
Alternative Lengthening of Telomeres and Mediated Telomere Synthesis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. ALT and ALT-Related Phenotypes
2.1. ALT-Associated PML Bodies
2.2. Extrachromosomal Telomeric Repeats
2.3. Telomere Sister Chromatid Exchange (T-SCE)
2.4. Chromatin Remodeling and Gene Mutations
2.5. Heterochromatin at Telomeres
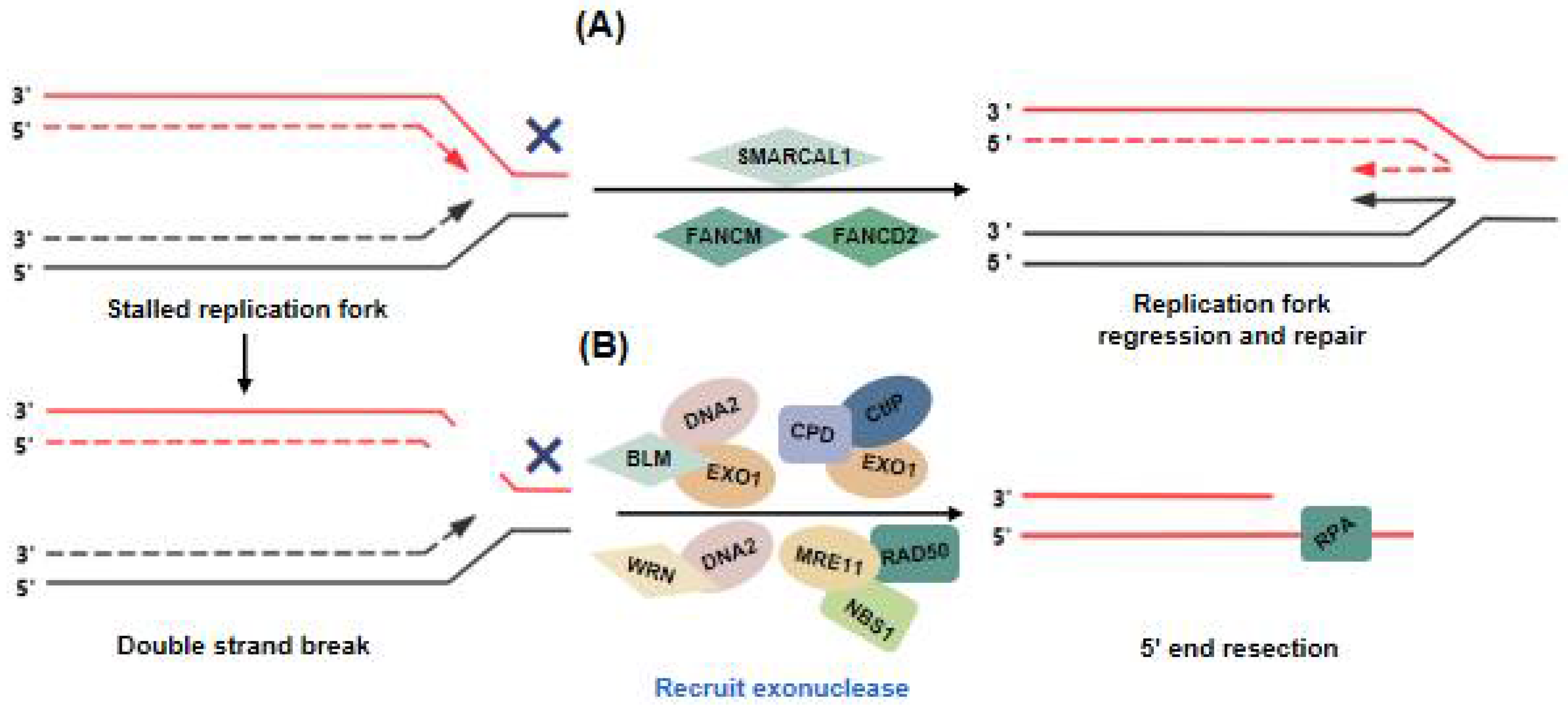
3. SMARCAL1 and FA Proteins Balance Replication Stress of ALT Telomeres to Ensure Telomere Synthesis
3.1. SMARCAL1
3.2. FA Proteins
4. Initiation of the ALT Pathway: DNA Damage at ALT Telomeres
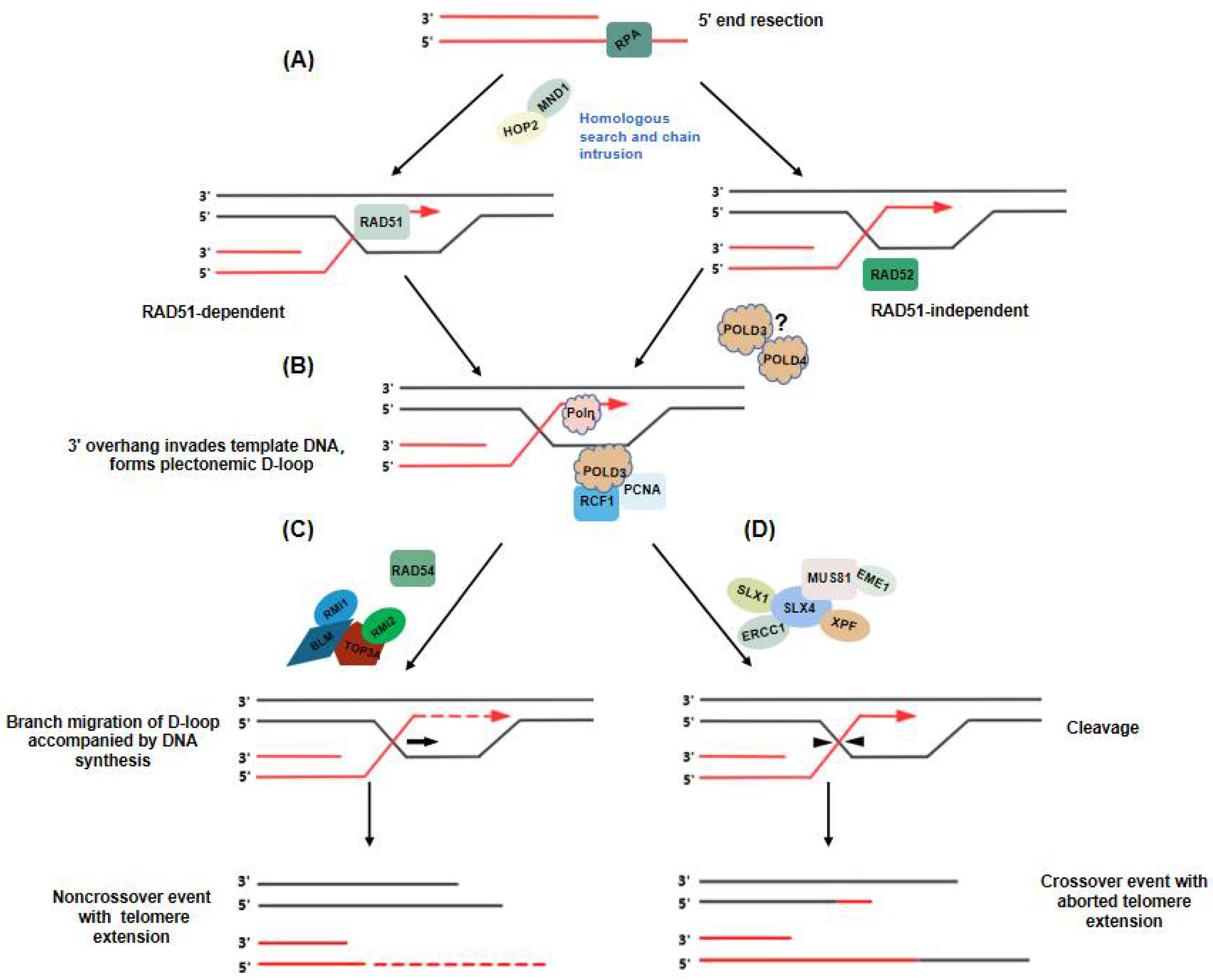
5. Telomere Extension in ALT: Two Distinct Break-Induced Replication Pathways
6. Unraveling Recombinant Intermediates in ALT+ Cells
7. Conclusions
Targeting ALT-Related Cancers
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Meaning |
| Alt-NHEJ | Alternative non-homologous end joining |
| ALT | Alternative lengthening of telomeres |
| ALT+ | ALT pathway-positive |
| ATRX | Alpha-thalassemia/mental retardation X-linked chromatin remodeler |
| APB | ALT -associated PML body |
| APBs | ALT-associated PML bodies |
| BIR | Break-induced replication |
| BTR | BLM-TOP3α-RMI1-RMI2 |
| c-NHEJ | Classical non-homologous end joining |
| CST | CTC1-STN1-TEN1 |
| DAXX | Death domain-associated protein |
| DDR | DNA damage response |
| DSBs | Double-strand breaks |
| D-loop | DNA displacement loop |
| ECTRs | Extrachromosomal telomeric repeats |
| HJ | Holliday junction |
| HP1α | Heterochromatin protein 1 |
| HR | Homologous recombination |
| I-loops | Internal loops |
| KAP1 | KRAB-associated protein 1 |
| LLPS | Liquid–liquid phase separation |
| MiDAS | Mitotic DNA synthesis |
| PARP | Poly (ADP-ribose) polymerase |
| PCNA | Proliferating cell nuclear antigen |
| PML | Promyelocytic leukemia |
| RFC | Replication factor C |
| SIM | SUMO-interacting motif |
| SMX | SLX1-SLX4, MUS81-EME1, and XPF-ERCC1 |
| ssDNA | Single-strand DNA |
| T-circle | Telomere circle |
| TERC | Telomerase RNA component |
| TERRA | Telomeric repeats containing RNA |
References
- Shin, J.S.; Hong, A.; Solomon, M.J.; Soon Lee, C. The role of telomeres and telomerase in the pathology of human cancer and aging. Pathology 2006, 38, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Dilley, R.L.; Greenberg, R.A. ALTernative Telomere Maintenance and Cancer. Trends Cancer 2015, 1, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1259. [Google Scholar] [CrossRef]
- Artandi, S.E.; DePinho, R.A. Telomeres and telomerase in cancer. Carcinogenesis 2010, 31, 9–18. [Google Scholar] [CrossRef]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, R.J.; Almouzni, G. Assembly of telomeric chromatin to create ALTernative endings. Trends Cell Biol. 2014, 24, 675–685. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Allen, J.A.M.; Neumann, A.A.; Yang, S.F.; Walsh, M.E.; Henson, J.D.; Reddel, R.R.; Pickett, H.A. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. Embo. J. 2017, 36, 2907–2919. [Google Scholar] [CrossRef]
- Acharya, S.; Kaul, Z.; Gocha, A.S.; Martinez, A.R.; Harris, J.; Parvin, J.D.; Groden, J. Association of BLM and BRCA1 during Telomere Maintenance in ALT Cells. PLoS ONE 2014, 9, e103819. [Google Scholar]
- Sobinoff, A.P.; Pickett, H.A. Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends Genet. 2017, 33, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.W.; Dilley, R.L.; Lampson, M.A.; Greenberg, R.A. Interchromosomal Homology Searches Drive Directional ALT Telomere Movement and Synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Episkopou, H.; Draskovic, I.; Van Beneden, A.; Tilman, G.; Mattiussi, M.; Gobin, M.; Arnoult, N.; Londono-Vallejo, A.; Decottignies, A. Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014, 42, 4391–4405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, I.; Leonhardt, H.; Rippe, K. De novo assembly of a PML nuclear subcompartment occurs through multiple pathways and induces telomere elongation. J. Cell Sci. 2011, 124, 3603–3618. [Google Scholar] [CrossRef] [Green Version]
- Fonin, A.V.; Silonov, S.A.; Shpironok, O.G.; Antifeeva, I.A.; Petukhov, A.V.; Romanovich, A.E.; Kuznetsova, I.M.; Uversky, V.N.; Turoverov, K.K. The Role of Non-Specific Interactions in Canonical and ALT-Associated PML-Bodies Formation and Dynamics. Int. J. Mol. Sci. 2021, 22, 5821. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Zhao, R.W.; Tones, J.; Liu, M.C.; Dilley, R.L.; Chenoweth, D.M.; Greenberg, R.A.; Lampson, M.A. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol. Biol. Cell 2020, 31, 2048–2056. [Google Scholar] [CrossRef]
- Zhang, J.M.; Genois, M.M.; Ouyang, J.; Lan, L.; Zou, L. Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Mol. Cell 2021, 81, 1027–1042.e4. [Google Scholar] [CrossRef]
- Loe, T.K.; Li, J.L.S.Z.; Zhang, Y.X.; Azeroglu, B.; Boddy, M.N.; Denchi, E.L. Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes Dev. 2020, 34, 650–662. [Google Scholar] [CrossRef]
- Potts, P.R.; Yu, H.T. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 2007, 14, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Wright, W.E.; Shay, J.W. Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev. 2019, 33, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Fasching, C.L.; Bower, K.; Reddel, R.R. Merase-independent telomere length maintenance in the absence of alternative lengthening of telomeres-associated promyelocytic leukemia bodies. Cancer Res. 2005, 65, 2722–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerone, M.A.; Autexier, C.; Londono-Vallejo, J.A.; Bacchetti, S. A human cell line that maintains telomeres in the absence of telomerase and of key markers of ALT. Oncogene 2005, 24, 7893–7901. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.P.; Zhang, Z.P.; Gong, S.Z.; Li, X.C.; Liu, H.Y.; Zhao, Y. Strand break-induced replication fork collapse leads to C-circles, C-overhangs and telomeric recombination. PLoS Genet. 2019, 15, e1007925. [Google Scholar] [CrossRef] [Green Version]
- Henson, J.D.; Cao, Y.; Huschtscha, L.I.; Chang, A.C.; Au, A.Y.M.; Pickett, H.A.; Reddel, R.R. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009, 27, 1181–1185. [Google Scholar] [CrossRef]
- Huang, C.H.; Jia, P.P.; Chastain, M.; Shiva, O.; Chai, W.H. The human CTC1/STN1/TEN1 complex regulates telomere maintenance in ALT cancer cells. Exp. Cell Res. 2017, 355, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Cox, K.E.; Marechal, A.; Flynn, R.L. SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell Rep. 2016, 14, 1032–1040. [Google Scholar] [CrossRef] [Green Version]
- Tomaska, L.; Nosek, J.; Kramara, J.; Griffith, J.D. Telomeric circles: Universal players in telomere maintenance? Nat. Struct. Mol. Biol. 2009, 16, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Compton, S.A.; Choi, J.H.; Cesare, A.J.; Ozgur, S.; Griffith, J.D. Xrcc3 and Nbs1 are required for the production of extrachromosomal telomeric circles in human alternative lengthening of telomere cells. Cancer Res. 2007, 67, 1513–1519. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.C.; Smogorzewska, A.; de Lange, T. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell 2004, 119, 355–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Root, H.; Larsen, A.; Komosa, M.; Al-Azri, F.; Li, R.; Bazett-Jones, D.P.; Meyn, M.S. FANCD2 limits BLM-dependent telomere instability in the alternative lengthening of telomeres pathway. Hum. Mol. Genet. 2016, 25, 3255–3268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavropoulos, D.J.; Bradshaw, P.S.; Li, X.B.; Pasic, I.; Truong, K.; Ikura, M.; Ungrin, M.; Meyn, M.S. The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum. Mol. Genet. 2002, 11, 3135–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzucco, G.; Huda, A.; Galli, M.; Piccini, D.; Giannattasio, M.; Pessina, F.; Doksani, Y. Telomere damage induces internal loops that generate telomeric circles. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tilman, G.; Loriot, A.; Van Beneden, A.; Arnoult, N.; Londono-Vallejo, J.A.; De Smet, C.; Decottignies, A. Subtelomeric DNA hypomethylation is not required for telomeric sister chromatid exchanges in ALT cells. Oncogene 2009, 28, 1682–1693. [Google Scholar] [CrossRef] [Green Version]
- Bailey, S.M.; Brenneman, M.A.; Goodwin, E.H. Frequent recombination in telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res. 2004, 32, 3743–3751. [Google Scholar] [CrossRef]
- Celli, G.B.; Denchi, E.L.; de Lange, T. Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat. Cell Biol. 2006, 8, 885–890. [Google Scholar] [CrossRef]
- Palm, W.; Hockemeyer, D.; Kibe, T.; de Lange, T. Functional Dissection of Human and Mouse POT1 Proteins. Mol. Cell. Biol. 2009, 29, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Londono-Vallejo, J.A.; Der-Sarkissian, H.; Cazes, L.; Bacchetti, S.; Reddel, R.R. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004, 64, 2324–2327. [Google Scholar] [CrossRef] [Green Version]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.C.; Klein, A.P.; Edil, B.H.; Shi, C.J.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered Telomeres in Tumors with ATRX and DAXX Mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, C.A.; Takai, K.; Huh, M.S.; Picketts, D.J.; de Lange, T. ATRX affects the repair of telomeric DSBs by promoting cohesion and a DAXX-dependent activity. PLoS Biol. 2020, 18, e3000594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, L.H.; McGhie, J.D.; Sim, M.; Anderson, M.A.; Ahn, S.; Hannan, R.D.; George, A.J.; Morgan, K.A.; Mann, J.R.; Choo, K.H.A. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010, 20, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKenzie, D.; Watters, A.K.; To, J.T.; Young, M.W.; Muratori, J.; Wilkoff, M.H.; Abraham, R.G.; Plummer, M.M.; Zhang, D. ALT Positivity in Human Cancers: Prevalence and Clinical Insights. Cancers 2021, 13, 2384. [Google Scholar] [CrossRef] [PubMed]
- Gauchier, M.; Kan, S.; Barral, A.; Sauzet, S.; Agirre, E.; Bonnell, E.; Saksouk, N.; Barth, T.K.; Ide, S.; Urbach, S.; et al. SETDB1-dependent heterochromatin stimulates alternative lengthening of telomeres. Sci. Adv. 2019, 5, eaav3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538. [Google Scholar] [CrossRef] [PubMed]
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX represses alternative lengthening of telomeres. Oncotarget 2015, 6, 16543–16558. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, R.J.; Arnoult, N.; Lackner, D.H.; Oganesian, L.; Haggblom, C.; Corpet, A.; Almouzni, G.; Karlseder, J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014, 21, 167–174. [Google Scholar] [CrossRef]
- Cubiles, M.D.; Barroso, S.; Vaquero-Sedas, M.I.; Enguix, A.; Aguilera, A.; Vega-Palas, M.A. Epigenetic features of human telomeres. Nucleic Acids Res. 2018, 46, 2347–2355. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, C.A.; Li, W.D.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.J.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, Genome Instability, and an Altered DNA Damage Response Are Hallmarks of the Alternative Lengthening of Telomeres Pathway. PLoS Genet 2012, 8, e1002772. [Google Scholar] [CrossRef]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.H.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.P.; Li, E.; Jenuwein, T.; Peters, A.H.F.M. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003, 13, 1192–1200. [Google Scholar] [CrossRef] [Green Version]
- Tardat, M.; Dejardin, J. Telomere chromatin establishment and its maintenance during mammalian development. Chromosoma 2018, 127, 3–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, R.V.; Feretzaki, M.; Lingner, J. The makings of TERRA R-loops at chromosome ends. Cell Cycle 2021, 20, 1745–1759. [Google Scholar] [CrossRef] [PubMed]
- Kappei, D.; Scheibe, M.; Paszkowski-Rogacz, M.; Bluhm, A.; Gossmann, T.I.; Dietz, S.; Dejung, M.; Herlyn, H.; Buchholz, F.; Mann, M.; et al. Phylointeractomics reconstructs functional evolution of protein binding. Nat. Commun. 2017, 8, 14334. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.L.; Zhou, S.Y.; Huang, Y. TRIM28 inhibits alternative lengthening of telomere phenotypes by protecting SETDB1 from degradation. Cell Biosci. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian Telomeres Resemble Fragile Sites and Require TRF1 for Efficient Replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Tarsounas, M.; Tijsterman, M. Genomes and G-Quadruplexes: For Better or for Worse. J. Mol. Biol. 2013, 425, 4782–4789. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.L.; Drosopoulos, W.C.; Sethi, L.; Madireddy, A.; Schildkraut, C.L.; Zhang, D. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc. Natl. Acad. Sci. USA 2017, 114, E5940–E5949. [Google Scholar] [CrossRef] [Green Version]
- Betous, R.; Mason, A.C.; Rambo, R.P.; Bansbach, C.E.; Badu-Nkansah, A.; Sirbu, B.M.; Eichman, B.F.; Cortez, D. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012, 26, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Wild, P.; Matos, J. Cell cycle control of DNA joint molecule resolution. Curr. Opin. Cell Biol. 2016, 40, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Poole, L.A.; Zhao, R.X.; Glick, G.G.; Lovejoy, C.A.; Eischen, C.M.; Cortez, D. SMARCAL1 maintains telomere integrity during DNA replication. Proc. Natl. Acad. Sci. USA 2015, 112, 14864–14869. [Google Scholar] [CrossRef] [Green Version]
- Feng, E.; Batenburg, N.L.; Walker, J.R.; Ho, A.; Mitchell, T.R.H.; Qin, J.; Zhu, X.D. CSB cooperates with SMARCAL1 to maintain telomere stability in ALT cells. J. Cell Sci. 2020, 133, jcs234914. [Google Scholar] [CrossRef] [PubMed]
- Fekairi, S.; Scaglione, S.; Chahwan, C.; Taylor, E.R.; Tissier, A.; Coulon, S.; Dong, M.Q.; Ruse, C.; Yates, J.R.; Russell, P.; et al. Human SLX4 Is a Holliday Junction Resolvase Subunit that Binds Multiple DNA Repair/Recombination Endonucleases. Cell 2009, 138, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, K.; Budzowska, M.; Davies, S.L.; van Drunen, E.; Onizawa, H.; Beverloo, H.B.; Maas, A.; Essers, J.; Hickson, I.D.; Kanaar, R. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol. 2007, 14, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Singh, T.R.; Meetei, A.R. FANCM-FAAP24 and FANCJ: FA proteins that metabolize DNA. Mutat. Res. Fund. Mol. Mech. Mutagenesis 2009, 668, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kais, Z.; Rondinelli, B.; Holmes, A.; O’Leary, C.; Kozono, D.; D’Andrea, A.D.; Ceccaldi, R. FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair. Cell Rep. 2016, 15, 2488–2499. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; O’Rourke, J.J.; Sobinoff, A.P.; Allen, J.A.M.; Nelson, C.B.; Tomlinson, C.G.; Lee, M.; Reddel, R.R.; Deans, A.J.; Pickett, H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Silva, B.; Pentz, R.; Figueira, A.M.; Arora, R.; Lee, Y.W.; Hodson, C.; Wischnewski, H.; Deans, A.J.; Azzalin, C.M. FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Pan, X.; Chen, Y.; Biju, B.; Ahmed, N.; Kong, J.; Goldenberg, M.; Huang, J.; Mohan, N.; Klosek, S.; Parsa, K.; et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol. Cell. Biol. 2017, 37, e00226-17. [Google Scholar] [CrossRef] [Green Version]
- Tye, S.; Ronson, G.E.; Morris, J.R. A fork in the road: Where homologous recombination and stalled replication fork protection part ways. Semin. Cell. Dev. Biol. 2021, 113, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Bermudez, A.; Hidalgo-Bravo, A.; Cotton, V.E.; Gravani, A.; Jeyapalan, J.N.; Royle, N.J. The roles of WRN and BLM RecQ helicases in the Alternative Lengthening of Telomeres. Nucleic Acids Res. 2012, 40, 10809–10820. [Google Scholar] [CrossRef] [PubMed]
- Sturzenegger, A.; Burdova, K.; Kanagaraj, R.; Levikova, M.; Pinto, C.; Cejka, P.; Janscak, P. DNA2 Cooperates with the WRN and BLM RecQ Helicases to Mediate Long-range DNA End Resection in Human Cells. J. Biol. Chem. 2014, 289, 27314–27326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafrance-Vanasse, J.; Williams, G.J.; Tainer, J.A. Envisioning the dynamics and flexibility of Mre11-Rad50-Nbs1 complex to decipher its roles in DNA replication and repair. Prog. Biophys. Mol. Bio. 2015, 117, 182–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribes-Zamora, A.; Indiviglio, S.M.; Mihalek, I.; Williams, C.L.; Bertuch, A.A. TRF2 Interaction with Ku Heterotetramerization Interface Gives Insight into c-NHEJ Prevention at Human Telomeres. Cell Rep. 2013, 5, 194–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazzerini-Denchi, E.; Sfeir, A. Stop pulling my strings—What telomeres taught us about the DNA damage response. Nat. Rev. Mol. Cell Bio. 2016, 17, 364–378. [Google Scholar]
- Sadhukhan, R.; Ghosh, U. PARP1 modulates telomere sister chromatid exchange and telomere length homeostasis by regulating telomere localization of SLX4 in U2OS cells. Life Sci. 2021, 277, 119556. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [CrossRef]
- Daley, J.M.; Niu, H.Y.; Miller, A.S.; Sung, P. Biochemical mechanism of DSB end resection and its regulation. DNA Repair 2015, 32, 66–74. [Google Scholar] [CrossRef] [Green Version]
- Bugreev, D.V.; Huang, F.; Mazina, O.M.; Pezza, R.J.; Voloshin, O.N.; Camerini-Otero, R.D.; Mazin, A.V. HOP2-MND1 modulates RAD51 binding to nucleotides and DNA. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Doksani, Y.; Wu, J.Y.; de Lange, T.; Zhuang, X.W. Super-Resolution Fluorescence Imaging of Telomeres Reveals TRF2-Dependent T-loop Formation. Cell 2013, 155, 345–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, Y.; Chaudhuri, A.R.; Lopes, M.; Costanzo, V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010, 17, 1305–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, P.; Dilley, R.L.; Zhang, T.P.; Gyparaki, M.T.; Li, Y.W.; Greenberg, R.A. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Gene Dev. 2019, 33, 221–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Exposito, L.; Bournique, E.; Bergoglio, V.; Bose, A.; Barroso-Gonzalez, J.; Zhang, S.F.; Roncaioli, J.L.; Lee, M.; Wallace, C.T.; Watkins, S.C.; et al. Proteomic Profiling Reveals a Specific Role for Translesion DNA Polymerase eta in the Alternative Lengthening of Telomeres. Cell Rep. 2016, 17, 1858–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majka, J.; Burgers, P.M.J. The PCNA-RFC families of DNA clamps and clamp loaders. Prog. Nucleic Acid Res. 2004, 78, 227–260. [Google Scholar]
- Roumelioti, F.M.; Sotiriou, S.K.; Katsini, V.; Chiourea, M.; Halazonetis, T.D.; Gagos, S. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. Embo. Rep. 2016, 17, 1731–1737. [Google Scholar] [CrossRef]
- Chan, Y.W.; Fugger, K.; West, S.C. Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat. Cell Biol. 2018, 20, 92–103. [Google Scholar] [CrossRef]
- Mason-Osann, E.; Terranova, K.; Lupo, N.; Lock, Y.J.; Carson, L.M.; Flynn, R.L. RAD54 promotes alternative lengthening of telomeres by mediating branch migration. Embo. Rep. 2020, 21, e49495. [Google Scholar] [CrossRef]
- Sarkar, J.; Wan, B.B.; Yin, J.H.; Vallabhaneni, H.; Horvath, K.; Kulikowicz, T.; Bohr, V.A.; Zhang, Y.B.; Lei, M.; Liu, Y. SLX4 contributes to telomere preservation and regulated processing of telomeric joint molecule intermediates. Nucleic Acids Res. 2015, 43, 5912–5923. [Google Scholar] [CrossRef] [Green Version]
- McEachern, M.J.; Haber, J.E. Break-induced replication and recombinational telomere elongation in yeast. Annu. Rev. Biochem. 2006, 75, 111–135. [Google Scholar] [CrossRef]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the Alternative Lengthening of Telomeres Telomere Maintenance Mechanism in Human Cancer Subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suva, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.H.; Nie, X.; Fang, Y.M.; Zhang, Z.P.; Xiao, Y.N.; Mao, Z.W.; Liu, H.Y.; Ren, J.; Wang, F.; Xia, L.X.; et al. A Cisplatin Derivative Tetra-Pt(bpy) as an Oncotherapeutic Agent for Targeting ALT Cancer. JNCI J. Natl. Cancer Inst. 2017, 109, djx061. [Google Scholar] [CrossRef] [PubMed]
- Pompili, L.; Leonetti, C.; Biroccio, A.; Salvati, E. Diagnosis and treatment of ALT tumors: Is Trabectedin a new therapeutic option? J. Exp. Clin. Cancer Res. 2017, 36, 189. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, J.; Pandita, A.; Kamalakar, C.; Johannessen, T.C.; Ohba, S.; Tang, Y.J.; Dalle-Ore, C.L.; Bjerkvig, R.; Pieper, R.O. A subset of PARP inhibitors induces lethal telomere fusion in ALT-dependent tumor cells. Sci. Transl. Med. 2021, 13, eabc7211. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, K.; Yu, Y.; Li, D.; Zhang, Y.; Zhang, K.; Tong, J.; Yang, K.; Jia, S. Alternative Lengthening of Telomeres and Mediated Telomere Synthesis. Cancers 2022, 14, 2194. https://doi.org/10.3390/cancers14092194
Hou K, Yu Y, Li D, Zhang Y, Zhang K, Tong J, Yang K, Jia S. Alternative Lengthening of Telomeres and Mediated Telomere Synthesis. Cancers. 2022; 14(9):2194. https://doi.org/10.3390/cancers14092194
Chicago/Turabian StyleHou, Kailong, Yuyang Yu, Duda Li, Yanduo Zhang, Ke Zhang, Jinkai Tong, Kunxian Yang, and Shuting Jia. 2022. "Alternative Lengthening of Telomeres and Mediated Telomere Synthesis" Cancers 14, no. 9: 2194. https://doi.org/10.3390/cancers14092194