
Inhibition of PI3K Class IA Kinases Using GDC-0941 Overcomes Cytoprotection of Multiple Myeloma Cells in the Osteoclastic Bone Marrow Microenvironment Enhancing the Efficacy of Current Clinical Therapeutics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Determination of Cell Proliferation in eGFP-MM Cell Lines in Co-Culture with m-Cherry-HS5 Cells
2.3. Determination of MM Cell Viability in Co-Culture with OCs
2.4. Cell Adhesion Assays
2.5. Analysis of Cell Area and Formation of F-Actin Rings in OCs
2.6. In Vitro Bone Resorption Analysis
2.7. In Vivo Testing of Drug Efficacy Using the GFP-5T33 and C57BL/KaLwRij Mouse Model
2.8. Statistics
3. Results
3.1. PI3K Class IA Kinases Regulate a Signalling Node Involved in MM Drug Resistance to Dexamethasone Mediated by the BM Mesenchymal/Fibroblastic and OC Niches
3.2. Inhibition of Single PI3K Class IA Isoforms Fails to Prevent Resistance to Dexamethasone Mediated by BM Mesenchymal Cells and OCs
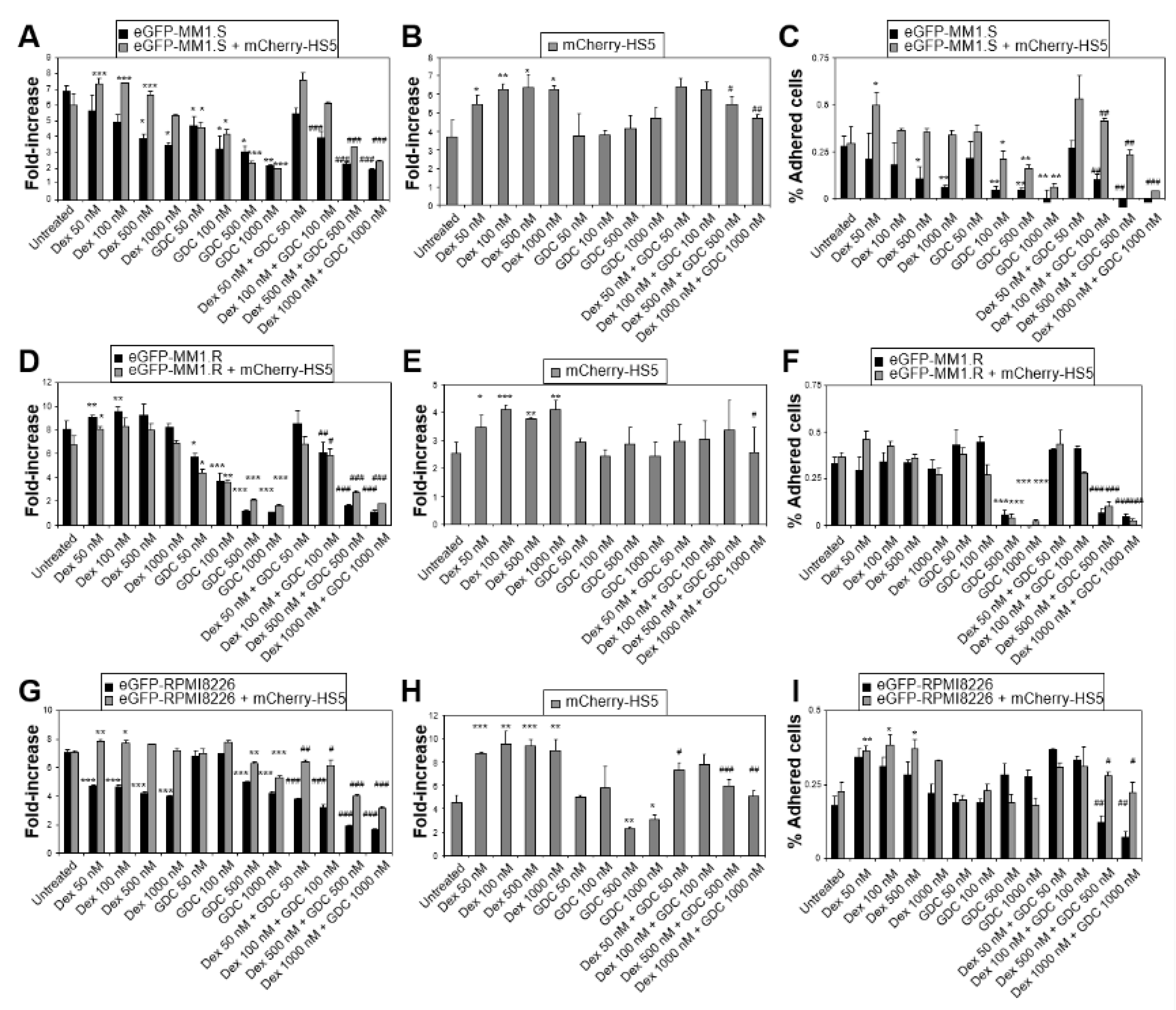
3.3. Simultaneous Targeting of All PI3K Class IA Isoforms with GDC-0941 Overcomes Resistance to Dexamethasone Mediated by Both BM Mesenchymal Cells and OCs
3.4. GDC-0941 Inhibits Bone Resorption by Osteoclasts by Repressing OC Differentiation and the Organisation of Sealing Zones
3.5. GDC-0941 Enhances the In Vivo Efficacy of Dexamethasone in the GFP-5T33/C57Rawji MM Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramasamy, K.; Gay, F.; Weisel, K.; Zweegman, S.; Mateos, M.V.; Richardson, P. Improving outcomes for patients with relapsed multiple myeloma: Challenges and considerations of current and emerging treatment options. Blood Rev. 2021, 49, 100808. [Google Scholar] [CrossRef] [PubMed]
- Davies, F.E.; Pawlyn, C.; Usmani, S.Z.; San-Miguel, J.F.; Einsele, H.; Boyle, E.M.; Corre, J.; Auclair, D.; Cho, H.J.; Lonial, S.; et al. Perspectives on the Risk-Stratified Treatment of Multiple Myeloma. Blood Cancer Discov. 2022, 3, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Durer, C.; Durer, S.; Lee, S.; Chakraborty, R.; Malik, M.N.; Rafae, A.; Zar, M.A.; Kamal, A.; Rosko, N.; Samaras, C.; et al. Treatment of relapsed multiple myeloma: Evidence-based recommendations. Blood Rev. 2020, 39, 100616. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Hudecek, M.; Einsele, H. What is the future of immunotherapy in multiple myeloma? Blood 2020, 136, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Solimando, A.G.; Da Via, M.C.; Cicco, S.; Leone, P.; Di Lernia, G.; Giannico, D.; Desantis, V.; Frassanito, M.A.; Morizio, A.; Delgado Tascon, J.; et al. High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. J. Clin. Med. 2019, 8, 997. [Google Scholar] [CrossRef] [Green Version]
- Ghobrial, I.M.; Detappe, A.; Anderson, K.C.; Steensma, D.P. The bone-marrow niche in MDS and MGUS: Implications for AML and MM. Nat. Rev. Clin. Oncol. 2018, 15, 219–233. [Google Scholar] [CrossRef]
- Solimando, A.G.; Malerba, E.; Leone, P.; Prete, M.; Terragna, C.; Cavo, M.; Racanelli, V. Drug resistance in multiple myeloma: Soldiers and weapons in the bone marrow niche. Front. Oncol. 2022, 12, 973836. [Google Scholar] [CrossRef]
- Dadzie, T.G.; Green, A.C. The role of the bone microenvironment in regulating myeloma residual disease and treatment. Front. Oncol. 2022, 12, 999939. [Google Scholar] [CrossRef]
- Danziger, S.A.; McConnell, M.; Gockley, J.; Young, M.H.; Rosenthal, A.; Schmitz, F.; Reiss, D.J.; Farmer, P.; Alapat, D.V.; Singh, A.; et al. Bone marrow microenvironments that contribute to patient outcomes in newly diagnosed multiple myeloma: A cohort study of patients in the Total Therapy clinical trials. PLoS Med. 2020, 17, e1003323. [Google Scholar] [CrossRef]
- Visram, A.; Dasari, S.; Anderson, E.; Kumar, S.; Kourelis, T.V. Relapsed multiple myeloma demonstrates distinct patterns of immune microenvironment and malignant cell-mediated immunosuppression. Blood Cancer J. 2021, 11, 45. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Hemann, M.T. DNA damage-mediated induction of a chemoresistant niche. Cell 2010, 143, 355–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 Paracrine Network Links Cancer Chemoresistance and Metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramasamy, K.; Khatun, H.; Macpherson, L.; Caley, M.P.; Sturge, J.; Mufti, G.J.; Schey, S.A.; Calle, Y. Fluorescence-based experimental model to evaluate the concomitant effect of drugs on the tumour microenvironment and cancer cells. Br. J. Haematol. 2012, 157, 564–579. [Google Scholar] [CrossRef]
- McMillin, D.W.; Delmore, J.; Weisberg, E.; Negri, J.M.; Geer, D.C.; Klippel, S.; Mitsiades, N.; Schlossman, R.L.; Munshi, N.C.; Kung, A.L.; et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat. Med. 2010, 16, 483–489. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Rao, L.; Moschetta, M.; Ria, R.; Di, M.L.; De, L.A.; Racanelli, V.; Catacchio, I.; Berardi, S.; Basile, A.; et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: In vitro and in vivo studies. Leukemia 2014, 28, 904–916. [Google Scholar] [CrossRef] [Green Version]
- Slany, A.; Haudek-Prinz, V.; Meshcheryakova, A.; Bileck, A.; Lamm, W.; Zielinski, C.; Gerner, C.; Drach, J. Extracellular Matrix Remodeling by Bone Marrow Fibroblast-like Cells Correlates with Disease Progression in Multiple Myeloma. J. Proteome. Res. 2013, 13, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Babarovic, E.; Valkovic, T.; Stifter, S.; Budisavljevic, I.; Seili-Bekafigo, I.; Duletic-Nacinovic, A.; Lucin, K.; Jonjic, N. Assessment of bone marrow fibrosis and angiogenesis in monitoring patients with multiple myeloma. Am. J. Clin. Pathol. 2012, 137, 870–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasch, S.; Lund, T.; Asmussen, J.T.; Lerberg Nielsen, A.; Faebo Larsen, R.; Osterheden Andersen, M.; Abildgaard, N. Multiple Myeloma Associated Bone Disease. Cancers (Basel) 2020, 12, 2113. [Google Scholar] [CrossRef]
- Abe, M.; Hiura, K.; Wilde, J.; Shioyasono, A.; Moriyama, K.; Hashimoto, T.; Kido, S.; Oshima, T.; Shibata, H.; Ozaki, S.; et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: A vicious cycle between bone destruction and myeloma expansion. Blood 2004, 104, 2484–2491. [Google Scholar] [CrossRef]
- Moreaux, J.; Hose, D.; Kassambara, A.; Reme, T.; Moine, P.; Requirand, G.; Goldschmidt, H.; Klein, B. Osteoclast-gene expression profiling reveals osteoclast-derived CCR2 chemokines promoting myeloma cell migration. Blood 2011, 117, 1280–1290. [Google Scholar] [CrossRef]
- Alici, E.; Konstantinidis, K.V.; Aints, A.; Dilber, M.S.; Abedi-Valugerdi, M. Visualization of 5T33 myeloma cells in the C57BL/KaLwRij mouse: Establishment of a new syngeneic murine model of multiple myeloma. Exp. Hematol. 2004, 32, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Gries, M.; Kurihara, N.; Honjo, T.; Anderson, J.; Donnenberg, V.; Donnenberg, A.; Ghobrial, I.; Mapara, M.Y.; Stirling, D.; et al. Thalidomide derivative CC-4047 inhibits osteoclast formation by down-regulation of PU.1. Blood 2006, 107, 3098–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, D.M.; Chen, C.; Niesvizky, R.; Wang, M.; Belch, A.; Stadtmauer, E.A.; Siegel, D.; Borrello, I.; Rajkumar, S.V.; Chanan-Khan, A.A.; et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N. Engl. J. Med. 2007, 357, 2133–2142. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Oriol, A.; Beksac, M.; Liberati, A.M.; Galli, M.; Schjesvold, F.; Lindsay, J.; Weisel, K.; White, D.; Facon, T.; et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed or refractory multiple myeloma previously treated with lenalidomide (OPTIMISMM): A randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 781–794. [Google Scholar] [CrossRef]
- Maouche, N.; Kishore, B.; Bhatti, Z.; Basu, S.; Karim, F.; Sundararaman, S.; Collings, F.; Tseu, B.; Leary, H.; Ryman, N.; et al. Panobinostat in combination with bortezomib and dexamethasone in multiply relapsed and refractory myeloma; UK routine care cohort. PLoS ONE 2022, 17, e0270854. [Google Scholar] [CrossRef]
- McCaughan, G.J.; Gandolfi, S.; Moore, J.J.; Richardson, P.G. Lenalidomide, bortezomib and dexamethasone induction therapy for the treatment of newly diagnosed multiple myeloma: A practical review. Br. J. Haematol. 2022, 199, 190–204. [Google Scholar] [CrossRef]
- O’Sullivan, B.T.; Cutler, D.J.; Hunt, G.E.; Walters, C.; Johnson, G.F.; Caterson, I.D. Pharmacokinetics of dexamethasone and its relationship to dexamethasone suppression test outcome in depressed patients and healthy control subjects. Biol. Psychiatry 1997, 41, 574–584. [Google Scholar] [CrossRef]
- Hofmann, C.; Stuhmer, T.; Schmiedl, N.; Wetzker, R.; Mottok, A.; Rosenwald, A.; Langer, C.; Zovko, J.; Chatterjee, M.; Einsele, H.; et al. PI3K-dependent multiple myeloma cell survival is mediated by the PIK3CA isoform. Br. J. Haematol. 2014, 166, 529–539. [Google Scholar] [CrossRef]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Perrone, G.; Miura, N.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Santo, L.; Vallet, S.; et al. PI3K/p110{delta} is a novel therapeutic target in multiple myeloma. Blood 2010, 116, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Shugg, R.P.; Thomson, A.; Tanabe, N.; Kashishian, A.; Steiner, B.H.; Puri, K.D.; Pereverzev, A.; Lannutti, B.J.; Jirik, F.R.; Dixon, S.J.; et al. Effects of isoform-selective phosphatidylinositol 3-kinase inhibitors on osteoclasts: Actions on cytoskeletal organization, survival, and resorption. J. Biol. Chem. 2013, 288, 35346–35357. [Google Scholar] [CrossRef]
- Munugalavadla, V.; Mariathasan, S.; Slaga, D.; Du, C.; Berry, L.; Del, R.G.; Yan, Y.; Boe, M.; Sun, L.; Friedman, L.S.; et al. The PI3K inhibitor GDC-0941 combines with existing clinical regimens for superior activity in multiple myeloma. Oncogene 2014, 33, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Han, S.; Sun, Y. An IL6-STAT3 loop mediates resistance to PI3K inhibitors by inducing epithelial-mesenchymal transition and cancer stem cell expansion in human breast cancer cells. Biochem. Biophys. Res. Commun. 2014, 453, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.W.; Gao, J.; Zheng, T.; Rui, C.; Li, T.; Bian, X.; Cheng, H.; Liu, P.; Luo, F. Macrophages confer resistance to PI3K inhibitor GDC-0941 in breast cancer through the activation of NF-kappaB signaling. Cell Death Dis. 2018, 9, 809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sezer, O.; Jakob, C.; Eucker, J.; Niemoller, K.; Gatz, F.; Wernecke, K.; Possinger, K. Serum levels of the angiogenic cytokines basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF) in multiple myeloma. Eur. J. Haematol. 2001, 66, 83–88. [Google Scholar] [CrossRef]
- Saltarella, I.; Morabito, F.; Giuliani, N.; Terragna, C.; Omede, P.; Palumbo, A.; Bringhen, S.; De Paoli, L.; Martino, E.; Larocca, A.; et al. Prognostic or predictive value of circulating cytokines and angiogenic factors for initial treatment of multiple myeloma in the GIMEMA MM0305 randomized controlled trial. J. Hematol. Oncol. 2019, 12, 4. [Google Scholar] [CrossRef]
- Chellaiah, M.A.; Soga, N.; Swanson, S.; McAllister, S.; Alvarez, U.; Wang, D.; Dowdy, S.F.; Hruska, K.A. Rho-A is critical for osteoclast podosome organization, motility, and bone resorption. J. Biol. Chem. 2000, 275, 11993–12002. [Google Scholar] [CrossRef] [Green Version]
- Lakkakorpi, P.T.; Wesolowski, G.; Zimolo, Z.; Rodan, G.A.; Rodan, S.B. Phosphatidylinositol 3-kinase association with the osteoclast cytoskeleton, and its involvement in osteoclast attachment and spreading. Exp. Cell Res. 1997, 237, 296–306. [Google Scholar] [CrossRef]
- Teti, A.; Colucci, S.; Grano, M.; Argentino, L.; Zambonin Zallone, A. Protein kinase C affects microfilaments, bone resorption, and [Ca2+]o sensing in cultured osteoclasts. Am. J. Physiol. 1992, 263, C130–C139. [Google Scholar] [CrossRef]
- Xin, Y.; Liu, Y.; Liu, D.; Li, J.; Zhang, C.; Wang, Y.; Zheng, S. New Function of RUNX2 in Regulating Osteoclast Differentiation via the AKT/NFATc1/CTSK Axis. Calcif. Tissue Int. 2020, 106, 553–566. [Google Scholar] [CrossRef]
- Asosingh, K. Migration, adhesion and differentiation of malignant plasma cells in the 5T murine model of myeloma. Verh. K. Acad. Geneeskd. Belg. 2003, 65, 127–134. [Google Scholar]
- Harada, T.; Hiasa, M.; Teramachi, J.; Abe, M. Myeloma-Bone Interaction: A Vicious Cycle via TAK1-PIM2 Signaling. Cancers (Basel) 2021, 13, 4441. [Google Scholar] [CrossRef] [PubMed]
- Amachi, R.; Hiasa, M.; Teramachi, J.; Harada, T.; Oda, A.; Nakamura, S.; Hanson, D.; Watanabe, K.; Fujii, S.; Miki, H.; et al. A vicious cycle between acid sensing and survival signaling in myeloma cells: Acid-induced epigenetic alteration. Oncotarget 2016, 7, 70447–70461. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Bat-Erdene, A.; Tenshin, H.; Cui, Q.; Teramachi, J.; Hiasa, M.; Oda, A.; Harada, T.; Miki, H.; Sogabe, K.; et al. Reveromycin A, a novel acid-seeking agent, ameliorates bone destruction and tumor growth in multiple myeloma. Haematologica 2021, 106, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Abe, M.; Hiasa, M.; Oda, A.; Amou, H.; Nakano, A.; Takeuchi, K.; Kitazoe, K.; Kido, S.; Inoue, D.; et al. Myeloma cell-osteoclast interaction enhances angiogenesis together with bone resorption: A role for vascular endothelial cell growth factor and osteopontin. Clin. Cancer Res. 2007, 13, 816–823. [Google Scholar] [CrossRef] [Green Version]
- Noll, J.E.; Williams, S.A.; Tong, C.M.; Wang, H.; Quach, J.M.; Purton, L.E.; Pilkington, K.; To, L.B.; Evdokiou, A.; Gronthos, S.; et al. Myeloma plasma cells alter the bone marrow microenvironment by stimulating the proliferation of mesenchymal stromal cells. Haematologica 2014, 99, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrabel, D.; Pour, L.; Sevcikova, S. The impact of NF-kappaB signaling on pathogenesis and current treatment strategies in multiple myeloma. Blood Rev. 2019, 34, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Bolzoni, M.; Storti, P.; Bonomini, S.; Todoerti, K.; Guasco, D.; Toscani, D.; Agnelli, L.; Neri, A.; Rizzoli, V.; Giuliani, N. Immunomodulatory drugs lenalidomide and pomalidomide inhibit multiple myeloma-induced osteoclast formation and the RANKL/OPG ratio in the myeloma microenvironment targeting the expression of adhesion molecules. Exp. Hematol. 2013, 41, 387–397 e381. [Google Scholar] [CrossRef]
- Breitkreutz, I.; Raab, M.S.; Vallet, S.; Hideshima, T.; Raje, N.; Mitsiades, C.; Chauhan, D.; Okawa, Y.; Munshi, N.C.; Richardson, P.G.; et al. Lenalidomide inhibits osteoclastogenesis, survival factors and bone-remodeling markers in multiple myeloma. Leukemia 2008, 22, 1925–1932. [Google Scholar] [CrossRef] [Green Version]
- Piva, R.; Ruggeri, B.; Williams, M.; Costa, G.; Tamagno, I.; Ferrero, D.; Giai, V.; Coscia, M.; Peola, S.; Massaia, M.; et al. CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood 2008, 111, 2765–2775. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Hideshima, T.; Xing, L.; Wang, S.; Zhou, W.; Samur, M.K.; Sewastianik, T.; Ogiya, D.; An, G.; Gao, S.; et al. ERK signaling mediates resistance to immunomodulatory drugs in the bone marrow microenvironment. Sci. Adv. 2021, 7, eabg2697. [Google Scholar] [CrossRef] [PubMed]
- Keane, N.A.; Glavey, S.V.; Krawczyk, J.; O’Dwyer, M. AKT as a therapeutic target in multiple myeloma. Expert Opin. Targets 2014, 18, 897–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lin, N.; Li, Y. The PI3K/AKT signaling pathway regulates ABCG2 expression and confers resistance to chemotherapy in human multiple myeloma. Oncol. Rep. 2019, 41, 1678–1690. [Google Scholar] [CrossRef] [PubMed]
- Zollinger, A.; Stuhmer, T.; Chatterjee, M.; Gattenlohner, S.; Haralambieva, E.; Muller-Hermelink, H.K.; Andrulis, M.; Greiner, A.; Wesemeier, C.; Rath, J.C.; et al. Combined functional and molecular analysis of tumor cell signaling defines 2 distinct myeloma subgroups: Akt-dependent and Akt-independent multiple myeloma. Blood 2008, 112, 3403–3411. [Google Scholar] [CrossRef]
- Heinemann, L.; Mollers, K.M.; Ahmed, H.M.M.; Wei, L.; Sun, K.; Nimmagadda, S.C.; Frank, D.; Baumann, A.; Poos, A.M.; Dugas, M.; et al. Inhibiting PI3K-AKT-mTOR Signaling in Multiple Myeloma-Associated Mesenchymal Stem Cells Impedes the Proliferation of Multiple Myeloma Cells. Front. Oncol. 2022, 12, 874325. [Google Scholar] [CrossRef]
- Garcia, V.M.; Baird, R.D.; Shah, K.J.; Basu, B.; Tunariu, N.; Blanco, M.; Cassier, P.A.; Pedersen, J.V.; Puglisi, M.; Sarker, D.; et al. A phase I study evaluating GDC-0941, an oral phosphoinositide-3 kinase (PI3K) inhibitor, in patients with advanced solid tumors or multiple myeloma. J. Clin. Oncol. 2011, 29, 3021. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Khong, T.; Spencer, A. Overcoming inherent resistance to histone deacetylase inhibitors in multiple myeloma cells by targeting pathways integral to the actin cytoskeleton. Cell Death Dis. 2014, 5, e1134. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, S.; Ri, M.; Kanamori, T.; Aoki, S.; Yoshida, T.; Narita, T.; Totani, H.; Ito, A.; Kusumoto, S.; Ishida, T.; et al. Potent antitumor effect of combination therapy with sub-optimal doses of Akt inhibitors and pomalidomide plus dexamethasone in multiple myeloma. Oncol. Lett. 2018, 15, 9450–9456. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.J.; Mishima, Y.; Shi, J.; Sklavenitis-Pistofidis, R.; Redd, R.A.; Moschetta, M.; Manier, S.; Roccaro, A.M.; Sacco, A.; Tai, Y.T.; et al. Progression signature underlies clonal evolution and dissemination of multiple myeloma. Blood 2021, 137, 2360–2372. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kikuchi, H.; Amofa, E.; Mcenery, M.; Schey, S.A.; Ramasamy, K.; Farzaneh, F.; Calle, Y. Inhibition of PI3K Class IA Kinases Using GDC-0941 Overcomes Cytoprotection of Multiple Myeloma Cells in the Osteoclastic Bone Marrow Microenvironment Enhancing the Efficacy of Current Clinical Therapeutics. Cancers 2023, 15, 462. https://doi.org/10.3390/cancers15020462
Kikuchi H, Amofa E, Mcenery M, Schey SA, Ramasamy K, Farzaneh F, Calle Y. Inhibition of PI3K Class IA Kinases Using GDC-0941 Overcomes Cytoprotection of Multiple Myeloma Cells in the Osteoclastic Bone Marrow Microenvironment Enhancing the Efficacy of Current Clinical Therapeutics. Cancers. 2023; 15(2):462. https://doi.org/10.3390/cancers15020462
Chicago/Turabian StyleKikuchi, Hugh, Eunice Amofa, Maeve Mcenery, Steve Arthur Schey, Karthik Ramasamy, Farzin Farzaneh, and Yolanda Calle. 2023. "Inhibition of PI3K Class IA Kinases Using GDC-0941 Overcomes Cytoprotection of Multiple Myeloma Cells in the Osteoclastic Bone Marrow Microenvironment Enhancing the Efficacy of Current Clinical Therapeutics" Cancers 15, no. 2: 462. https://doi.org/10.3390/cancers15020462
APA StyleKikuchi, H., Amofa, E., Mcenery, M., Schey, S. A., Ramasamy, K., Farzaneh, F., & Calle, Y. (2023). Inhibition of PI3K Class IA Kinases Using GDC-0941 Overcomes Cytoprotection of Multiple Myeloma Cells in the Osteoclastic Bone Marrow Microenvironment Enhancing the Efficacy of Current Clinical Therapeutics. Cancers, 15(2), 462. https://doi.org/10.3390/cancers15020462