that this article focused on, with a final concentration greater than 2.5 μM in H2227 cells (Figure 3B), and inhibit the proliferation and migration of H2227 cells (Figure S4). Furthermore, the results of Flow Cytometry showed that Entinostat could up-regulate the MFI of MICA/B on H2227 cells (Figure 3C). Similar results were obtained from H446 cells (Figure 3D,E and Figure S4). Further, the results of cytotoxicity assay showed that Entinostat distinctly increased the susceptibility of H2227 cells and H446 cells to the killing by NK-92MI cells (Figure 3F,G). Intriguingly, the MFI of MICA/B and the susceptibility to NK cell killing of H196 cells, the SCLC-Y cells, were not significantly altered by Entinostat treatment (Figure 3H,I). The above results indicated that the class Ⅰ HDAC inhibitor, Entinostat, could induce the expression of MICA/B and improve the killing by NK cells of H2227 and H446 cells.
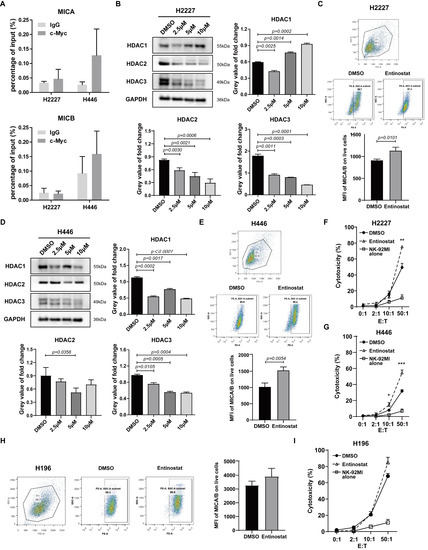
Figure 3.
HDAC inhibitor modulated NKG2DL expression and susceptibility to NK cell killing of SCLC-N cells. (A) ChIP-qPCR analysis of c-Myc enrichment at MICA and MICB promoters in H2227 and H446 cells. (B,D) Western blotting analysis of HDAC1, HDAC2, and HDAC3 in H2227 and H446 cells treated with or without Entinostat (2.5 μM, 5 μM, and 10 μM). (C,E,H) Flow cytometry analysis of MICA/B expression levels on H2227, H446, and H196 cells treated with or without Entinostat (10 μM). (F,G,I) LDH analysis of the susceptibility to NK-92MI cell killing of H2227, H446, and H196 cells treated with or without Entinostat (10 μM). Data are represented as mean ± SD (n = 3). * p < 0.05, ** p < 0.01, *** p < 0.001 vs. DMSO group.
3.4. c-Myc Regulated NKG2DL Expression on SCLC-N Cells through HDAC3
To clarify whether and which subtype of HDAC participated the modulation of c-Myc on NKG2DL expression in SCLC-N cells, we firstly detected the combination of c-Myc and HDAC1, HDAC2, or HDAC3 by Co-IP and found that only HDAC3 substantially interacted with c-Myc in H2227 and H446 cells (Figure 4A and Figure S5A). Further, the ChIP-qPCR analysis performed that HDAC3 could directly bind the promoter of MICA and MICB in control H2227 cells, and the binding was prevented by the transfection of siMYC (Figure 4B). Next, to confirm that c-Myc was indeed inhibiting the NKG2DL expression in SCLC-N cells through HDAC3, a highly selective HDAC3 inhibitor, RGFP966, was added to block the HDAC3 in H2227 cells after the transfection of MYC-OE. Since there is no effective inhibition of any other HDACs at concentrations of up to 15 μm [31,32], we chose the relatively high concentration, 10 μm, of RGFP966 to treat H2227 cells. The results of flow cytometry and LDH analysis showed that the percentage of MICA/B+ cells, MFI of MICA/B, and the susceptibility to NK-92MI cell killing of H2227 cells were decreased by the transfection of MYC-OE and recovered by RGFP966 (Figure 4C,D), suggesting that HDAC3 was the key molecule of c-Myc to suppress the expression of MICA/B and the susceptibility to NK cell killing of H2227 cells. Furthermore, we found RGFP966 could increase the acetylation of histone H3K9, H3K14, and H3K27 (Figure 4E), the possible sites of HDAC3 deacetylation [33,34,35], in H2227 cells. Importantly, the ChIP-qPCR assay confirmed that RGFP966 markedly increased the enrichment of H3K9ac, but not H3K14ac or H3K27ac, at the promoter of MICA and MICB in H2227 cells, which was recovered by the transfecting of siMYC (Figure 4F). The similar results were obtained from H446 cells (Figure S5). Collectively, these findings indicated that c-Myc suppressed the expression of NKG2DL in SCLC-N cells by recruiting HDAC3 to deacetylate H3K9ac at the promoter of NKG2DL.
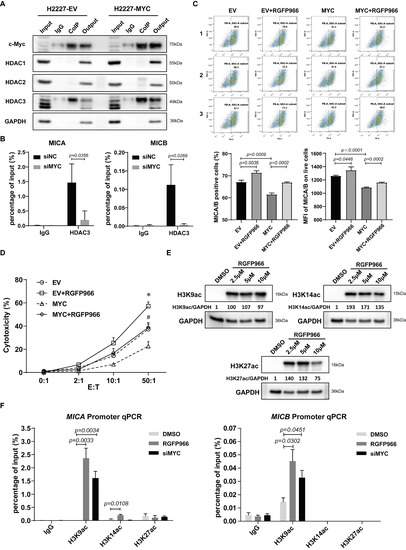
Figure 4.
c-Myc modulated HDAC3 to deacetylate histone H3K9ac at MICA and MICB promoters. (A) Co-IP assay of the binding of c-Myc with HDAC1, HDAC2, or HDAC3 in H2227 cells transfected with or without MYC-OE. (B) ChIP-qPCR analysis of the binding of HDAC3 and MICA/B promoter in MYC-silencing H2227 cells. (C) Flow cytometry analysis of MICA/B expression in H2227 cells transfected with EV or MYC-OE followed the treatment of RGFP966 (10 μM) or not. (D) LDH analysis of the susceptibility to NK-92MI cell killing of H2227 cells transfected with EV or MYC-OE followed by the treatment of RGFP966 (10 μM) or not. (E) Western blotting analysis of H3K9ac, H3K14ac, and H3K27ac in H2227 cells treated with or without RGFP966 (2.5 μM, 5 μM, and 10 μM). (F) ChIP-qPCR analysis of H3K9ac, H3K14ac, and H3K27ac enrichment at MICA promoter and MICB promoter in H2227 cells treated with or without RGFP966 (10 μM) or transfected with siMYC. Data are represented as mean ± SD (n = 3). *, #, p < 0.05; * p: EV + RGFP966 group vs. EV group; # p: MYC + RGFP966 group vs. MYC group.
3.5. Correlation between MYC and NKG2DL Expression in SCLC Cells and Lung Cancer Patients
In order to further determine the universality of the negative correlation between MYC and NKG2DL expression levels in SCLC cells, we analyzed the relationship between MYC and NKG2DL expression levels in online SCLC cell line data. CCLE data analysis showed that MYC mRNA expression levels were negatively associated with MICA and MICB mRNA expression levels in 18 SCLC-N cell lines [25] (p = 0.0167 and p = 0.0033, respectively) (Figure 5A, Table S3), but not in total 71 SCLC cell lines (Figure S6), indicating that the negative correlation between MYC and NKG2DL expression levels may only exist in SCLC-N. Due to the lack of studies on SCLC samples in the database, we analyzed the prognostic significance of MYC and MICA/B expression levels in lung cancer using Kaplan-Meier plotter to investigate the clinical significance of MYC and NKG2DL. We found that the patients with high MYC mRNA expression had worse OS (HR 1.38 [1.02–1.86], p = 0.0035), and the patients with high MICA mRNA expression had better OS (HR 0.7 [0.52–0.95], p = 0.019) (Figure 5B) which also suggested the negative correlation between MYC and MICA in lung cancer.
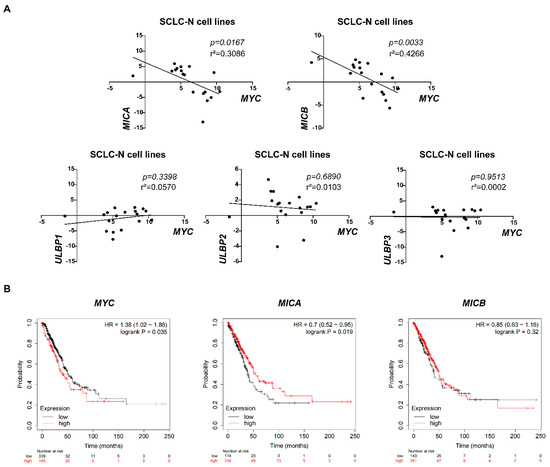
Figure 5.
The negative correlation between MYC and NKG2DL expression in SCLC-N cells and lung cancer patients. (A) CCLE data analysis of the correlation between MYC and MICA, MICB, ULBP1, ULBP2, or ULBP3 in SCLC-N cells lines, respectively (n = 18). (B) Kaplan-Meier survival curve of MYC, MICA, and MICB expression groups, respectively (n = 504).
4. Discussion
NKG2DL has been considered as an activating signal on the surface of cancer cells to trigger the cytotoxicity cells including NK cells and CD8+ T cells and was the substantial mechanism of immune surveillance to initiate the immune response to find and eliminate NKG2DL+ tumor cells. In this study, we found that compared to the SCLC-Y cells, which was reported to express high level of inflammation gene and be infiltrated by more immune cells [8,36], SCLC-N cells expressed a lower level of NKG2DL, especially MICA/B (Figure 1B), which may be one of the reasons that SCLC-N was infiltrated with less immune cells [26]. Further, we found MICA and MICB, the most widely expressed NKG2DL, on SCLC-N cells were suppressed by MYC (Figure 2 and Figure S2), whose encoded protein, c-Myc, was higher expressed in SCLC-N cells than in other types of SCLC cells [8], indicating the key role of MYC in the immune characteristics of SCLC. In addition, previous studies reported that H2227 and H446 cells were SCLC-N cells [25] while H196 cells were SCLC-Y cells [24], which was consistent with our results that H2227 and H446 cells expressed higher level of c-Myc than H196 cells (Figure 1A,D). Since the structure and regulatory mechanism of ULBP family was quite different from MIC family [14,37,38,39], ULBPs may not be mainly regulated by MYC, which is consistent with the increased level of ULBP3 in both MYC-overexpressed and -silenced cells (Figure 2). Notably, among the five major NKG2DL, MICB was the main NKG2DL regulated by MYC in H2227 cells (Figure 2C), while MICA was the main one regulated by MYC in H446 cells (Figure S2C), indicating that the regulatory pathways of the same NKG2DL may be different in distinct tumor cells, which required to be further studied.
Additionally, the study on the mechanism revealed that c-Myc recruited HDAC3 to deacetylate histone H3K9ac, which was bound to the promoter region of NKG2DL (Figure 4), thereby suppressing the transcription of NKG2DL in SCLC-N cells and inhibiting the cytotoxic effect of NK cells (Figure 2 and Figure 6). Consistently, Entinostat up-regulated NKG2DL expression and susceptibility to NK cell killing in SCLC-N, but not in SCLC-Y (Figure 3). Supporting our finding, rising evidence of RNA-seq showed the HDAC inhibitor sensitive gene signature of NEUROD1-subtype SCLC, but the immune system-enriched HDAC inhibitor resistant gene signature of SCLC-Y [40], which was consistent with the enrichment of immune signaling pathways in SCLC-Y [8,41,42]. However, it appeared that c-Myc did not up-regulate the expression of HDAC3 in SCLC-N, as the HDAC3 level in input was not up-regulated by overexpression of MYC (Figure 4A). The specific inhibitor of HDAC3, RGFP966, could recover the expression levels of NKG2DL and the susceptibility to the killing by NK cells that down-regulated by the overexpression of MYC (Figure 4), which was consistent with the reports that NKG2DL in SCLC-A subtype cells was suppressed by deacetylation [18]. Unexpectedly, we found that high concentration (10 µM) of RGFP966 did not significantly alter the acetylation levels of H3K9 and H3K14, but slightly down-regulated the level of H3K27 (Figure 4E), indicating that the compensatory mechanisms may be activated. Despite both the promoters of MICA and MICB could be regulated by HDAC3 (Figure 4B,F), only MICB mRNA was regulated by MYC (Figure 2C,G), indicating that MICA might be regulated by the unknown specific mechanism which needed further study. Moreover, SCLC-I tumors, which obtained the highest total immune infiltrate and cytolytic activity score [8], expressed higher level of NKG2DL. Whether the highly expressed NKG2DL are regulated by acetylation and the distinction in the acetylation level of NKG2DL in different types of SCLC is what we will focus on next. Considering for the increased susceptibility of SCLC-N cells to the killing of NK cells by both c-Myc inhibitor and HDAC inhibitor, we speculated that the application of HDAC inhibitor, especially HDAC3 inhibitor, might be an effective immunotherapy strategy targeting innate immunity for MYC-amplificated SCLC-N and could solve the problem that c-Myc is largely unstructured, falling in the category of ontrinsically disordered proteins [43] and is difficult to be targeted by small molecule inhibitors. Similarly, the effect of HDAC inhibitors and MYC on the cytotoxicity of NK cells is worthy of further study.
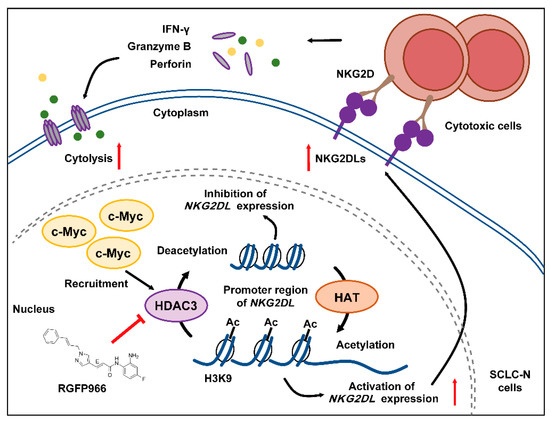
Figure 6.
Schematic model of c-Myc regulating the expression of NKG2DL and the cytolysis in SCLC-N. c-Myc recruited and combined with HDAC3, which may facilitate the deacetylation of histone H3K9ac at the NKG2DL promoter region of SCLC-N cells, resulting in the decreased transcription level of NKG2DL and the inability to activate cytotoxic cells and the reduced cell lysis. HDAC3 inhibitor could reverse the decline of the cytotoxic cell killing effect by up-regulating the expression of NKG2DL.
Here, it was found that c-Myc in SCLC-N cells could combined with HDAC3, but not HDAC1 or HDAC2 (Figure 4A), indicating that HDAC3 in SCLC-N cells might be combined with and recruited by c-Myc due to its special structure. In Non-Hodgkin B-cell lymphoma and acute myeloid leukemia, c-Myc was also reported to regulate the gene transcription by recruiting HDAC3 [44,45], while HDAC1/2 were most likely to be recruited to the promoter of MYC and regulated the transcription of MYC [46,47]. The above results were consistent with that HDAC1 and HDAC2 formed the functional complex with mSin3A, NuRD, and RCOR1/Co-REST, while HDAC3 formed a functional complex with N-CoR [48]. Due to the particularity of HDAC3, the mechanism of HDAC3 regulating NKG2DL transcription deserved further study. We found that HDAC3 may regulate the accessibility of the promoter and inhibit the transcription of NKG2DL by deacetylating histone H3K9ac and the treatment with HDAC3 specific inhibitors increased the acetylation level of H3K9 (Figure 4F) and up-regulated NKG2DL expression in SCLC-N cells (Figure 4C). Since the binding of HDAC3 and the promoters of MICA/B and the level of H3K9ac were prevented by the silencing of MYC (Figure 4B,F), it is further demonstrated the c-Myc/HDAC3 axis in the modulating of NKG2DL expression. This study proved the role and mechanism of HDAC3 in c-Myc-regulated SCLC-N immune characteristics, implying that HDAC3 may be a new target for the treatment of SCLC-N.
5. Conclusions
To date, evidence for c-Myc to participate in the regulation and mechanism of SCLC immune microenvironment and immune characteristics is still lacking. Based on our study, we confirmed that in MYC-amplificated SCLC cells, c-Myc was involved in regulating the expression of NKG2DL, the activating ligand for cytotoxic cells such as NK cells. The main mechanism might be that c-Myc recruited HDAC3 to deacetylate the histone H3K9ac, which bind to the promoter region of NKG2DL, thereby inhibiting the transcription of NKG2DL. Our findings suggested that the role of c-Myc in SCLC-N cells as a key modulator for immune characteristics and immunotherapy should be noted; compared with c-Myc, HDAC3 might be a better target for SCLC immunotherapy.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers14030457/s1, Figure S1: Expression levels of c-Myc in the transfected H2227 and H446 cells. Figure S2: c-Myc was a negative regulator of NKG2DL in H446 cells. Figure S3: The inhibition of c-Myc in SCLC-N cells enhanced the killing ability of NK-92MI cells in the co-culture system. Figure S4: Effect of Entinostat on proliferation and metastasis of H2227 and H446 cells. Figure S5: c-Myc modulated HDAC3 to deacetylate histone H3K9ac at NKG2DL promoter in H446 cells. Figure S6: Correlation between MYC and NKG2DL expression levels in SCLC cells. Table S1: Primers of specific genes used for qRT-PCR. Table S2: Primers used for ChIP-qPCR. Table S3: The expression levels of MYC, MICA, MICB, ULBP1-3 mRNA in 18 SCLC-N cell lines (data form CCLE). File S1: Original Western blots figures.
Author Contributions
Conceptualization: P.Z. and Y.C. (Ying Cheng); Data curation: P.Z., X.S. and Y.C. (Yanan Cui); Formal analysis: P.Z., X.S. and Y.L.; Funding acquisition: P.Z., H.L. and Y.C. (Ying Cheng); Methodology: P.Z., X.S. and Y.L.; Project administration: H.L. and Y.L.; Resources: Y.C. (Ying Cheng); Software: Y.C. (Yanan Cui); Supervision: L.T.; Validation: L.T.; Writing—Original draft: P.Z.; Writing—Review and editing: P.Z. and Y.C. (Ying Cheng). All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (Grant Number: 82103343), Health Commission of Jilin Province (Grant Number: 2021JC097), Jilin Province Development and Reform Commission (Grant Number: 2021C043-1), Science and Technology Agency of Jilin Provincial (Grant Number: 20200201518JC).
Institutional Review Board Statement
Not applicable for no studies involving humans or animals.
Informed Consent Statement
Not applicable for no studies involving humans.
Data Availability Statement
Not applicable.
Acknowledgments
The authors appreciate Shaowei Lan and Chenchen Tang for technical assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nicholson, A.G.; Chansky, K.; Crowley, J.; Beyruti, R.; Kubota, K.; Turrisi, A.; Eberhardt, W.E.E.; van Meerbeeck, J.; Rami-Porta, R. The International Association for the Study of Lung Cancer Lung Cancer Staging Project: Proposals for the Revision of the Clinical and Pathologic Staging of Small Cell Lung Cancer in the Forthcoming Eighth Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016, 11, 300–311. [Google Scholar] [CrossRef]
- Rossi, A.; di Maio, M.; Chiodini, P.; Rudd, R.M.; Okamoto, H.; Skarlos, D.V.; Früh, M.; Qian, W.; Tamura, T.; Samantas, E.; et al. Carboplatin- or cisplatin-based chemotherapy in first-line treatment of small-cell lung cancer: The COCIS meta-analysis of individual patient data. J. Clin. Oncol. 2012, 30, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef]
- Chung, H.C.; Piha-Paul, S.A.; Lopez-Martin, J.; Schellens, J.H.M.; Kao, S.; Miller, W.H., Jr.; Delord, J.P.; Gao, B.; Planchard, D.; Gottfried, M.; et al. Pembrolizumab After Two or More Lines of Previous Therapy in Patients with Recurrent or Metastatic SCLC: Results From the KEYNOTE-028 and KEYNOTE-158 Studies. J. Thorac. Oncol. 2020, 15, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Hamilton, G.; Rath, B. Immunotherapy for small cell lung cancer: Mechanisms of resistance. Expert Opin. Biol. Ther. 2019, 19, 423–432. [Google Scholar] [CrossRef]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021. [Google Scholar] [CrossRef]
- Busch, S.E.; Hanke, M.L.; Kargl, J.; Metz, H.E.; MacPherson, D.; Houghton, A.M. Lung Cancer Subtypes Generate Unique Immune Responses. J. Immunol. 2016, 197, 4493–4503. [Google Scholar] [CrossRef]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar] [CrossRef]
- Cong, J.; Wang, X.; Zheng, X.; Wang, D.; Fu, B.; Sun, R.; Tian, Z.; Wei, H. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab. 2018, 28, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Best, S.A.; Hess, J.B.; Souza-Fonseca-Guimaraes, F.; Cursons, J.; Kersbergen, A.; Dong, X.; Rautela, J.; Hyslop, S.R.; Ritchie, M.E.; Davis, M.J.; et al. Harnessing Natural Killer Immunity in Metastatic SCLC. J. Thorac. Oncol. 2020, 15, 1507–1521. [Google Scholar] [CrossRef]
- Lowry, L.E.; Zehring, W.A. Potentiation of Natural Killer Cells for Cancer Immunotherapy: A Review of Literature. Front. Immunol. 2017, 8, 1061. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Verneris, M.R.; Karimi, M.; Baker, J.; Jayaswal, A.; Negrin, R.S. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood 2004, 103, 3065–3072. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Yang, L.; Li, X.; Lu, W.; Lu, F.; Wang, S.; Wang, Y.; Hua, L.; Cui, C.; Dong, B.; et al. Rae1 drives NKG2D binding-dependent tumor development in mice by activating mTOR and STAT3 pathways in tumor cells. Cancer Sci. 2020, 111, 2234–2247. [Google Scholar] [CrossRef]
- Zhu, M.; Huang, Y.; Bender, M.E.; Girard, L.; Kollipara, R.; Eglenen-Polat, B.; Naito, Y.; Savage, T.K.; Huffman, K.E.; Koyama, S.; et al. Evasion of Innate Immunity Contributes to Small Cell Lung Cancer Progression and Metastasis. Cancer Res. 2021, 81, 1813–1826. [Google Scholar] [CrossRef]
- Brägelmann, J.; Böhm, S.; Guthrie, M.R.; Mollaoglu, G.; Oliver, T.G.; Sos, M.L. Family matters: How MYC family oncogenes impact small cell lung cancer. Cell Cycle 2017, 16, 1489–1498. [Google Scholar] [CrossRef]
- Dammert, M.A.; Bragelmann, J.; Olsen, R.R.; Bohm, S.; Monhasery, N.; Whitney, C.P.; Chalishazar, M.D.; Tumbrink, H.L.; Guthrie, M.R.; Klein, S.; et al. MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat. Commun. 2019, 10, 3485. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef]
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Soltero, D.; et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020, 38, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Quintanal-Villalonga, Á.; Gao, V.R.; Xie, Y.; Allaj, V.; Chaudhary, O.; Masilionis, I.; Egger, J.; Chow, A.; Walle, T.; et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell 2021. [Google Scholar] [CrossRef]
- Liu, M.; Meng, Y.; Zhang, L.; Han, Z.; Feng, X. High-efficient generation of natural killer cells from peripheral blood with preferable cell vitality and enhanced cytotoxicity by combination of IL-2, IL-15 and IL-18. Biochem. Biophys. Res. Commun. 2021, 534, 149–156. [Google Scholar] [CrossRef]
- Kloss, S.; Oberschmidt, O.; Morgan, M.; Dahlke, J.; Arseniev, L.; Huppert, V.; Granzin, M.; Gardlowski, T.; Matthies, N.; Soltenborn, S.; et al. Optimization of Human NK Cell Manufacturing: Fully Automated Separation, Improved Ex Vivo Expansion Using IL-21 with Autologous Feeder Cells, and Generation of Anti-CD123-CAR-Expressing Effector Cells. Hum. Gene Ther. 2017, 28, 897–913. [Google Scholar] [CrossRef]
- Wang, J.; Elahi, A.; Ajidahun, A.; Clark, W.; Hernandez, J.; Achille, A.; Hao, J.-h.; Seto, E.; Shibata, D. The interplay between histone deacetylases and c-Myc in the transcriptional suppression of HPP1 in colon cancer. Cancer Biol. Ther. 2014, 15, 1198–1207. [Google Scholar] [CrossRef]
- Weichert, W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [PubMed]
- Bieszczad, K.M.; Bechay, K.; Rusche, J.R.; Jacques, V.; Kudugunti, S.; Miao, W.; Weinberger, N.M.; McGaugh, J.L.; Wood, M.A. Histone Deacetylase Inhibition via RGFP966 Releases the Brakes on Sensory Cortical Plasticity and the Specificity of Memory Formation. J. Neurosci. 2015, 35, 13124–13132. [Google Scholar] [CrossRef] [PubMed]
- Price, A.J.; Manjegowda, M.C.; Kain, J.; Anandh, S.; Bochkis, I.M. Hdac3, Setdb1, and Kap1 mark H3K9me3/H3K14ac bivalent regions in young and aged liver. Aging Cell 2020, 19, e13092. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, K.; Niida, H.; Uchida, C.; Ohhata, T.; Kitagawa, K.; Motegi, A.; Suda, T.; Kitagawa, M. HDAC3 Is Required for XPC Recruitment and Nucleotide Excision Repair of DNA Damage Induced by UV Irradiation. Mol. Cancer Res. 2020, 18, 1367–1378. [Google Scholar] [CrossRef]
- Yuliana, A.; Jheng, H.-F.; Kawarasaki, S.; Nomura, W.; Takahashi, H.; Ara, T.; Kawada, T.; Goto, T. β-adrenergic Receptor Stimulation Revealed a Novel Regulatory Pathway via Suppressing Histone Deacetylase 3 to Induce Uncoupling Protein 1 Expression in Mice Beige Adipocyte. Int. J. Mol. Sci. 2018, 19, 2436. [Google Scholar] [CrossRef]
- Dora, D.; Rivard, C.; Yu, H.; Bunn, P.; Suda, K.; Ren, S.; Lueke Pickard, S.; Laszlo, V.; Harko, T.; Megyesfalvi, Z.; et al. Neuroendocrine subtypes of small cell lung cancer differ in terms of immune microenvironment and checkpoint molecule distribution. Mol. Oncol. 2020, 14, 1947–1965. [Google Scholar] [CrossRef]
- Textor, S.; Fiegler, N.; Arnold, A.; Porgador, A.; Hofmann, T.G.; Cerwenka, A. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 2011, 71, 5998–6009. [Google Scholar] [CrossRef]
- Li, H.; Lakshmikanth, T.; Garofalo, C.; Enge, M.; Spinnler, C.; Anichini, A.; Szekely, L.; Kärre, K.; Carbone, E.; Selivanova, G. Pharmacological activation of p53 triggers anticancer innate immune response through induction of ULBP2. Cell Cycle 2011, 10, 3346–3358. [Google Scholar] [CrossRef]
- Soriani, A.; Borrelli, C.; Ricci, B.; Molfetta, R.; Zingoni, A.; Fionda, C.; Carnevale, S.; Abruzzese, M.P.; Petrucci, M.T.; Ricciardi, M.R.; et al. p38 MAPK differentially controls NK activating ligands at transcriptional and post-transcriptional level on multiple myeloma cells. Oncoimmunology 2017, 6, e1264564. [Google Scholar] [CrossRef]
- Yang, H.; Sun, B.; Xu, K.; He, Y.; Zhang, T.; Hall, S.R.R.; Tan, S.T.; Schmid, R.A.; Peng, R.W.; Hu, G.; et al. Pharmaco-transcriptomic correlation analysis reveals novel responsive signatures to HDAC inhibitors and identifies Dasatinib as a synergistic interactor in small-cell lung cancer. eBioMedicine 2021, 69, 103457. [Google Scholar] [CrossRef]
- Tlemsani, C.; Pongor, L.; Elloumi, F.; Girard, L.; Huffman, K.E.; Roper, N.; Varma, S.; Luna, A.; Rajapakse, V.N.; Sebastian, R.; et al. SCLC-CellMiner: A Resource for Small Cell Lung Cancer Cell Line Genomics and Pharmacology Based on Genomic Signatures. Cell Rep. 2020, 33, 108296. [Google Scholar] [CrossRef] [PubMed]
- Owonikoko, T.K.; Dwivedi, B.; Chen, Z.; Zhang, C.; Barwick, B.; Ernani, V.; Zhang, G.; Gilbert-Ross, M.; Carlisle, J.; Khuri, F.R.; et al. YAP1 Expression in Small Cell Lung Cancer Defines a Distinct Subtype with T-cell Inflamed Phenotype. J. Thorac. Oncol. 2020, 16, 464–476. [Google Scholar] [CrossRef]
- Masso-Valles, D.; Beaulieu, M.E.; Soucek, L. MYC, MYCL, and MYCN as therapeutic targets in lung cancer. Expert Opin. Ther. Targets 2020, 24, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, X.; Lin, J.; Lwin, T.; Wright, G.; Moscinski, L.C.; Dalton, W.S.; Seto, E.; Wright, K.; Sotomayor, E.; et al. Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene 2012, 31, 3002–3008. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Gong, J.-N.; Chen, M.-T.; Song, L.; Shen, C.; Zhang, X.-H.; Yin, X.-L.; Ning, H.-M.; Liu, B.; Wang, F.; et al. c-Myc suppresses miR-451⊣YWTAZ/AKT axis via recruiting HDAC3 in acute myeloid leukemia. Oncotarget 2016, 7, 77430–77443. [Google Scholar] [CrossRef][Green Version]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Torrini, C.; Zhao, J.; Bianchetti, E.; Mela, A.; Humala, N.; Mahajan, A.; et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Investig. 2020, 130, 3699–3716. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.-Y.; Roe, J.-S.; Seol, J.-H.; Kim, H.-R.; Cho, E.-J.; Youn, H.-D. pVHL-mediated transcriptional repression of c-Myc by recruitment of histone deacetylases. Mol. Cells 2012, 33, 195–201. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).