
Development and Characterization of MYB-NFIB Fusion Expression in Adenoid Cystic Carcinoma

 , and
, and 
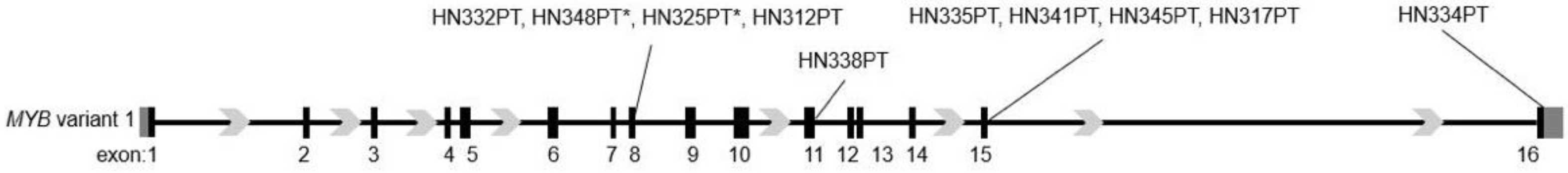{kind=link}
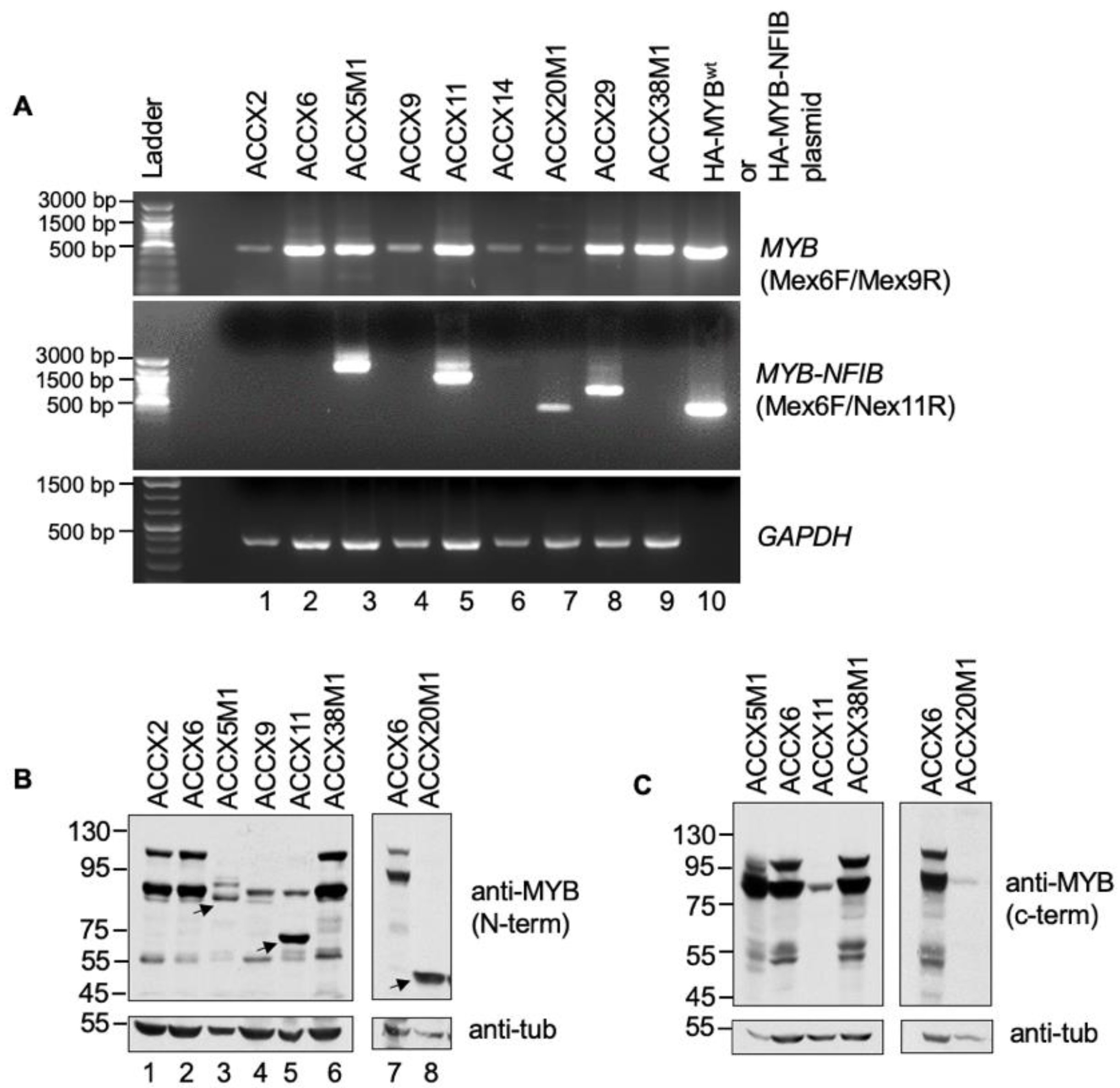{kind=link}
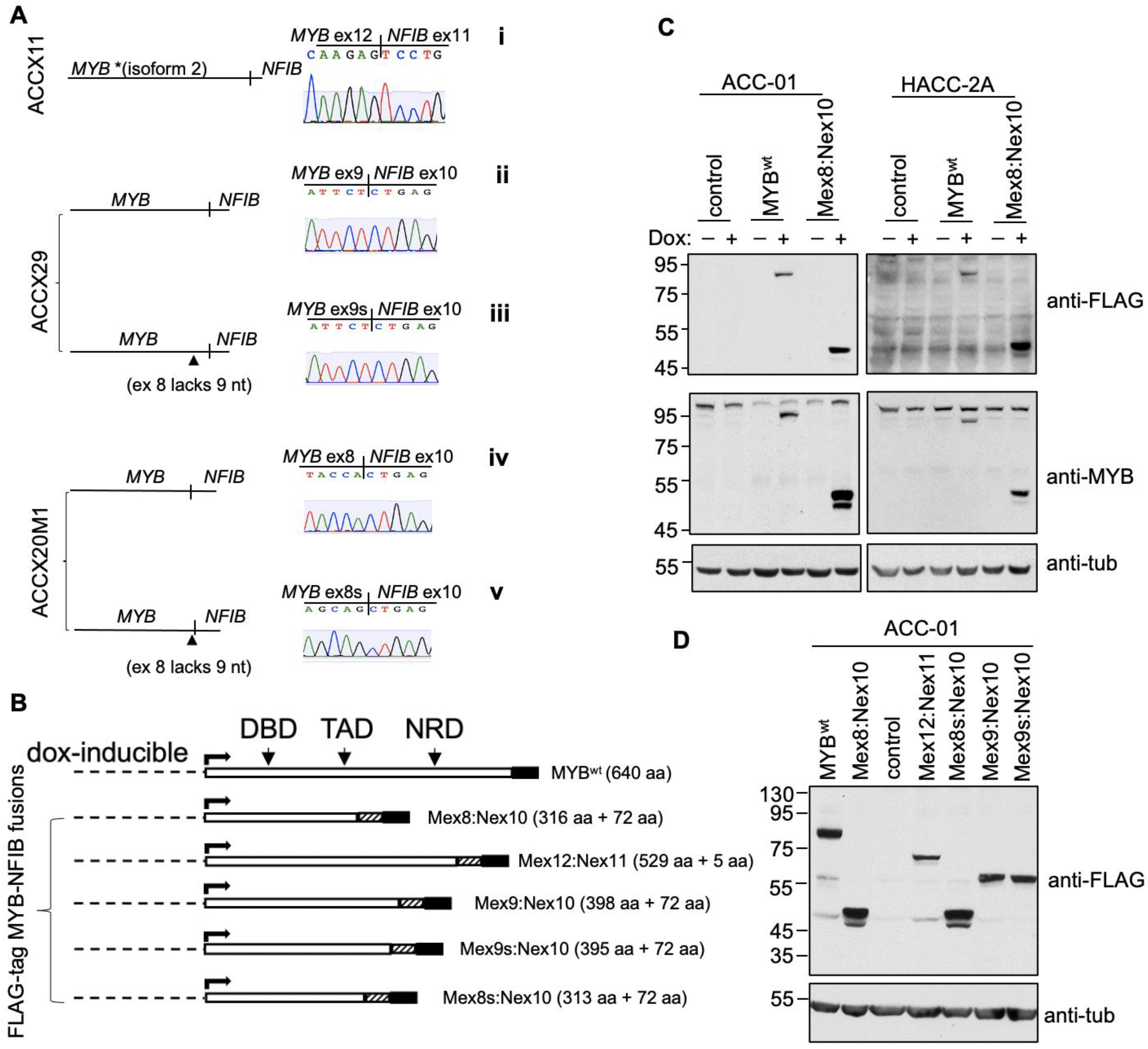{kind=link}
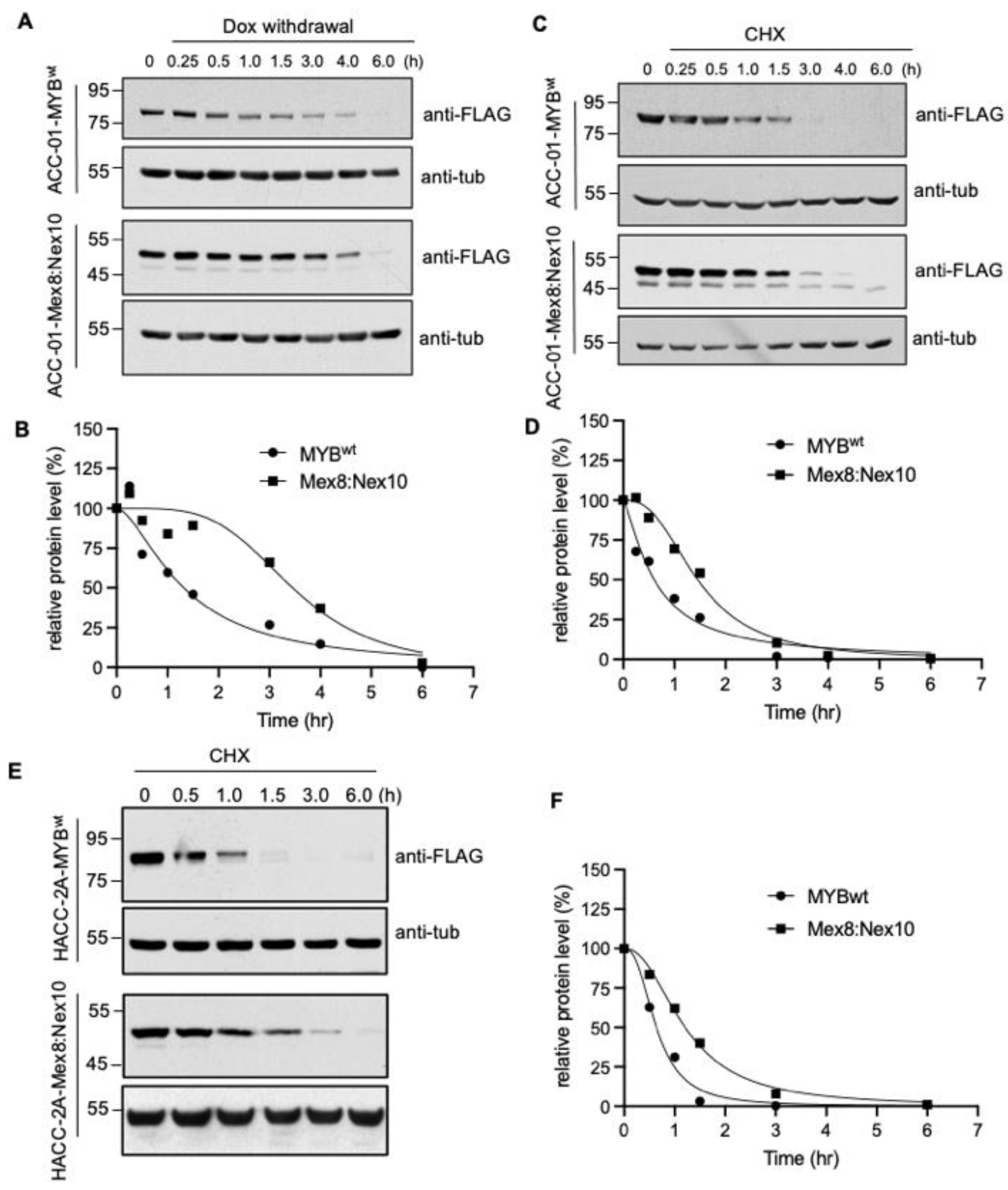{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. ACC Patient-Derived Xenografts and Cell Lines
2.2. Patient Samples and MYB Breakpoint Identification
2.3. MYB-NFIB Expression Analysis, Cloning and Generation of MYB-NFIB Fusion Constructs
2.4. Biochemical Analysis
2.5. RNA Library Generation, Sequencing and Alignment
2.6. RNA-Seq Analysis
2.7. Gene Expression Validation by RT-qPCR
2.8. In Vivo Xenografts Assay
2.9. Additional Statistical Analysis
3. Results
3.1. Heterogenous MYB Breakpoint Loci in MYB-NFIB Fusion Positive ACC
3.2. MYB-NFIB Fusion Expression in ACC Patient-Derived-Xenograft (ACCX)
3.3. Identification and Generation of MYB-NFIB Fusion from ACCXs
3.4. Increased Stability of MYB-NFIB Fusion in ACC Cells
3.5. MYB-NFIB Fusion Expression Enriches Genes Associated in Interferon Signaling Pathway
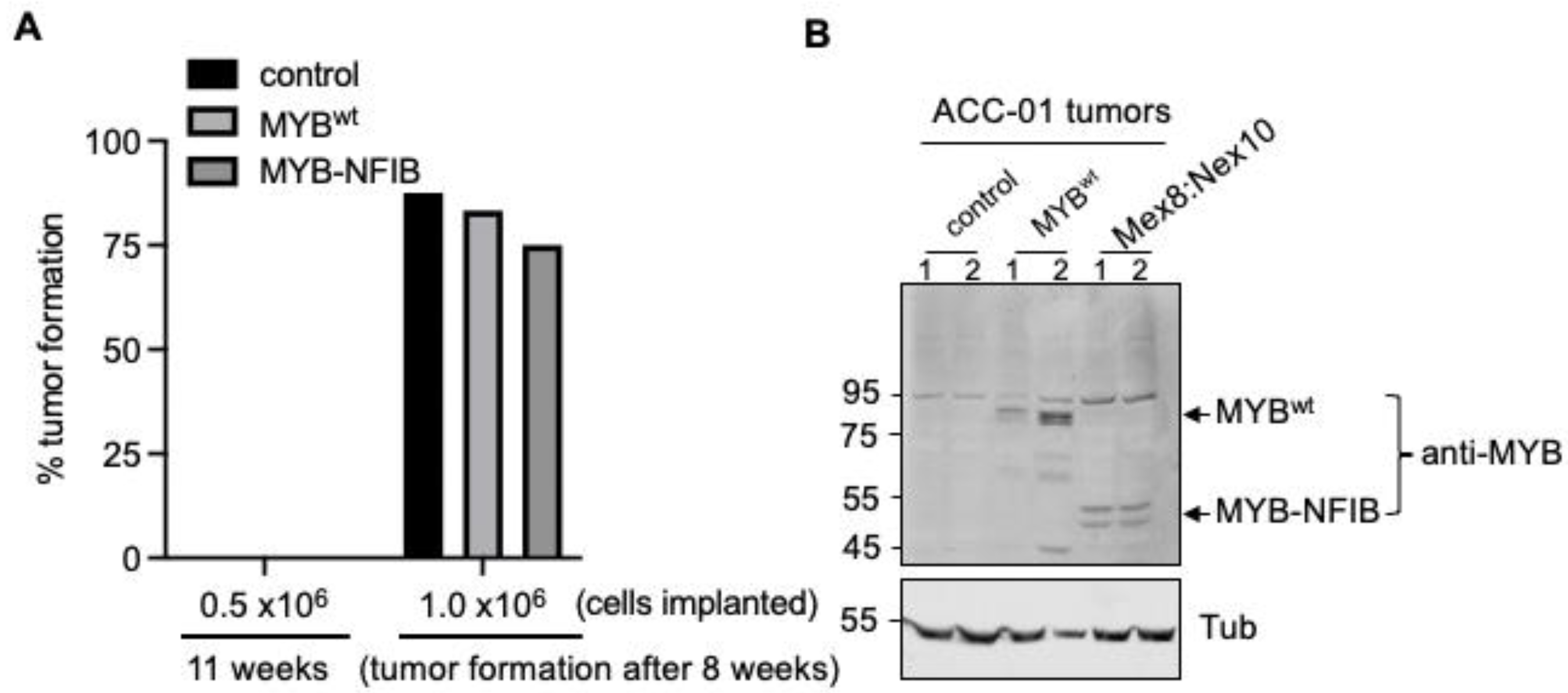
3.6. Development of MYB-NFIB Fusion Expressing ACC Cells for In Vivo Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seethala, R.R.; Stenman, G. Update from the 4th Edition of the World Health Organization Classification of Head and Neck Tumours: Tumors of the Salivary Gland. Head Neck Pathol. 2017, 11, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Okuyemi, O.T. Malignant Salivary Gland Tumors; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Spiro, R.H. Distant metastasis in adenoid cystic carcinoma of salivary origin. Am. J. Surg. 1997, 174, 495–498. [Google Scholar] [CrossRef]
- Park, G.; Roh, J.; Cho, K.; Jin, M.; Choi, S.; Nam, S.; Kim, S. Incidence and risk factors of late recurrence in patients with salivary gland cancer. Clin. Otolaryngol. 2017, 42, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Matsuda, S.; Okada, A.; Sasaki, M.; Imamura, Y.; Yoshimura, H. Adenoid cystic carcinoma of the sublingual gland developing lung metastasis 20 years after primary treatment. Medicine 2021, 100, e28098. [Google Scholar] [CrossRef] [PubMed]
- Sahara, S.; Herzog, A.E.; Nor, J.E. Systemic therapies for salivary gland adenoid cystic carcinoma. Am. J. Cancer Res. 2021, 11, 4092–4110. [Google Scholar] [PubMed]
- Geiger, J.L.; Ismaila, N.; Beadle, B.; Caudell, J.J.; Chau, N.; Deschler, D.; Glastonbury, C.; Kaufman, M.; Lamarre, E.; Lau, H.Y.; et al. Management of Salivary Gland Malignancy: ASCO Guideline. J. Clin. Oncol. 2021, 39, 1909–1941. [Google Scholar] [CrossRef]
- Mueller, S.K.; Haderlein, M.; Lettmaier, S.; Agaimy, A.; Haller, F.; Hecht, M.; Fietkau, R.; Iro, H.; Mantsopoulos, K. Targeted Therapy, Chemotherapy, Immunotherapy and Novel Treatment Options for Different Subtypes of Salivary Gland Cancer. J. Clin. Med. 2022, 11, 720. [Google Scholar] [CrossRef]
- Zheng, J. Oncogenic chromosomal translocations and human cancer (Review). Oncol. Rep. 2013, 30, 2011–2019. [Google Scholar] [CrossRef]
- Nambiar, M.; Kari, V.; Raghavan, S.C. Chromosomal translocations in cancer. Biochim. Biophys. Acta 2008, 1786, 139–152. [Google Scholar] [CrossRef]
- Fröhling, S.; Döhner, H. Chromosomal Abnormalities in Cancer. N. Engl. J. Med. 2008, 359, 722–734. [Google Scholar] [CrossRef]
- Meyer, M.; Watermann, C.; Dreyer, T.; Ergün, S.; Karnati, S. 2021 Update on Diagnostic Markers and Translocation in Salivary Gland Tumors. Int. J. Mol. Sci. 2021, 22, 6771. [Google Scholar] [CrossRef] [PubMed]
- Wysocki, P.; Izumchenko, E.; Meir, J.; Ha, P.K.; Sidransky, D.; Brait, M. Adenoid cystic carcinoma: Emerging role of translocations and gene fusions. Oncotarget 2016, 7, 66239–66254. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Andren, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef] [PubMed]
- Brill, L.B.; Kanner, W.; Fehr, A.; Andrén, Y.; Moskaluk, C.; Löning, T.; Stenman, G.; Frierson Jr, H.F. Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Mod. Pathol. 2011, 24, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Drier, Y.; Cotton, M.J.; Williamson, K.E.; Gillespie, S.; Ryan, R.; Kluk, M.J.; Carey, C.D.; Rodig, S.J.; Sholl, L.M.; Afrogheh, A.H.; et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 2016, 48, 265–272. [Google Scholar] [CrossRef]
- George, O.L.; Ness, S.A. Situational Awareness: Regulation of the Myb Transcription Factor in Differentiation, the Cell Cycle and Oncogenesis. Cancers 2014, 6, 2049–2071. [Google Scholar] [CrossRef]
- Moskaluk, C.; Baras, A.S.; Mancuso, S.; Fan, H.; Davidson, R.J.; Dirks, D.C.; Golden, W.L.; Jr, H.F.F. Development and characterization of xenograft model systems for adenoid cystic carcinoma. Lab. Investig. 2011, 91, 1480–1490. [Google Scholar] [CrossRef]
- ACCRF. PDX Models and Screening Program. Available online: https://accrf.org/tools-for-researchers/pdx-models-and-screening-program/ (accessed on 15 March 2022).
- Humtsoe, J.O.; Kim, H.-S.; Leonard, B.; Ling, S.; Keam, B.; Marchionni, L.; Afsari, B.; Considine, M.; Favorov, A.V.; Fertig, E.J.; et al. Newly Identified Members of FGFR1 Splice Variants Engage in Cross-talk with AXL/AKT Axis in Salivary Adenoid Cystic Carcinoma. Cancer Res. 2021, 81, 1001–1013. [Google Scholar] [CrossRef]
- Li, J.; Perlaky, L.; Rao, P.; Weber, R.S.; El-Naggar, A.K. Development and characterization of salivary adenoid cystic carcinoma cell line. Oral Oncol. 2014, 50, 991–999. [Google Scholar] [CrossRef]
- Warner, K.A.; Oklejas, A.E.; Pearson, A.; Zhang, Z.; Wu, W.; Divi, V.; Rodriguez-Ramirez, C.; Castilho, R.M.; Polverini, P.J.; Nör, J.E. UM-HACC-2A: MYB-NFIB fusion-positive human adenoid cystic carcinoma cell line. Oral Oncol. 2018, 87, 21–28. [Google Scholar] [CrossRef]
- Rettig, E.M.; Talbot, C.C.; Sausen, M.; Jones, S.; Bishop, J.A.; Wood, L.D.; Tokheim, C.; Niknafs, N.; Karchin, R.; Fertig, E.; et al. Whole-Genome Sequencing of Salivary Gland Adenoid Cystic Carcinoma. Cancer Prev. Res. 2016, 9, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Fotakis, G.; Rieder, D.; Haider, M.; Trajanoski, Z.; Finotello, F. NeoFuse: Predicting fusion neoantigens from RNA sequencing data. Bioinformatics 2019, 36, 2260–2261. [Google Scholar] [CrossRef] [PubMed]
- Uhrig, S.; Ellermann, J.; Walther, T.; Burkhardt, P.; Fröhlich, M.; Hutter, B.; Toprak, U.H.; Neumann, O.; Stenzinger, A.; Scholl, C.; et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Genome Res. 2021, 31, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Brayer, K.J.; Frerich, C.A.; Kang, H.; Ness, S.A. Recurrent Fusions in MYB and MYBL1 Define a Common, Transcription Factor–Driven Oncogenic Pathway in Salivary Gland Adenoid Cystic Carcinoma. Cancer Discov. 2015, 6, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Zhu, A.; Ibrahim, J.G.; Love, M. Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2018, 35, 2084–2092. [Google Scholar] [CrossRef]
- Croft, D.; Mundo, A.F.; Haw, R.; Orlic-Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.V.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2013, 42, D472–D477. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. BioSyst. 2015, 12, 477–479. [Google Scholar] [CrossRef]
- Panaccione, A.; Zhang, Y.; Ryan, M.; Moskaluk, C.A.; Anderson, K.S.; Yarbrough, W.G.; Ivanov, S.V. MYB fusions and CD markers as tools for authentication and purification of cancer stem cells from salivary adenoid cystic carcinoma. Stem Cell Res. 2017, 21, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Farzan, M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013, 13, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Mitani, Y.; Li, J.; Rao, P.H.; Zhao, Y.-J.; Bell, D.; Lippman, S.M.; Weber, R.S.; Caulin, C.; El-Naggar, A.K. Comprehensive Analysis of the MYB-NFIB Gene Fusion in Salivary Adenoid Cystic Carcinoma: Incidence, Variability, and Clinicopathologic Significance. Clin. Cancer Res. 2010, 16, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Mitani, Y.; Liu, B.; Rao, P.H.; Borra, V.J.; Zafereo, M.; Weber, R.S.; Kies, M.; Lozano, G.; Futreal, P.A.; Caulin, C.; et al. Novel MYBL1 Gene Rearrangements with Recurrent MYBL1–NFIB Fusions in Salivary Adenoid Cystic Carcinomas Lacking t(6;9) Translocations. Clin. Cancer Res. 2015, 22, 725–733. [Google Scholar] [CrossRef]
- Togashi, Y.; Dobashi, A.; Sakata, S.; Sato, Y.; Baba, S.; Seto, A.; Mitani, H.; Kawabata, K.; Takeuchi, K. MYB and MYBL1 in adenoid cystic carcinoma: Diversity in the mode of genomic rearrangement and transcripts. Mod. Pathol. 2018, 31, 934–946. [Google Scholar] [CrossRef]
- Yang, W.; Lee, K.-W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef]
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef]
- Ness, S. Myb binding proteins: Regulators and cohorts in transformation. Oncogene 1999, 18, 3039–3046. [Google Scholar] [CrossRef]
- Wang, X.; Angelis, N.; Thein, S.L. MYB—A regulatory factor in hematopoiesis. Gene 2018, 665, 6–17. [Google Scholar] [CrossRef]
- Kitagawa, K.; Hiramatsu, Y.; Uchida, C.; Isobe, T.; Hattori, T.; Oda, T.; Shibata, K.; Nakamura, S.; Kikuchi, A. Fbw7 promotes ubiquitin-dependent degradation of c-Myb: Involvement of GSK3-mediated phosphorylation of Thr-572 in mouse c-Myb. Oncogene 2009, 28, 2393–2405. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bies, J.; Feiková, S.; Markus, J.; Wolff, L. Phosphorylation-Dependent Conformation and Proteolytic Stability of c-Myb. Blood Cells Mol. Dis. 2001, 27, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Bies, J.; Wolff, L. Oncogenic activation of c-Myb by carboxyl-terminal truncation leads to decreased proteolysis by the ubiquitin-26S proteasome pathway. Oncogene 1997, 14, 203–212. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lei, W.; Rushton, J.J.; Davis, L.M.; Liu, F.; Ness, S. Positive and Negative Determinants of Target Gene Specificity in Myb Transcription Factors. J. Biol. Chem. 2004, 279, 29519–29527. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lei, W.; O’Rourke, J.P.; Ness, S. Oncogenic mutations cause dramatic, qualitative changes in the transcriptional activity of c-Myb. Oncogene 2005, 25, 795–805. [Google Scholar] [CrossRef]
- Frerich, C.A.; Sedam, H.N.; Kang, H.; Mitani, Y.; El-Naggar, A.K.; Ness, S.A. N-Terminal Truncated Myb with New Transcriptional Activity Produced Through Use of an Alternative MYB Promoter in Salivary Gland Adenoid Cystic Carcinoma. Cancers 2019, 12, 45. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.A.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef]
- Pidugu, V.K.; Wu, M.-M.; Yen, A.-H.; Pidugu, H.B.; Chang, K.-W.; Liu, C.-J.; Lee, T.-C. IFIT1 and IFIT3 promote oral squamous cell carcinoma metastasis and contribute to the anti-tumor effect of gefitinib via enhancing p-EGFR recycling. Oncogene 2019, 38, 3232–3247. [Google Scholar] [CrossRef]
- Andersson, M.K.; Mangiapane, G.; Nevado, P.T.; Tsakaneli, A.; Carlsson, T.; Corda, G.; Nieddu, V.; Abrahamian, C.; Chayka, O.; Rai, L.; et al. ATR is a MYB regulated gene and potential therapeutic target in adenoid cystic carcinoma. Oncogenesis 2020, 9, 5–10. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Humtsoe, J.O.; Kim, H.-S.; Jones, L.; Cevallos, J.; Boileau, P.; Kuo, F.; Morris, L.G.T.; Ha, P. Development and Characterization of MYB-NFIB Fusion Expression in Adenoid Cystic Carcinoma. Cancers 2022, 14, 2263. https://doi.org/10.3390/cancers14092263
Humtsoe JO, Kim H-S, Jones L, Cevallos J, Boileau P, Kuo F, Morris LGT, Ha P. Development and Characterization of MYB-NFIB Fusion Expression in Adenoid Cystic Carcinoma. Cancers. 2022; 14(9):2263. https://doi.org/10.3390/cancers14092263
Chicago/Turabian StyleHumtsoe, Joseph O., Hyun-Su Kim, Leilani Jones, James Cevallos, Philippe Boileau, Fengshen Kuo, Luc G. T. Morris, and Patrick Ha. 2022. "Development and Characterization of MYB-NFIB Fusion Expression in Adenoid Cystic Carcinoma" Cancers 14, no. 9: 2263. https://doi.org/10.3390/cancers14092263
APA StyleHumtsoe, J. O., Kim, H.-S., Jones, L., Cevallos, J., Boileau, P., Kuo, F., Morris, L. G. T., & Ha, P. (2022). Development and Characterization of MYB-NFIB Fusion Expression in Adenoid Cystic Carcinoma. Cancers, 14(9), 2263. https://doi.org/10.3390/cancers14092263