
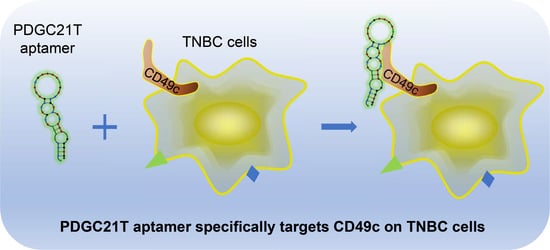
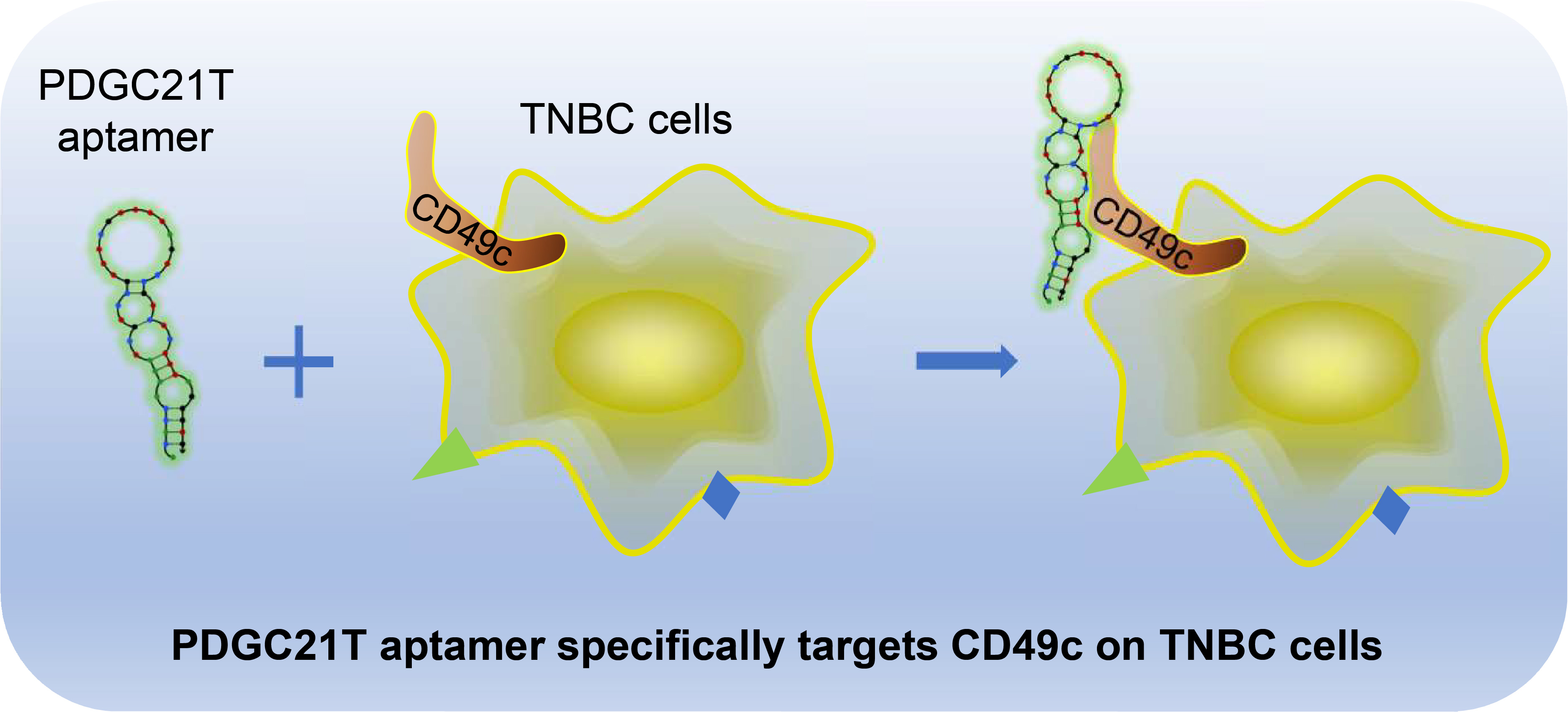
Aptamer Targets Triple-Negative Breast Cancer through Specific Binding to Surface CD49c

 ,
, 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
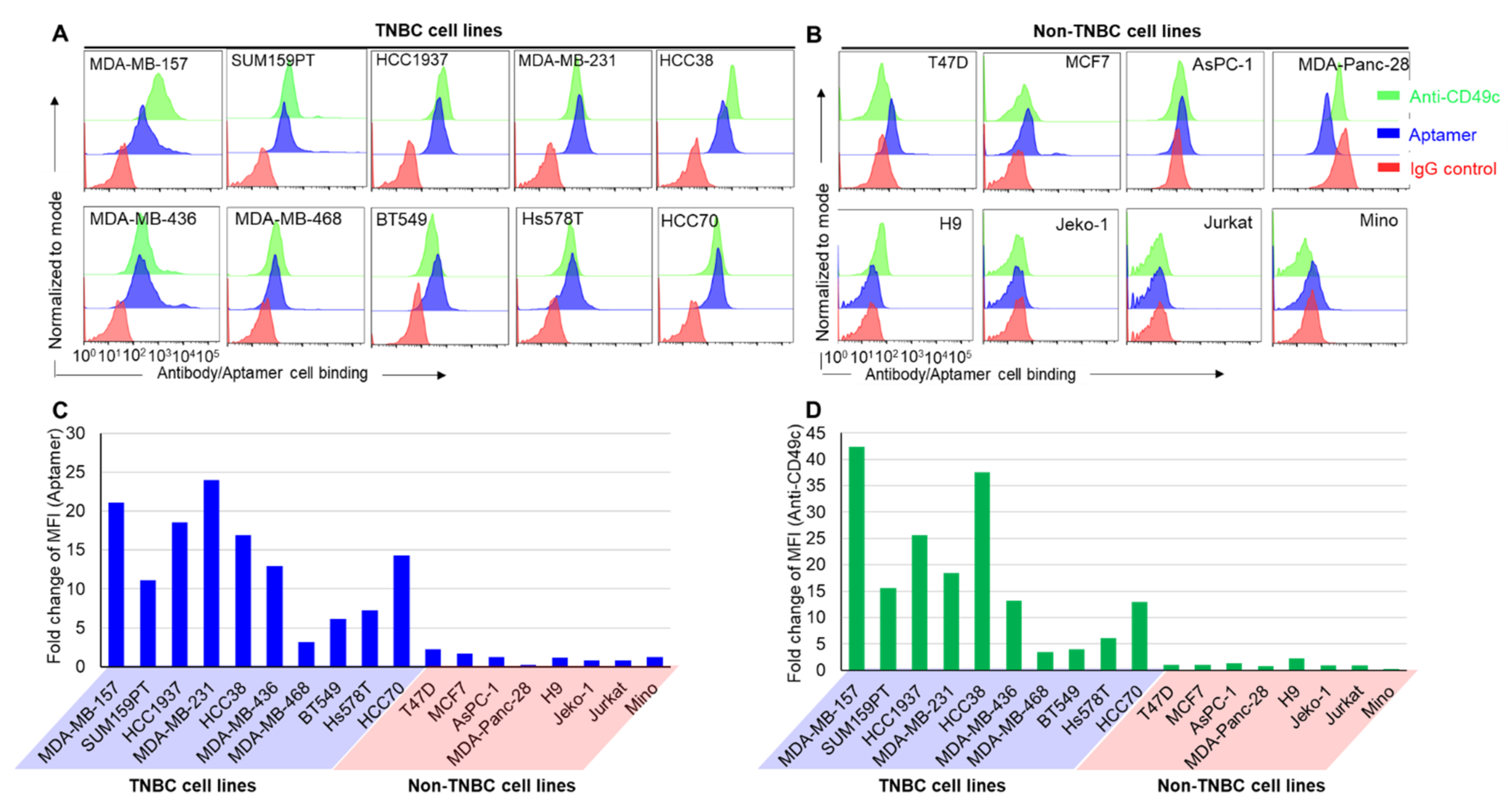
2.2. Aptamer Binding Assay
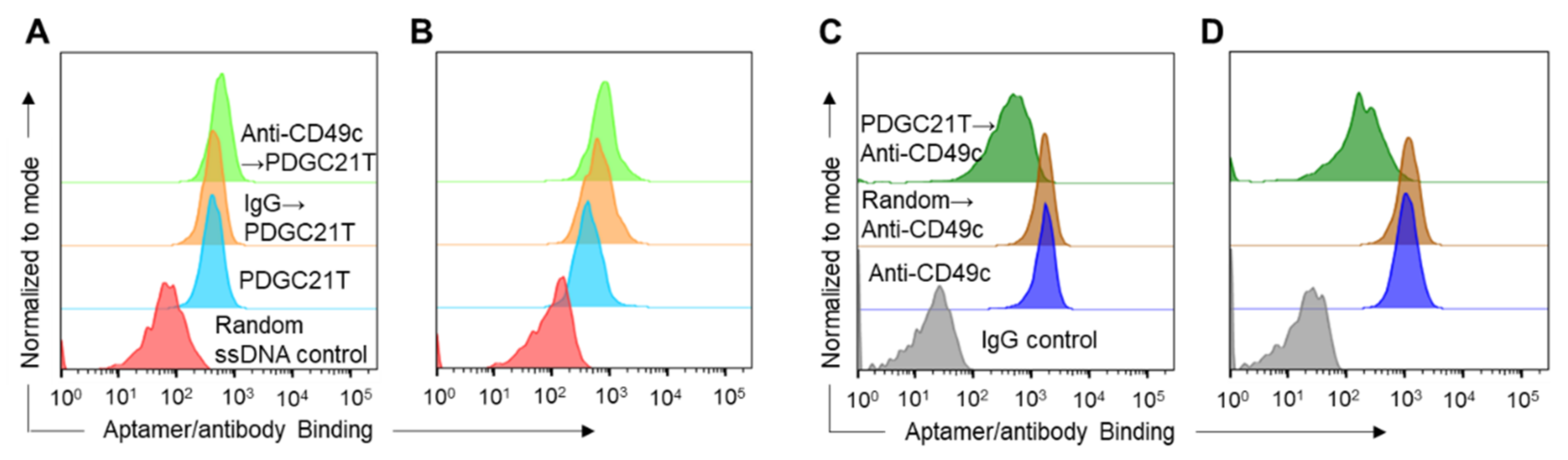
2.3. Aptamer-Antibody Competition Binding Assay
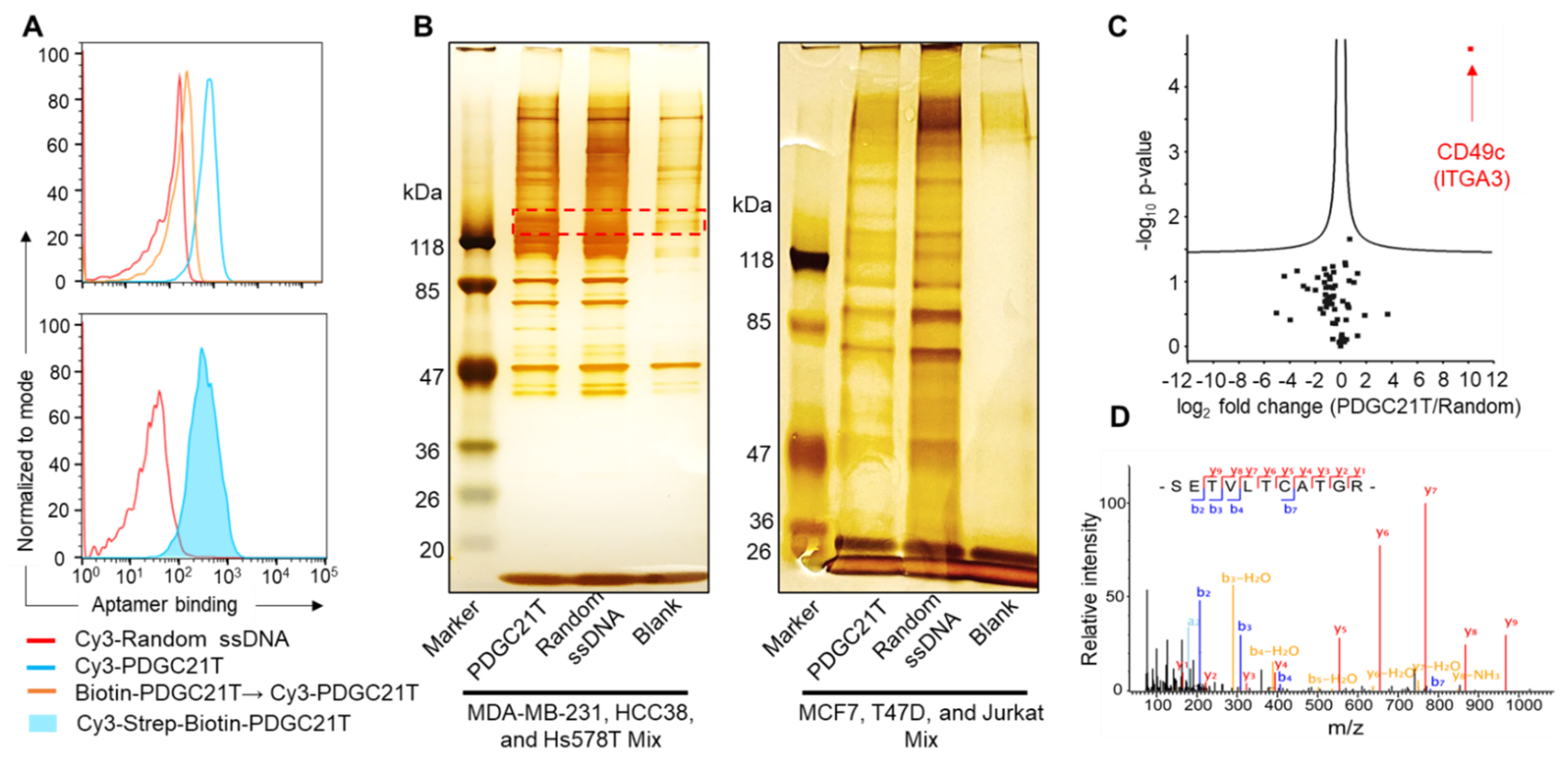
2.4. PDGC21T Pull-Down Assay
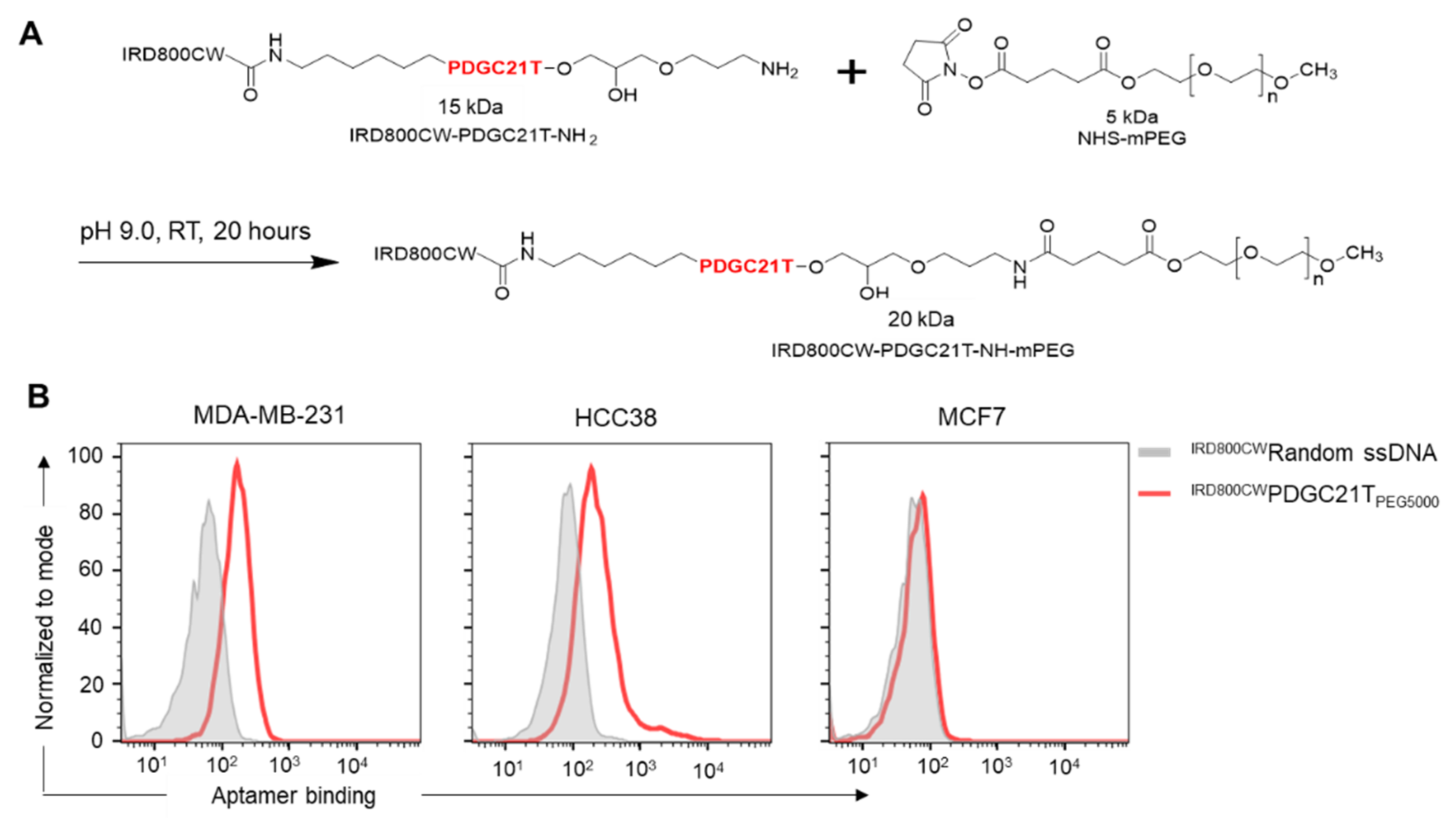
2.5. PDGC21T PEGylation
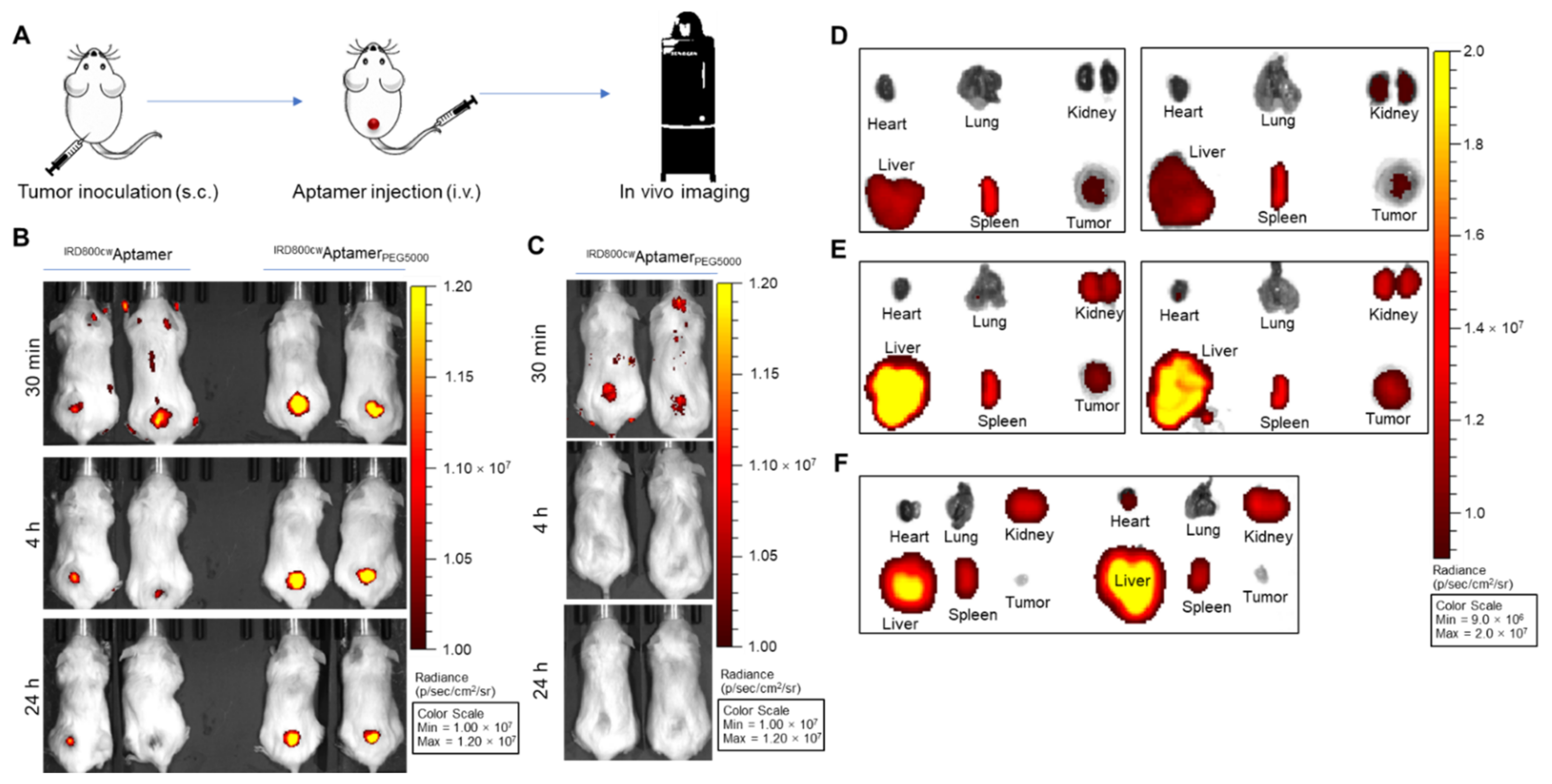
2.6. Tumor Xenograft and In Vivo and Ex Vivo PDGC21T Targeting
2.7. Xenograft Tumor Cells Isolation
3. Results
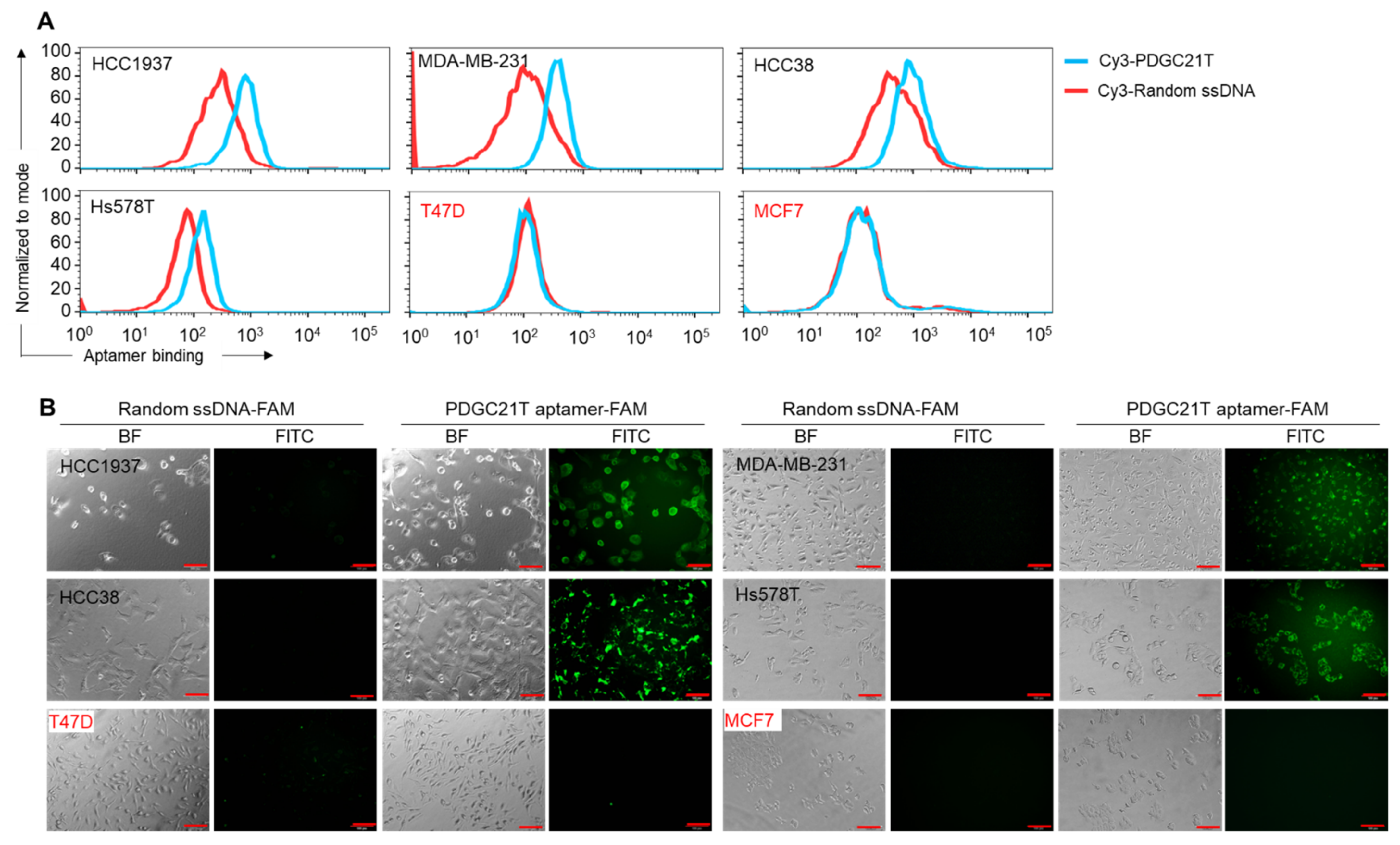
3.1. Synthetic PDGC21T Aptamer Specifically Binds to TNBC Cells
3.2. PDGC21T Aptamer Targets Xenograft TNBC Tumors
3.3. PDGC21T Targets Human CD49c
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast cancer treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Zhao, J.; Li, J.; Liao, X.; Chen, F. Advances of aptamers screened by Cell-SELEX in selection procedure, cancer diagnostics and therapeutics. Anal. Biochem. 2020, 598, 113620. [Google Scholar] [CrossRef]
- Pang, X.; Cui, C.; Wan, S.; Jiang, Y.; Zhang, L.; Xia, L.; Li, L.; Li, X.; Tan, W. Bioapplications of Cell-SELEX-generated aptamers in cancer diagnostics, therapeutics, theranostics and biomarker discovery: A comprehensive review. Cancers 2018, 10, 47. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Yu, Y.; Jiang, F.; Zhou, J.; Li, Y.; Liang, C.; Dang, L.; Lu, A.; Zhang, G. Development of Cell-SELEX technology and its application in cancer diagnosis and therapy. Int. J. Mol. Sci. 2016, 17, 2079. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Tang, Z.; Kim, Y.; Nie, H.; Huang, Y.F.; He, X.; Deng, K.; Wang, K.; Tan, W. In vivo fluorescence imaging of tumors using molecular aptamers generated by cell-SELEX. Chem. Asian J. 2010, 5, 2209–2213. [Google Scholar] [CrossRef]
- Yuan, B.; Jiang, X.; Chen, Y.; Guo, Q.; Wang, K.; Meng, X.; Huang, Z.; Wen, X. Metastatic cancer cell and tissue-specific fluorescence imaging using a new DNA aptamer developed by Cell-SELEX. Talanta 2017, 170, 56–62. [Google Scholar] [CrossRef]
- Shi, H.; Cui, W.; He, X.; Guo, Q.; Wang, K.; Ye, X.; Tang, J. Whole cell-SELEX aptamers for highly specific fluorescence molecular imaging of carcinomas in vivo. PLoS ONE 2013, 8, e70476. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Rossi, J.J. Cell-specific aptamer-mediated targeted drug delivery. Oligonucleotides 2011, 21, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zueva, E.; Rubio, L.I.; Duconge, F.; Tavitian, B. Metastasis-focused cell-based SELEX generates aptamers inhibiting cell migration and invasion. Int. J. Cancer 2011, 128, 797–804. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Hsieh, I.S.; Tung, C.-H.; Weng, C.-H.; Wu, J.-E.; Yu, J.-S.; Hong, T.-M.; Chen, Y.-L. A novel DNA aptamer targeting lung cancer stem cells exerts a therapeutic effect by binding and neutralizing Annexin A2. Mol. Ther. Nucleic Acids 2022, 27, 956–968. [Google Scholar] [CrossRef]
- Berezovski, M.V.; Lechmann, M.; Musheev, M.U.; Mak, T.W.; Krylov, S.N. Aptamer-facilitated biomarker discovery (AptaBiD). J. Am. Chem. Soc. 2008, 130, 9137–9143. [Google Scholar] [CrossRef]
- Li, W.; Wang, S.; Zhou, L.; Cheng, Y.; Fang, J. An ssDNA aptamer selected by Cell-SELEX for the targeted imaging of poorly differentiated gastric cancer tissue. Talanta 2019, 199, 634–642. [Google Scholar] [CrossRef]
- Wu, X.; Liu, H.; Han, D.; Peng, B.; Zhang, H.; Zhang, L.; Li, J.; Liu, J.; Cui, C.; Fang, S.; et al. Elucidation and structural modeling of CD71 as a molecular target for cell-specific aptamer binding. J. Am. Chem. Soc. 2019, 141, 10760–10769. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, H.; Zhang, J.; Brasier, A.R.; Zhao, Y. Quantitative Assessment of the Effects of Trypsin Digestion Methods on Affinity Purification–Mass Spectrometry-based Protein–Protein Interaction Analysis. J. Proteome Res. 2017, 16, 3068–3082. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [Green Version]
- Prodeus, A.; Abdul-Wahid, A.; Fischer, N.W.; Huang, E.H.; Cydzik, M.; Gariepy, J. Targeting the PD-1/PD-L1 immune evasion axis with DNA aptamers as a novel therapeutic strategy for the treatment of disseminated cancers. Mol. Ther. Nucleic Acids 2015, 4, e237. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, K. PEGylation of therapeutic oligonucletides: From linear to highly branched PEG architectures. Nano Res. 2018, 11, 5519–5534. [Google Scholar] [CrossRef] [PubMed]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Srinivasarao, M.; Low, P.S. Ligand-targeted drug delivery. Chem. Rev. 2017, 117, 12133–12164. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Parslow, A.C.; Gan, H.K.; Scott, A.M. Antibody-mediated delivery of therapeutics for cancer therapy. Expert Opin. Drug Deliv. 2016, 13, 401–419. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, A.; Vidula, N.; Ellisen, L.; Bardia, A. Novel antibody-drug conjugates for triple negative breast cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920915980. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhu, X.; Lu, P.Y.; Rosato, R.R.; Tan, W.; Zu, Y. Oligonucleotide aptamers: New tools for targeted cancer therapy. Mol. Ther. Nucleic Acids 2014, 3, e182. [Google Scholar] [CrossRef] [PubMed]
- Sefah, K.; Shangguan, D.; Xiong, X.; O’Donoghue, M.B.; Tan, W. Development of DNA aptamers using cell-SELEX. Nat. Protoc. 2010, 5, 1169–1185. [Google Scholar] [CrossRef]
- Cohen, R.; Stammes, M.A.; de Roos, I.H.; Stigter-van Walsum, M.; Visser, G.W.; van Dongen, G.A. Inert coupling of IRDye800CW to monoclonal antibodies for clinical optical imaging of tumor targets. EJNMMI Res. 2011, 1, 31. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 181–202. [Google Scholar] [CrossRef] [Green Version]
- Ng, E.W.; Shima, D.T.; Calias, P.; Cunningham, E.T., Jr.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef]
- Jin, C.; Qiu, L.; Li, J.; Fu, T.; Zhang, X.; Tan, W. Cancer biomarker discovery using DNA aptamers. Analyst 2016, 141, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Chen, X.; Fu, X.; Li, Z.; Huang, Y.; Liang, C. Advances in aptamer-based biomarker discovery. Front. Cell Dev. Biol. 2021, 9, 659760. [Google Scholar] [CrossRef]
- Yen, T.Y.; Macher, B.A.; McDonald, C.A.; Alleyne-Chin, C.; Timpe, L.C. Glycoprotein profiles of human breast cells demonstrate a clear clustering of normal/benign versus malignant cell lines and basal versus luminal cell lines. J. Proteome Res. 2012, 11, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Yang, J.; Liu, D.; Huang, L.; Fell, G.; Huang, J.; Moses, M.A.; Auguste, D.T. Dual complementary liposomes inhibit triple-negative breast tumor progression and metastasis. Sci. Adv. 2019, 5, eaav5010. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.; Li, Y.; Liu, S.; Chen, Q.; Liu, Y. ITGA3 serves as a diagnostic and prognostic biomarker for pancreatic cancer. OncoTargets Ther. 2019, 12, 4141–4152. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, M.; Kukkurainen, S.; Hytonen, V.P.; Wehrle-Haller, B. Cell adhesion by integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef]
- Michael, M.; Parsons, M. New perspectives on integrin-dependent adhesions. Curr. Opin. Cell Biol. 2020, 63, 31–37. [Google Scholar] [CrossRef]
- Melchiori, A.; Mortarini, R.; Carlone, S.; Marchisio, P.C.; Anichini, A.; Noonan, D.M.; Albini, A. The α3β1 integrin is involved in melanoma cell migration and invasion. Exp. Cell. Res. 1995, 219, 233–242. [Google Scholar] [CrossRef]
- Koshizuka, K.; Hanazawa, T.; Kikkawa, N.; Arai, T.; Okato, A.; Kurozumi, A.; Kato, M.; Katada, K.; Okamoto, Y.; Seki, N. Regulation of ITGA3 by the anti-tumor miR-199 family inhibits cancer cell migration and invasion in head and neck cancer. Cancer Sci. 2017, 108, 1681–1692. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lee, J.; Choi, C.; Kim, J.H. Blockade of integrin alpha3 attenuates human pancreatic cancer via inhibition of EGFR signalling. Sci. Rep. 2019, 9, 2793. [Google Scholar] [CrossRef]
- Huang, Y.; Kong, Y.; Zhang, L.; He, T.; Zhou, X.; Yan, Y.; Zhang, L.; Zhou, D.; Lu, S.; Zhou, J.; et al. High expression of ITGA3 promotes proliferation and cell cycle progression and indicates poor prognosis in intrahepatic cholangiocarcinoma. Biomed. Res. Int. 2018, 2018, 2352139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miskin, R.P.; Warren, J.S.A.; Ndoye, A.; Wu, L.; Lamar, J.M.; DiPersio, C.M. Integrin α3β1 promotes invasive and metastatic properties of breast cancer cells through induction of the Brn-2 transcription factor. Cancers 2021, 13, 480. [Google Scholar] [CrossRef] [PubMed]
- Nakada, M.; Nambu, E.; Furuyama, N.; Yoshida, Y.; Takino, T.; Hayashi, Y.; Sato, H.; Sai, Y.; Tsuji, T.; Miyamoto, K.I.; et al. Integrin α3 is overexpressed in glioma stem-like cells and promotes invasion. Br. J. Cancer 2013, 108, 2516–2524. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Secades, P.; van Hulst, L.; Kreft, M.; Song, J.Y.; Sonnenberg, A. Loss of integrin α3 prevents skin tumor formation by promoting epidermal turnover and depletion of slow-cycling cells. Proc. Natl. Acad. Sci. USA 2012, 109, 21468–21473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, M.; Noman, A.A.; Suzuki, K.; Kurita, H.; Ohnishi, M.; Ohyama, T.; Kitamura, N.; Kobayashi, T.; Uematsu, K.; Takahashi, K.; et al. ITGA3 and ITGB4 expression biomarkers estimate the risks of locoregional and hematogenous dissemination of oral squamous cell carcinoma. BMC Cancer 2013, 13, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Ma, W.; Chen, S.; Tian, E.C.; Wei, S.; Fan, R.R.; Wang, T.; Zhou, C.; Li, T. High integrin α3 expression is associated with poor prognosis in patients with non-small cell lung cancer. Transl. Lung Cancer Res. 2020, 9, 1361–1378. [Google Scholar] [CrossRef] [PubMed]
- Remsik, J.; Fedr, R.; Navratil, J.; Bino, L.; Slabakova, E.; Fabian, P.; Svoboda, M.; Soucek, K. Plasticity and intratumoural heterogeneity of cell surface antigen expression in breast cancer. Br. J. Cancer 2018, 118, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Shirakihara, T.; Kawasaki, T.; Fukagawa, A.; Semba, K.; Sakai, R.; Miyazono, K.; Miyazawa, K.; Saitoh, M. Identification of integrin alpha3 as a molecular marker of cells undergoing epithelial-mesenchymal transition and of cancer cells with aggressive phenotypes. Cancer Sci. 2013, 104, 1189–1197. [Google Scholar] [CrossRef]
- Li, C.; Yang, Z.; Du, Y.; Tang, H.; Chen, J.; Hu, D.; Fan, Z. BCMab1, a monoclonal antibody against aberrantly glycosylated integrin α3β1, has potent antitumor activity of bladder cancer in vivo. Clin. Cancer Res. 2014, 20, 4001–4013. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Kuttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.H.; Springer, T.A. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 2006, 18, 579–586. [Google Scholar] [CrossRef] [Green Version]
- Askari, J.A.; Buckley, P.A.; Mould, A.P.; Humphries, M.J. Linking integrin conformation to function. J. Cell Sci. 2009, 122, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Mezu-Ndubuisi, O.J.; Maheshwari, A. The role of integrins in inflammation and angiogenesis. Pediatr Res. 2021, 89, 1619–1626. [Google Scholar] [CrossRef]
- Khan, N.H.; Bui, A.A.; Xiao, Y.; Sutton, R.B.; Shaw, R.W.; Wylie, B.J.; Latham, M.P. A DNA aptamer reveals an allosteric site for inhibition in metallo-beta-lactamases. PLoS ONE 2019, 14, e0214440. [Google Scholar]
- Das, L.; Anderson, T.A.; Gard, J.M.; Sroka, I.C.; Strautman, S.R.; Nagle, R.B.; Morrissey, C.; Knudsen, B.S.; Cress, A.E. Characterization of laminin binding integrin internalization in prostate cancer cells. J. Cell. Biochem. 2017, 118, 1038–1049. [Google Scholar] [CrossRef] [Green Version]
- Larrain, J.; Brown, C.; De Robertis, E.M. Integrin-α3 mediates binding of Chordin to the cell surface and promotes its endocytosis. EMBO Rep. 2003, 4, 813–818. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, Q.; Zeng, Z.; Qi, J.; Zhao, Y.; Liu, X.; Chen, Z.; Zhou, H.; Zu, Y. Aptamer Targets Triple-Negative Breast Cancer through Specific Binding to Surface CD49c. Cancers 2022, 14, 1570. https://doi.org/10.3390/cancers14061570
Wan Q, Zeng Z, Qi J, Zhao Y, Liu X, Chen Z, Zhou H, Zu Y. Aptamer Targets Triple-Negative Breast Cancer through Specific Binding to Surface CD49c. Cancers. 2022; 14(6):1570. https://doi.org/10.3390/cancers14061570
Chicago/Turabian StyleWan, Quanyuan, Zihua Zeng, Jianjun Qi, Yingxin Zhao, Xiaohui Liu, Zhenghu Chen, Haijun Zhou, and Youli Zu. 2022. "Aptamer Targets Triple-Negative Breast Cancer through Specific Binding to Surface CD49c" Cancers 14, no. 6: 1570. https://doi.org/10.3390/cancers14061570
APA StyleWan, Q., Zeng, Z., Qi, J., Zhao, Y., Liu, X., Chen, Z., Zhou, H., & Zu, Y. (2022). Aptamer Targets Triple-Negative Breast Cancer through Specific Binding to Surface CD49c. Cancers, 14(6), 1570. https://doi.org/10.3390/cancers14061570