
Gene-Mutation-Based Algorithm for Prediction of Treatment Response in Colorectal Cancer Patients

 and
and
Abstract
:Simple Summary
Abstract
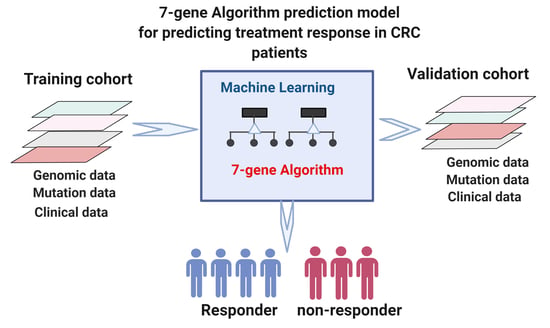
1. Introduction
2. Materials and Methods
2.1. CRC Patients Cohorts
2.2. Algorithms for Prediction of Cancer Progression after Treatment
2.3. Statistical Analysis
3. Results
3.1. Development of the 7-Gene Algorithm for Stratification of Responder and Nonresponder Patients to Predict Response to Treatment
3.2. Assessment of the 7-Gene Algorithm for Prediction of Progression-Free Survival after Treatment in the MSK Cohort
3.3. The 7-Gene Progression Algorithm for Prediction of Progression after Treatment
3.4. Validation of the 7-Gene Algorithm in the TCGA Cohort
3.5. Assessment of the 7-Gene Algorithm for Prediction of Treatment Response in mCRC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Dyba, T.; Randi, G.; Bray, F.; Martos, C.; Giusti, F.; Nicholson, N.; Gavin, A.; Flego, M.; Neamtiu, L.; Dimitrova, N. The European cancer burden in 2020: Incidence and mortality estimates for 40 countries and 25 major cancers. Eur. J. Cancer 2021, 157, 308–347. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Cancer Stat Facts: Colorectal Cancer. Available online: https://seer.cancer.gov/statfacts/html/colorect.html (accessed on 30 December 2021).
- Filip, S.; Vymetalkova, V.; Petera, J.; Vodickova, L.; Kubecek, O.; John, S.; Cecka, F.; Krupova, M.; Manethova, M.; Cervena, K. Distant Metastasis in Colorectal Cancer Patients—Do We Have New Predicting Clinicopathological and Molecular Biomarkers? A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 5255. [Google Scholar] [CrossRef] [PubMed]
- Zakaria, S.; Donohue, J.H.; Que, F.G.; Farnell, M.B.; Schleck, C.D.; Ilstrup, D.M.; Nagorney, D.M. Hepatic resection for colorectal metastases: Value for risk scoring systems? Ann. Surg. 2007, 246, 183. [Google Scholar] [CrossRef] [PubMed]
- Nathan, H.; de Jong, M.C.; Pulitano, C.; Ribero, D.; Strub, J.; Mentha, G.; Gigot, J.-F.; Schulick, R.D.; Choti, M.A.; Aldrighetti, L. Conditional survival after surgical resection of colorectal liver metastasis: An international multi-institutional analysis of 949 patients. J. Am. Coll. Surg. 2010, 210, 755–764. [Google Scholar] [CrossRef]
- Abdalla, E.; Hicks, M.; Vauthey, J. Portal vein embolization: Rationale, technique and future prospects. Br. J. Surg. 2001, 88, 165–175. [Google Scholar] [CrossRef]
- Fong, Y.; Fortner, J.; Sun, R.L.; Brennan, M.F.; Blumgart, L.H. Clinical score for predicting recurrence after hepatic resection for metastatic colorectal cancer: Analysis of 1001 consecutive cases. Ann. Surg. 1999, 230, 309. [Google Scholar] [CrossRef]
- Iwatsuki, S.; Dvorchik, I.; Madariaga, J.R.; Marsh, J.W.; Dodson, F.; Bonham, A.C.; Geller, D.A.; Gayowski, T.J.; Fung, J.J.; Starzl, T.E. Hepatic resection for metastatic colorectal adenocarcinoma: A proposal of a prognostic scoring system. J. Am. Coll. Surg. 1999, 189, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Rees, M.; Tekkis, P.P.; Welsh, F.K.; O’Rourke, T.; John, T.G. Evaluation of long-term survival after hepatic resection for metastatic colorectal cancer: A multifactorial model of 929 patients. Ann. Surg. 2008, 247, 125–135. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Network, C.G.A. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.Y.; Tolias, P. Recent advances in cancer drug discovery targeting RAS. Drug Discov. Today 2016, 21, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biller, L.H.; Schrag, D. Diagnosis and treatment of metastatic colorectal cancer: A review. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef]
- Cohen, R.; Cervera, P.; Svrcek, M.; Pellat, A.; Dreyer, C.; de Gramont, A.; André, T. BRAF-mutated colorectal cancer: What is the optimal strategy for treatment? Curr. Treat. Options Oncol. 2017, 18, 9. [Google Scholar] [CrossRef]
- Tol, J.; Nagtegaal, I.D.; Punt, C.J. BRAF mutation in metastatic colorectal cancer. N. Engl. J. Med. 2009, 361, 98–99. [Google Scholar] [CrossRef] [Green Version]
- Pectasides, E.; Bass, A.J. ERBB2 emerges as a new target for colorectal cancer. Cancer Discov. 2015, 5, 799–801. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.; Jiang, C.; Tse, P.; Achacoso, N.; Alexeeff, S.; Solorzano, A.V.; Chung, E.; Hu, W.; Truong, T.-G.; Arora, A. TP53 Gain-of-Function and Non–Gain-of-Function Mutations Are Differentially Associated With Sidedness-Dependent Prognosis in Metastatic Colorectal Cancer. J. Clin. Oncol. 2022, 40, 171–179. [Google Scholar] [CrossRef]
- Wang, C.; Ouyang, C.; Sandhu, J.S.; Kahn, M.; Fakih, M. Wild-type APC and prognosis in metastatic colorectal cancer. J. Clin. Oncol. 2020, 38, 223. [Google Scholar] [CrossRef]
- Reichling, C.; Taieb, J.; Derangere, V.; Klopfenstein, Q.; Le Malicot, K.; Gornet, J.-M.; Becheur, H.; Fein, F.; Cojocarasu, O.; Kaminsky, M.C. Artificial intelligence-guided tissue analysis combined with immune infiltrate assessment predicts stage III colon cancer outcomes in PETACC08 study. Gut 2020, 69, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Ichimasa, K.; Kudo, S.-e.; Mori, Y.; Misawa, M.; Matsudaira, S.; Kouyama, Y.; Baba, T.; Hidaka, E.; Wakamura, K.; Hayashi, T. Artificial intelligence may help in predicting the need for additional surgery after endoscopic resection of T1 colorectal cancer. Endoscopy 2018, 50, 230–240. [Google Scholar] [PubMed]
- Vera-Yunca, D.; Girard, P.; Parra-Guillen, Z.P.; Munafo, A.; Trocóniz, I.F.; Terranova, N. Machine learning analysis of individual tumor lesions in four metastatic colorectal cancer clinical studies: Linking tumor heterogeneity to overall survival. AAPS J. 2020, 22, 58. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-C.; Yeh, Y.-M.; Lin, B.-W.; Chan, R.-H.; Su, P.-F.; Liu, Y.-C.; Lee, C.-T.; Chen, S.-H.; Lin, P.-C. A prediction model for tumor recurrence in stage II–III colorectal cancer patients: From a machine learning model to genomic profiling. Biomedicines 2022, 10, 340. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Choi, S.; Lee, M.; Park, Y.S.; Sung, J.S.; Chang, W.J.; Kim, J.W.; Choi, Y.J.; Kim, J.; Kim, D.-S. Clinical implication of concordant or discordant genomic profiling between primary and matched metastatic tissues in patients with colorectal cancer. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2020, 52, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferroni, P.; Zanzotto, F.M.; Riondino, S.; Scarpato, N.; Guadagni, F.; Roselli, M. Breast cancer prognosis using a machine learning approach. Cancers 2019, 11, 328. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.W.; Song, C.S. Predicting adoption of colorectal cancer screening among Korean Americans using a decision tree model. Ethn. Health 2022, 1–15. [Google Scholar] [CrossRef]
- Johnson, H.; Guo, J.; Zhang, X.; Zhang, H.; Simoulis, A.; Wu, A.H.; Xia, T.; Li, F.; Tan, W.; Johnson, A. Development and validation of a 25-Gene Panel urine test for prostate cancer diagnosis and potential treatment follow-up. BMC Med. 2020, 18, 376. [Google Scholar] [CrossRef]
- Guo, J.; Liu, D.; Zhang, X.; Johnson, H.; Feng, X.; Zhang, H.; Wu, A.H.; Chen, L.; Fang, J.; Xiao, Z. Establishing a urine-based biomarker assay for prostate cancer risk stratification. Front. Cell Dev. Biol. 2020, 1448. [Google Scholar] [CrossRef]
- Guo, J.; Johnson, H.; Zhang, X.; Feng, X.; Zhang, H.; Simoulis, A.; Wu, A.H.; Xia, T.; Li, F.; Tan, W. A 23-Gene Classifier urine test for prostate cancer prognosis. Clin. Transl. Med. 2021, 11, e340. [Google Scholar] [CrossRef]
- Colorectal Cancer (MSK, Gastroenterology 2020). 2020. Available online: https://www.cbioportal.org/study/summary?id=crc_apc_impact_2020 (accessed on 30 December 2021).
- Mondaca, S.; Walch, H.; Nandakumar, S.; Chatila, W.K.; Schultz, N.; Yaeger, R. Specific mutations in APC, but not alterations in DNA damage response, associate with outcomes of patients with metastatic colorectal cancer. Gastroenterology 2020, 159, 1975.E4–1978.E4. [Google Scholar] [CrossRef]
- Misiakos, E.P.; Karidis, N.P.; Kouraklis, G. Current treatment for colorectal liver metastases. World J. Gastroenterol. WJG 2011, 17, 4067. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.; Jackson, R.; Dunne, D.; Malik, H.; Fenwick, S.; Poston, G.; Ghaneh, P. Systematic review and meta-analysis of follow-up after hepatectomy for colorectal liver metastases. J. Br. Surg. 2012, 99, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Nagase, M.; Tamagawa, H.; Ueda, S.; Tamura, T.; Murata, K.; Nakajima, T.E.; Baba, E.; Tsuda, M.; Moriwaki, T. Randomized phase III study of bevacizumab plus FOLFIRI and bevacizumab plus mFOLFOX6 as first-line treatment for patients with metastatic colorectal cancer (WJOG4407G). Ann. Oncol. 2016, 27, 1539–1546. [Google Scholar] [CrossRef]
- Tsilimigras, D.I.; Ntanasis-Stathopoulos, I.; Bagante, F.; Moris, D.; Cloyd, J.; Spartalis, E.; Pawlik, T.M. Clinical significance and prognostic relevance of KRAS, BRAF, PI3K and TP53 genetic mutation analysis for resectable and unresectable colorectal liver metastases: A systematic review of the current evidence. Surg. Oncol. 2018, 27, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Antoniotti, C.; Korn, W.M.; Marmorino, F.; Rossini, D.; Lonardi, S.; Masi, G.; Randon, G.; Conca, V.; Boccaccino, A.; Tomasello, G. Tumour mutational burden, microsatellite instability, and actionable alterations in metastatic colorectal cancer: Next-generation sequencing results of TRIBE2 study. Eur. J. Cancer 2021, 155, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Stahler, A.; Stintzing, S.; Jobst, C.; Westphalen, C.B.; Heinrich, K.; Krämer, N.; Michl, M.; Modest, D.P.; von Weikersthal, L.F.; Decker, T. Single-nucleotide variants, tumour mutational burden and microsatellite instability in patients with metastatic colorectal cancer: Next-generation sequencing results of the FIRE-3 trial. Eur. J. Cancer 2020, 137, 250–259. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MSK Cohort | TCGA Cohort | |
|---|---|---|
| No of patients | 471 | 191 |
| Gender (Female) (%) | 232 (49%) | 92 (48%) |
| Gender (male) (%) | 239 (51%) | 99 (52%) |
| Median age (Q1, Q3) | 59 (50, 68) | 69 (62, 78) |
| Distant metastasis (%) | 388 (82%) | 21 (11%) |
| Cancers stage at diagnosis (%) | ||
| Stage I | 8 (2%) | 8 (4%) |
| Stage II | 31 (7%) | 45 (24%) |
| Stage III | 90 (19%) | 125 (65%) |
| Stage IV | 342 (73%) | 13 (7%) |
| MSI type (%) | ||
| Stable | 428 (94%) | NA |
| Instable Prior adjuvant therapies (%) | 27 (6%) | NA |
| Yes | 370 (79%) | 2 (1%) |
| No | 101 (21%) | 189 (99%) |
| Surgery on primary tumor (%) | ||
| Yes | 258 (55%) | NA |
| No | 211 (45%) | NA |
| Overall survival (%) | ||
| Living | 160 (34%) | 182 (95%) |
| Diseased | 311 (66%) | 9 (5%) |
| Progression/disease-free survival (%) | ||
| Progressed | 447 (95%) | 161 (84%) |
| Non-progressed | 24 (5%) | 30 (16%) |
| Sensitivity (95% CI) | Specificity (95% CI) | PPV (95% CI) | NPV (95% CI) | |
|---|---|---|---|---|
| Prediction of Progression in the MSK Cohort (n = 471) | ||||
| 7-Gene Algorithm | 83% (68–98%) | 98% (97–100%) | 74% (58–91%) | 99% (98–100%) |
| Cancer stage | 0% (0–0%) | 100% (100–100%) | 0% (0–0%) | 95% (93–97%) |
| Adjuvant therapies | 0% (0–0%) | 100% (100–100%) | 0% (0–0%) | 95% (93–97%) |
| Surgery on primary tumor | 0% (0–0%) | 100% (100–100%) | 0% (0–0%) | 95% (93–97%) |
| MSI | 0% (0–0%) | 100% (100–100%) | 0% (0–0%) | 95% (93–97%) |
| Combination | 83% (68–98%) | 99% (97–100%) | 77% (61–93%) | 99% (98–100%) |
| Prediction of Progression in the TCGA Progression Cohort (n = 191) | ||||
| 7-Gene Algorithm | 96% (93–99%) | 77% (62–92%) | 96% (93–99%) | 79% (65–94%) |
| Cancer stage | 100% (100–100%) | 0% (0–0%) | 85% (79–89%) | 0% (0–0%) |
| Adjuvant therapies | 100% (100–100%) | 0% (0–0%) | 84% (79–89%) | 0% (0–0%) |
| Combination | 96% (93–99%) | 77% (62–92%) | 96% (93–99%) | 79% (65–94%) |
| Univariate | Multivariate | |||
|---|---|---|---|---|
| HR (95% CI) | p Value | HR (95% CI) | p Value | |
| Prediction of PFS in the MSK Cohort (n = 471) | ||||
| 7-Gene Algorithm | 7.5 (3.5–15.9) | <0.0001 | 8.9 (4.0–20.1) | <0.0001 |
| Cancer stage | 1.3 (0.9–1.9) | 0.128 | 1.1 (0.7–1.5) | 0.755 |
| Adjuvant therapies | 1.1 (0.8–1.3) | 0.877 | 1.1 (0.8–1.4) | 0.536 |
| Surgery on primary tumor | 0.8 (0–1.0) | 0.013 | 0.7 (0–0.9) | 0.002 |
| MSI | 0.7 (0.5–1.1) | 0.097 | 0.6 (0–0.9) | 0.009 |
| Prediction of PFS in the TCGA Cohort (n = 191) | ||||
| 7-Gene Algorithm | 16.9 (7.2–39.6) | <0.0001 | 16.9 (7.2–39.7) | <0.0001 |
| Cancer stage | 1.2 (0.5–2.7) | 0.723 | 1.3 (0.6–3.1) | 0.539 |
| Adjuvant therapies | 3.0 × 10−7 (0-Inf) | 0.997 | 1.7 × 10−6 (0-Inf) | 0.996 |
| Univariate | Multivariate | |||
|---|---|---|---|---|
| HR (95% CI) | p Value | HR (95% CI) | p Value | |
| 7-Gene Algorithm | 16.9 (4.2–68.0) | <0.0001 | 17.6 (4.4–70.8) | <0.0001 |
| Cancer stage | 1.3 (0.9–2.0) | 0.194 | 1.1 (0.7–1.7) | 0.735 |
| Adjuvant therapies | 1.1 (0.8–1.4) | 0.671 | 0.7 (0–1.6) | 0.317 |
| Surgery on primary tumor | 0.8 (0–1.0) | 0.044 | 0.7 (0–0.9) | 0.003 |
| MSI | 0.4 (0–0.7) | 0.002 | 0.4 (0–0.8) | 0.003 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, H.; El-Schich, Z.; Ali, A.; Zhang, X.; Simoulis, A.; Wingren, A.G.; Persson, J.L. Gene-Mutation-Based Algorithm for Prediction of Treatment Response in Colorectal Cancer Patients. Cancers 2022, 14, 2045. https://doi.org/10.3390/cancers14082045
Johnson H, El-Schich Z, Ali A, Zhang X, Simoulis A, Wingren AG, Persson JL. Gene-Mutation-Based Algorithm for Prediction of Treatment Response in Colorectal Cancer Patients. Cancers. 2022; 14(8):2045. https://doi.org/10.3390/cancers14082045
Chicago/Turabian StyleJohnson, Heather, Zahra El-Schich, Amjad Ali, Xuhui Zhang, Athanasios Simoulis, Anette Gjörloff Wingren, and Jenny L. Persson. 2022. "Gene-Mutation-Based Algorithm for Prediction of Treatment Response in Colorectal Cancer Patients" Cancers 14, no. 8: 2045. https://doi.org/10.3390/cancers14082045