
Atovaquone: An Inhibitor of Oxidative Phosphorylation as Studied in Gynecologic Cancers


 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Cell Lines
2.2. Cell Line Authentication
2.3. Cell Viability Assays
2.4. Monitoring Apoptosis, DNA Breaks and Oxygen Radicals
2.5. Western Blotting
2.6. Imaging Cytometry
2.7. Docking of Atovaquone with Mitochondrial Complexes
2.8. Oxygen Consumption Rate
2.9. Mitochondrial Respiratory Complex Activity
2.10. NMR Sample Preparation
2.11. NMR Acquisition and Processing
2.12. NMR Metabolomic Analysis
2.13. In Vivo Tumor Models and Therapy
3. Results
3.1. Atovaquone Reduces Viability and Induces Apoptosis in Gynecologic Cancer Cell Lines
3.2. Mepron Decreases Tumor Growth in an Orthotopic Model for Ovarian Cancer
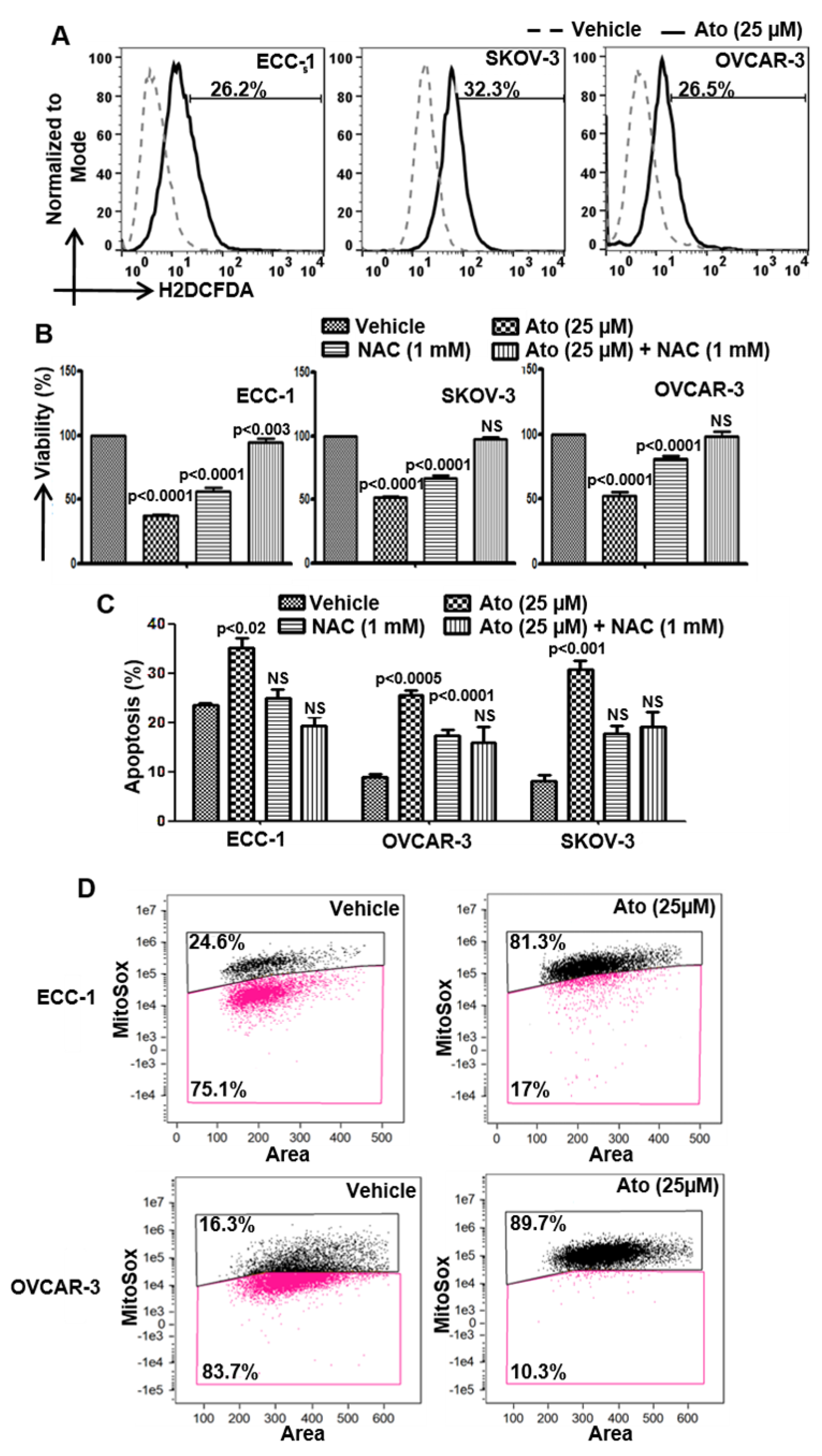
3.3. Atovaquone-Induced Cell Death Occurs through the Generation of Intracellular Oxygen Radical Flux
3.4. Atovaquone Activates p53
3.5. Atovaquone Mimics Ubiquinone When Docked to Q-sites of Mitochondrial Complexes I, II, and III
3.6. Atovaquone Inhibits Oxidative Phosphorylation
3.7. Atovaquone Treatment Alters Energy and Amino Acid Metabolism
3.8. Global Metabolic Perturbations in Response to Atovaquone Treatment of ECC-1 Cells
3.9. Atovaquone Inhibits In Vivo Proliferation of Patient-Derived Ovarian Cancer Stem Cell Initiated Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim. Biophys. Acta BBA Bioenerg. 2010, 1797, 1225–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Banerjee, V.; Czinn, S.; Blanchard, T. Increased reactive oxygen species levels cause ER stress and cytotoxicity in andrographolide treated colon cancer cells. Oncotarget 2017, 8, 26142–26153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ippolito, L.; Marini, A.; Cavallini, L.; Morandi, A.; Pietrovito, L.; Pintus, G.; Giannoni, E.; Schrader, T.; Puhr, M.; Chiarugi, P.; et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget 2016, 7, 61890–61904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakula, M.; Mikula-Pietrasik, J.; Stryczynski, L.; Uruski, P.; Szubert, S.; Moszynski, R.; Szpurek, D.; Sajdak, S.; Tykarski, A.; Ksiazek, K. Mitochondria-related oxidative stress contributes to ovarian cancer-promoting activity of mesothelial cells subjected to malignant ascites. Int. J. Biochem. Cell Biol. 2018, 98, 82–88. [Google Scholar] [CrossRef]
- Solaini, G.; Sgarbi, G.; Baracca, A. Oxidative phosphorylation in cancer cells. Biochim. Biophys. Acta BBA Bioenerg. 2011, 1807, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Yadav, N.; Kumar, S.; Marlowe, T.; Chaudhary, A.K.; Kumar, R.; Wang, J.; O’Malley, J.; Boland, P.M.; Jayanthi, S.; Kumar, T.K.; et al. Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell Death Dis. 2015, 6, e1969. [Google Scholar] [CrossRef]
- Kapur, A.; Beres, T.; Rathi, K.; Nayak, A.P.; Czarnecki, A.; Felder, M.; Gillette, A.; Ericksen, S.S.; Sampene, E.; Skala, M.C.; et al. Oxidative stress via inhibition of the mitochondrial electron transport and Nrf-2-mediated anti-oxidative response regulate the cytotoxic activity of plumbagin. Sci. Rep. 2018, 8, 1073. [Google Scholar] [CrossRef] [Green Version]
- Kapur, A.; Felder, M.; Fass, L.; Kaur, J.; Czarnecki, A.; Rathi, K.; Zeng, S.; Osowski, K.K.; Howell, C.; Xiong, M.P.; et al. Modulation of oxidative stress and subsequent induction of apoptosis and endoplasmic reticulum stress allows citral to decrease cancer cell proliferation. Sci. Rep. 2016, 6, 27530. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [Green Version]
- Mayak, A.P.; Kapur, A.; Barroilhet, L.; Patankar, M.S. Oxidative Phosphorylation: A target for novel therapeutic strategies against ovarian cancer. Cancers 2018, 10, 337. [Google Scholar]
- Alharbi, Y.; Kapur, A.; Felder, M.; Barroilhet, L.; Stein, T.; Pattnaik, B.R.; Patankar, M.S. Plumbagin-induced oxidative stress leads to inhibition of Na(+)/K(+)-ATPase (NKA) in canine cancer cells. Sci. Rep. 2019, 9, 11471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Mutti, L.; Krstic-Demonacos, M.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7, 34084–34099. [Google Scholar] [CrossRef] [Green Version]
- Ashton, T.M.; Fokas, E.; Kunz-Schughart, L.A.; Folkes, L.K.; Anbalagan, S.; Huether, M.; Kelly, C.J.; Pirovano, G.; Buffa, F.M.; Hammond, E.M.; et al. The anti-malarial atovaquone increases radiosensitivity by alleviating tumour hypoxia. Nat. Commun. 2016, 7, 12308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Raghavan, S.; Mehta, P.; Ward, M.R.; Bregenzer, M.E.; Fleck, E.M.A.; Tan, L.; McLean, K.; Buckanovich, R.J.; Mehta, G. Personalized Medicine-Based Approach to Model Patterns of Chemoresistance and Tumor Recurrence Using Ovarian Cancer Stem Cell Spheroids. Clin. Cancer Res. 2017, 23, 6934–6945. [Google Scholar] [CrossRef] [Green Version]
- Bregenzer, M.E.; Davis, C.; Horst, E.N.; Mehta, P.; Novak, C.M.; Raghavan, S.; Snyder, C.S.; Mehta, G. Physiologic Patient Derived 3D Spheroids for Anti-neoplastic Drug Screening to Target Cancer Stem Cells. JoVE J. Vis. Exp. 2019, 149, e59696. [Google Scholar] [CrossRef]
- Mehta, P.; Novak, C.; Raghavan, S.; Ward, M.; Mehta, G. Self-Renewal and CSCs In Vitro Enrichment: Growth as Floating Spheres. Methods Mol. Biol. 2018, 1692, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- PubChem Database. Atovaquone, CID = 74989. Available online: https://pubchem.ncbi.nlm.nih.gov/#query=atovaquone (accessed on 31 July 2021).
- PubChem Database. 1,4-Naphthoquinone, CID = 8530. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/1_4-Naphthoquinone (accessed on 31 July 2021).
- Fendel, U.; Tocilescu, M.A.; Kerscher, S.; Brandt, U. Exploring the inhibitor binding pocket of respiratory complex I. Biochim. Biophys. Acta BBA Bioenerg. 2008, 1777, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Bhute, V.J.; Palecek, S.P. Metabolic responses induced by DNA damage and poly (ADP-ribose) polymerase (PARP) inhibition in MCF-7 cells. Metabolomics 2015, 11, 1779–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robin, C.; Le, M.P.; Melica, G.; Massias, L.; Redjoul, R.; Khoudour, N.; Leclerc, M.; Beckerich, F.; Maury, S.; Hulin, A.; et al. Plasma concentrations of atovaquone given to immunocompromised patients to prevent Pneumocystis jirovecii. J. Antimicrob. Chemother. 2017, 72, 2602–2606. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Painter, H.J.; Morrisey, J.M.; Mather, M.W.; Vaidya, A.B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446, 88–91. [Google Scholar] [CrossRef]
- Painter, H.J.; Morrisey, J.M.; Vaidya, A.B. Mitochondrial electron transport inhibition and viability of intraerythrocytic Plasmodium falciparum. Antimicrob. Agents Chemother. 2010, 54, 5281–5287. [Google Scholar] [CrossRef] [Green Version]
- Pasto, A.; Bellio, C.; Pilotto, G.; Ciminale, V.; Silic-Benussi, M.; Guzzo, G.; Rasola, A.; Frasson, C.; Nardo, G.; Zulato, E.; et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget 2014, 5, 4305–4319. [Google Scholar] [CrossRef] [Green Version]
- Einstein, A.; Podolsky, B.; Rosen, N. Can Quantum-Mechanical Description of Physical Reality Be Considered Complete? Phys. Rev. 1935, 47, 777–780. [Google Scholar] [CrossRef] [Green Version]
- Ward Rashidi, M.R.; Mehta, P.; Bregenzer, M.; Raghavan, S.; Fleck, E.M.; Horst, E.N.; Harissa, Z.; Ravikumar, V.; Brady, S.; Bild, A.; et al. Engineered 3D Model of Cancer Stem Cell Enrichment and Chemoresistance. Neoplasia 2019, 21, 822–836. [Google Scholar] [CrossRef]
- Raghavan, S.; Ward, M.R.; Rowley, K.R.; Wold, R.M.; Takayama, S.; Buckanovich, R.J.; Mehta, G. Formation of stable small cell number three-dimensional ovarian cancer spheroids using hanging drop arrays for preclinical drug sensitivity assays. Gynecol. Oncol. 2015, 138, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Parkes, E.E.; Kennedy, R.D. Clinical Application of Poly(ADP-Ribose) Polymerase Inhibitors in High-Grade Serous Ovarian Cancer. Oncologist 2016, 21, 586–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.E.; Meghani, K.; Brault, M.E.; Leclerc, L.; He, Y.J.; Day, T.A.; Elias, K.M.; Drapkin, R.; Weinstock, D.M.; Dao, F.; et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell Rep. 2016, 14, 429–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Wang, L.; Chen, H.; Hao, J.; Ni, J.; Chang, L.; Duan, W.; Graham, P.; Li, Y. Targeting epithelial-mesenchymal transition and cancer stem cells for chemoresistant ovarian cancer. Oncotarget 2016, 7, 55771–55788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birth, D.; Kao, W.C.; Hunte, C. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat. Commun. 2014, 5, 4029. [Google Scholar] [CrossRef] [Green Version]
- Fabi, F.; Adam, P.; Vincent, K.; Demontigny, F.; Parent, S.; Joncas, F.H.; Asselin, E. Inhibition of CRM1 activity sensitizes endometrial and ovarian cell lines to TRAIL-induced cell death. Cell Commun. Signal. 2018, 16, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef]
- Felippe Goncalves-de-Albuquerque, C.; Ribeiro Silva, A.; Ignacio da Silva, C.; Caire Castro-Faria-Neto, H.; Burth, P. Na/K Pump and Beyond: Na/K-ATPase as a Modulator of Apoptosis and Autophagy. Molecules 2017, 22, 578. [Google Scholar] [CrossRef] [Green Version]
- Cloer, E.W.; Goldfarb, D.; Schrank, T.P.; Weissman, B.E.; Major, M.B. NRF2 Activation in Cancer: From DNA to Protein. Cancer Res. 2019, 79, 889–898. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapur, A.; Mehta, P.; Simmons, A.D.; Ericksen, S.S.; Mehta, G.; Palecek, S.P.; Felder, M.; Stenerson, Z.; Nayak, A.; Dominguez, J.M.A.; et al. Atovaquone: An Inhibitor of Oxidative Phosphorylation as Studied in Gynecologic Cancers. Cancers 2022, 14, 2297. https://doi.org/10.3390/cancers14092297
Kapur A, Mehta P, Simmons AD, Ericksen SS, Mehta G, Palecek SP, Felder M, Stenerson Z, Nayak A, Dominguez JMA, et al. Atovaquone: An Inhibitor of Oxidative Phosphorylation as Studied in Gynecologic Cancers. Cancers. 2022; 14(9):2297. https://doi.org/10.3390/cancers14092297
Chicago/Turabian StyleKapur, Arvinder, Pooja Mehta, Aaron D Simmons, Spencer S. Ericksen, Geeta Mehta, Sean P. Palecek, Mildred Felder, Zach Stenerson, Amruta Nayak, Jose Maria Ayuso Dominguez, and et al. 2022. "Atovaquone: An Inhibitor of Oxidative Phosphorylation as Studied in Gynecologic Cancers" Cancers 14, no. 9: 2297. https://doi.org/10.3390/cancers14092297
APA StyleKapur, A., Mehta, P., Simmons, A. D., Ericksen, S. S., Mehta, G., Palecek, S. P., Felder, M., Stenerson, Z., Nayak, A., Dominguez, J. M. A., Patankar, M., & Barroilhet, L. M. (2022). Atovaquone: An Inhibitor of Oxidative Phosphorylation as Studied in Gynecologic Cancers. Cancers, 14(9), 2297. https://doi.org/10.3390/cancers14092297