
The Combination of ATM and Chk1 Inhibitors Induces Synthetic Lethality in Colorectal Cancer Cells
 , ,
, ,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Drug Stimulation
2.2. Cell Viability Assay
2.3. Western Blotting Analyses
2.4. Cell Cycle Analysis
2.5. Immunofluorescence Staining
2.6. TdT-Mediated dUTP Nick-End Labeling (TUNEL)
2.7. In Vivo Experiment
2.8. Statistical Analysis
3. Results
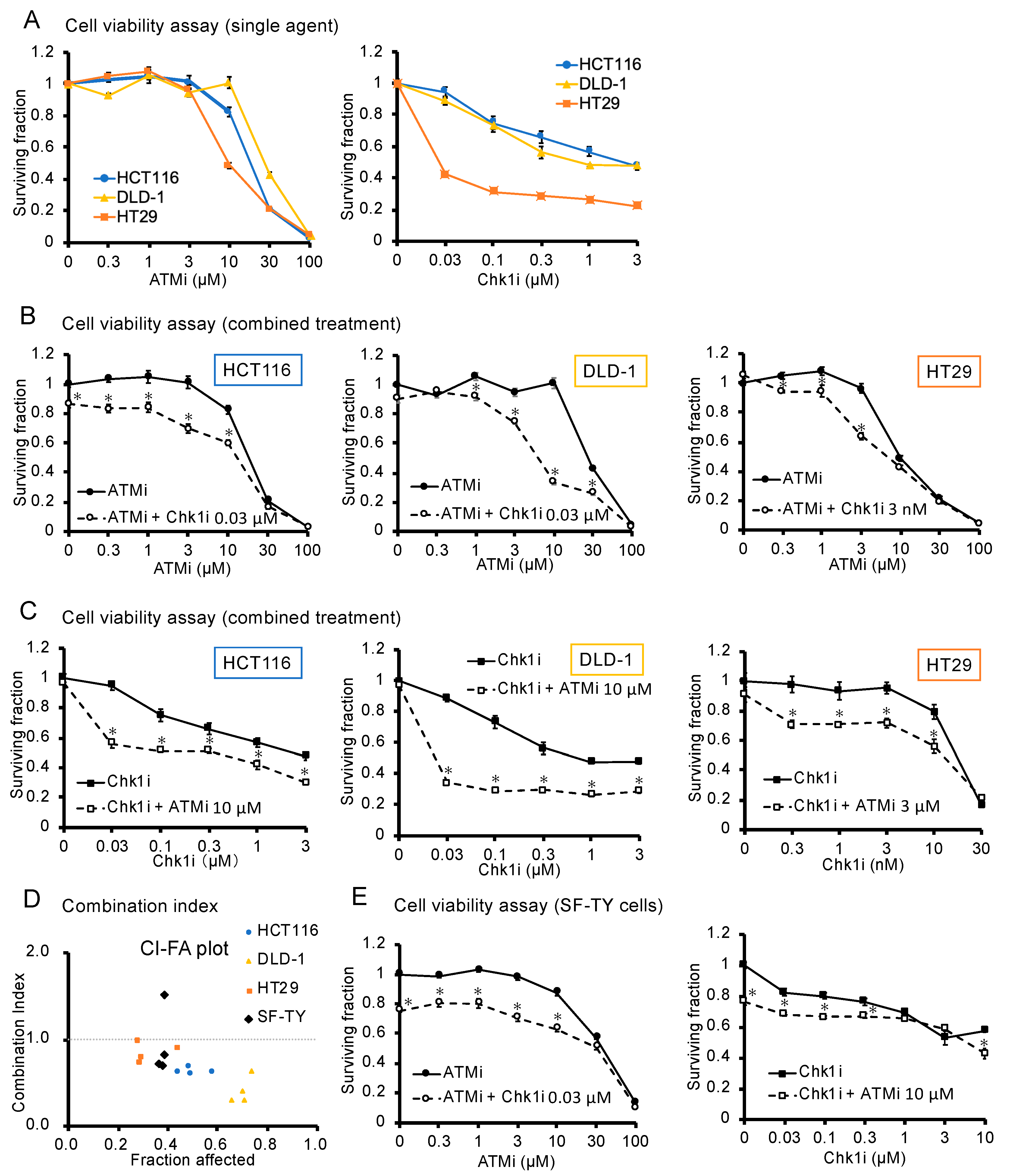
3.1. ATMi and Chk1i Exert Antitumor Effects as Single Agents
3.2. Combined ATMi and Chk1i Treatment Exhibits Synergistic Antitumor Effects
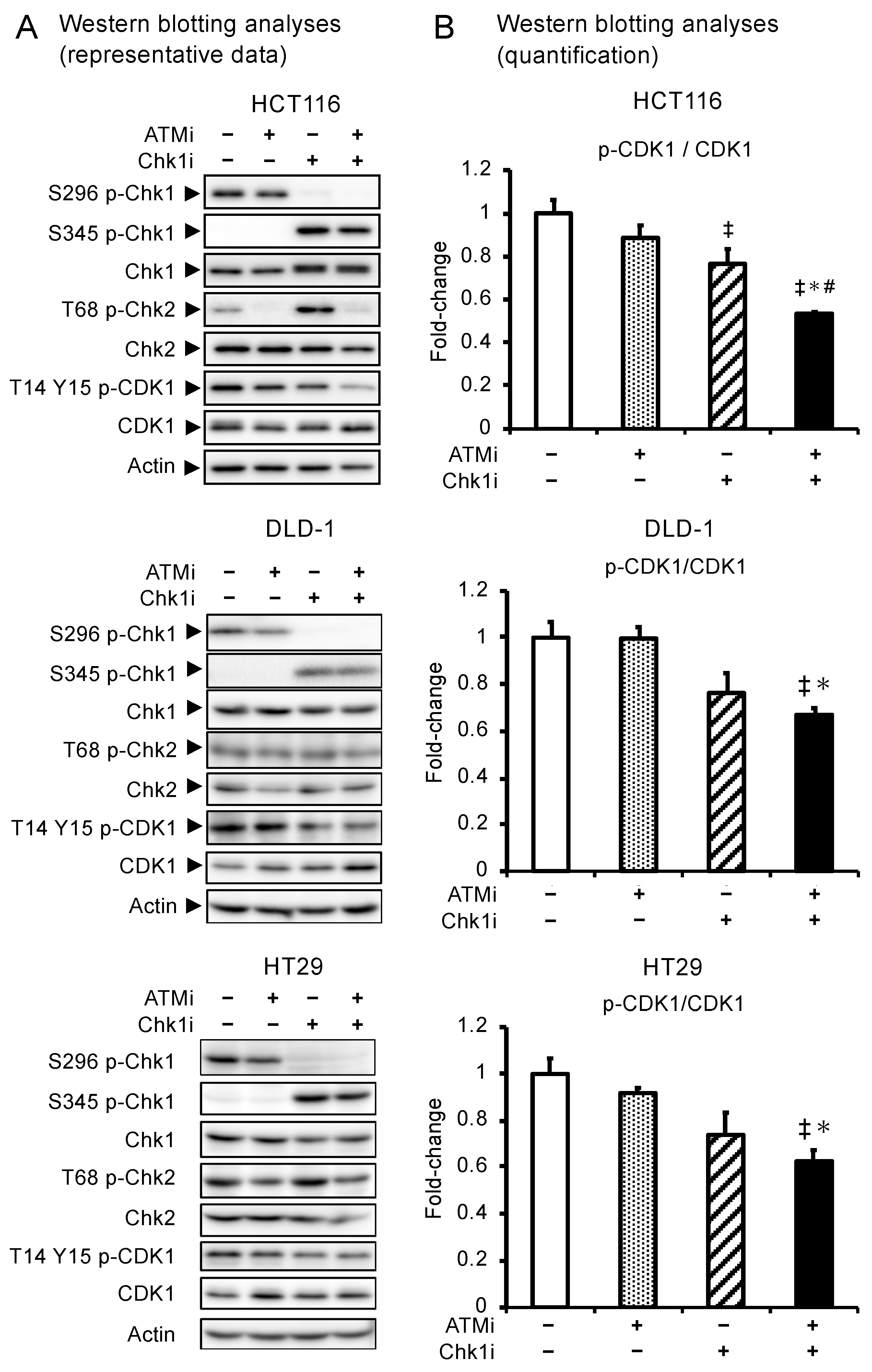
3.3. Combined ATMi and Chk1i Treatment Synergistically Decreases CDK1 Phosphorylation Levels
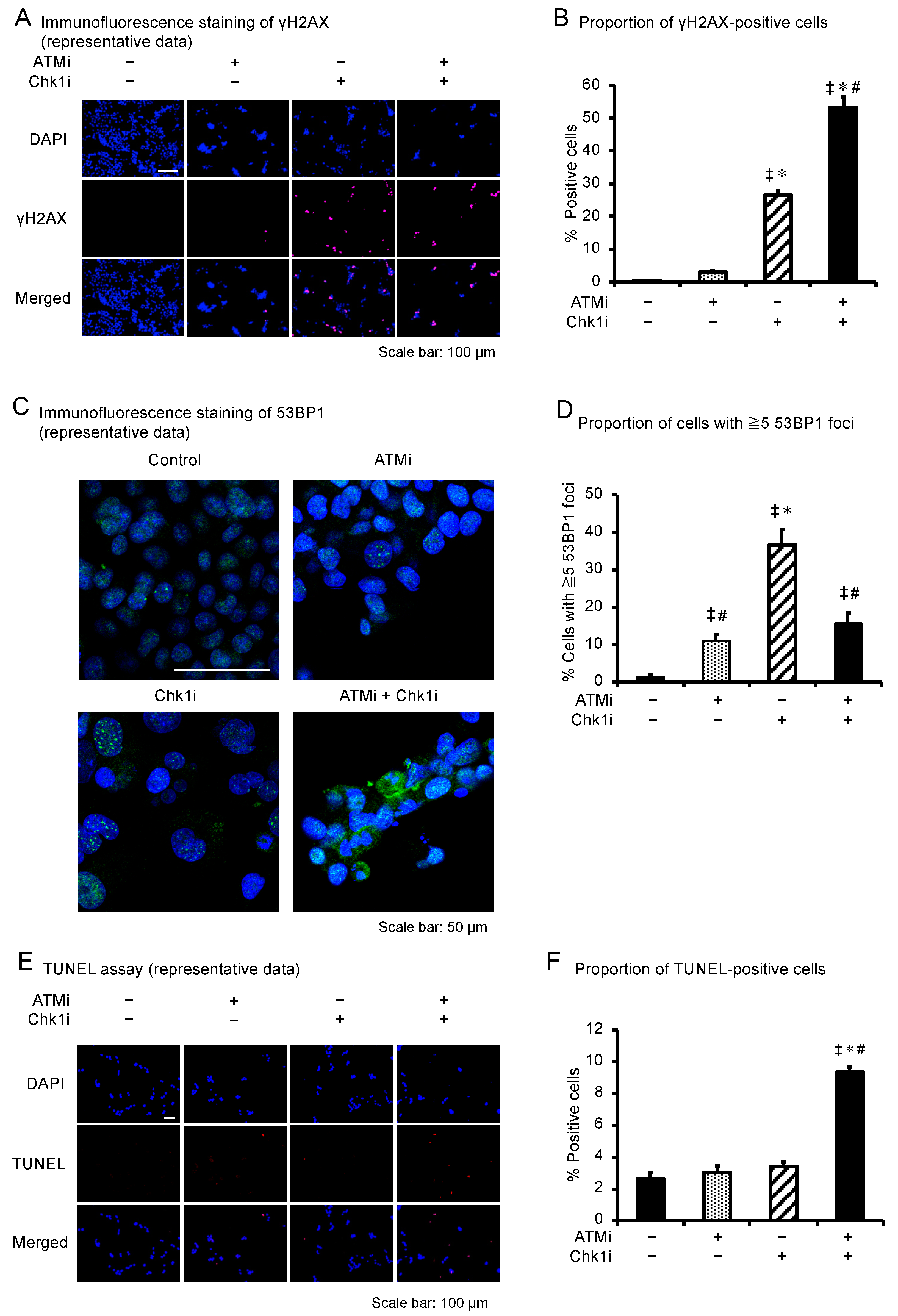
3.4. The ATMi and Chk1i Combination Increases the Number of Cells in the Sub-G1 Phase
3.5. The ATMi and Chk1i Combination Induces Apoptosis by Introducing DNA Damage
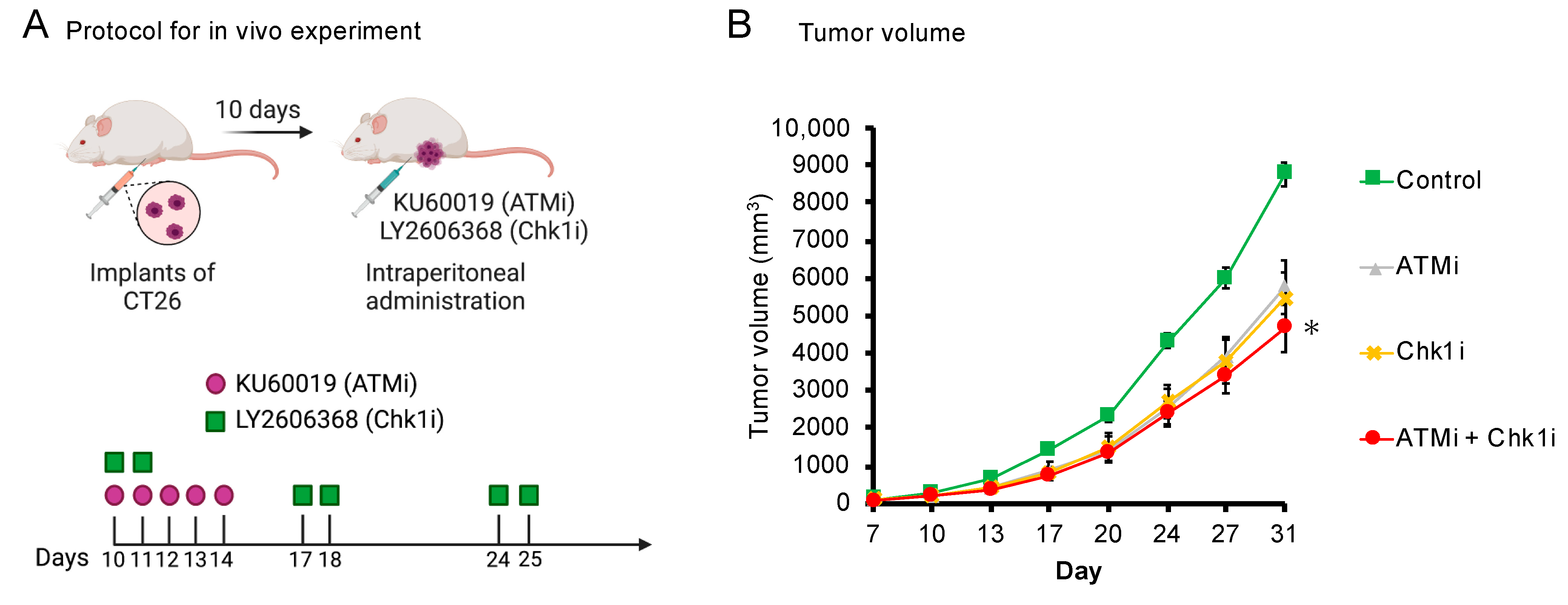
3.6. The ATMi and Chk1i Combination Significantly Reduces Tumor Volume in Syngeneic Tumor Model Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of Colorectal Cancer: Incidence, Mortality, Survival, and Risk Factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive Review of Targeted Therapy for Colorectal Cancer. Signal Transduct. Target. Ther. 2020, 5, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 Pathways in DNA Damage Signaling and Cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [PubMed]
- Karlsson-Rosenthal, C.; Millar, J.B.A. Cdc25: Mechanisms of Checkpoint Inhibition and Recovery. Trends Cell Biol. 2006, 16, 285–292. [Google Scholar] [CrossRef]
- Zhang, Y.; Hunter, T. Roles of Chk1 in Cell Biology and Cancer Therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [Green Version]
- Stover, E.H.; Konstantinopoulos, P.A.; Matulonis, U.A.; Swisher, E.M. Biomarkers of Response and Resistance to DNA Repair Targeted Therapies. Clin. Cancer Res. 2016, 22, 5651–5660. [Google Scholar] [CrossRef] [Green Version]
- Ronco, C.; Martin, A.R.; Demange, L.; Benhida, R. ATM, ATR, CHK1, CHK2 and WEE1 Inhibitors in Cancer and Cancer Stem Cells. MedChemComm 2017, 8, 295–319. [Google Scholar] [CrossRef] [PubMed]
- Merry, C.; Fu, K.; Wang, J.; Yeh, I.-J.; Zhang, Y. Targeting the Checkpoint Kinase Chk1 in Cancer Therapy. Cell Cycle 2010, 9, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, I.; Fleuren, E.D.G.; Williamson, C.T.; Lord, C.J. Directing the Use of DDR Kinase Inhibitors in Cancer Treatment. Expert Opin. Investig. Drugs 2017, 26, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Cremona, C.A.; Behrens, A. ATM Signalling and Cancer. Oncogene 2013, 33, 3351–3360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer Ther. 2020, 19, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; MacCormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective Killing of ATM- or p53-Deficient Cancer Cells through Inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; Artista, L.D.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting Oncogene-Induced Replicative Stress for the Selective Killing of Myc-Driven Tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef]
- Choi, S.; Toledo, L.I.; Fernandez-Capetillo, O.; Bakkenist, C.J. CGK733 Does Not Inhibit ATM or ATR Kinase Activity in H460 Human Lung Cancer Cells. DNA Repair 2011, 10, 1000–1001. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Reinhardt, H.C.; Bartkova, J.; Tommiska, J.; Blomqvist, C.; Nevanlinna, H.; Bartek, J.; Yaffe, M.B.; Hemann, M.T. The Combined Status of ATM and p53 Link Tumor Development with Therapeutic Response. Genes Dev. 2009, 23, 1895–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic Mutations Affect Key Pathways in Lung Adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Dent, P. Investigational CHK1 Inhibitors in Early Phase Clinical Trials for the Treatment of Cancer. Expert Opin. Investig. Drugs 2019, 28, 1095–1100. [Google Scholar] [CrossRef]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I Dose-Escalation Study of AZD7762, a Checkpoint Kinase Inhibitor, in Combination with Gemcitabine in US Patients with Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannini, S.; Weller, M.-C.; Repmann, S.; Moch, H.; Jiricny, J. Synthetic Lethality between BRCA1 Deficiency and poly(ADP-Ribose) Polymerase Inhibition Is Modulated by Processing of Endogenous Oxidative DNA Damage. Nucleic Acids Res. 2019, 47, 9132–9143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Lippman, S.M. An intermittent approach for cancer chemoprevention. Nat. Rev. Cancer. 2011, 11, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic Lethality as an Engine for Cancer Drug Target Discovery. Nat. Rev. Drug Discov. 2019, 19, 23–38. [Google Scholar] [CrossRef]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR Kinases Produces Tumor-Selective Synthetic Lethality and Suppresses Metastasis. J. Clin. Invest. 2019, 129, 1329–1344. [Google Scholar] [CrossRef] [Green Version]
- Sanjiv, K.; Hagenkort, A.; Calderón-Montaño, J.M.; Koolmeister, T.; Reaper, P.M.; Mortusewicz, O.; Jacques, S.A.; Kuiper, R.V.; Schultz, N.; Scobie, M.; et al. Cancer-Specific Synthetic Lethality between ATR and CHK1 Kinase Activities. Cell Rep. 2016, 14, 298–309. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR Inhibition Induces Synthetic Lethality and Overcomes Chemoresistance in TP53- or ATM-Defective Chronic Lymphocytic Leukemia Cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Tong, P.; Stewart, C.A.; Cristea, S.; Valliani, A.; Shames, D.S.; Redwood, A.B.; Fan, Y.H.; Li, L.; Glisson, B.S.; et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res. 2017, 77, 3870–3884. [Google Scholar] [CrossRef] [Green Version]
- McCabe, N.; Hanna, C.; Walker, S.M.; Gonda, D.; Li, J.; Wikstrom, K.; Savage, K.I.; Butterworth, K.T.; Chen, C.; Harkin, D.P.; et al. Mechanistic Rationale to Target PTEN-Deficient Tumor Cells with Inhibitors of the DNA Damage Response Kinase ATM. Cancer Res. 2015, 75, 2159–2165. [Google Scholar] [CrossRef] [Green Version]
- Kanda, Y. Investigation of the Freely Available Easy-to-Use Software “EZR” for Medical Statistics. Bone Marrow Transpl. 2013, 48, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Squire, C.J.; Dickson, J.M.; Ivanovic, I.; Baker, E.N. Structure and Inhibition of the Human Cell Cycle Checkpoint Kinase, Wee1A Kinase: An Atypical Tyrosine Kinase with a Key Role in CDK1 Regulation. Structure 2005, 13, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Plesca, D.; Mazumder, S.; Almasan, A. DNA Damage Response and Apoptosis. Methods Enzymol. 2008, 446, 107–122. [Google Scholar]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 2015, 14, 2004–2013. [Google Scholar] [CrossRef] [Green Version]
- Isono, M.; Niimi, A.; Oike, T.; Hagiwara, Y.; Sato, H.; Sekine, R.; Yoshida, Y.; Isobe, S.; Obuse, C.; Nishi, R.; et al. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017, 18, 520–532. [Google Scholar] [CrossRef] [Green Version]
- Sakasai, R.; Teraoka, H.; Takagi, M.; Tibbetts, R.S. Transcription-dependent activation of ataxia telangiectasia mutated prevents DNA-dependent protein kinase-mediated cell death in response to topoisomerase I poison. J. Biol. Chem. 2010, 285, 15201–15208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neizer-Ashuna, F.; Bhattacharya, R. Reality CHEK: Understanding the biology and clinical potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Wilsker, D.; Petermann, E.; Helleday, T.; Bunz, F. Essential Function of Chk1 Can Be Uncoupled from DNA Damage Checkpoint and Replication Control. Proc. Natl. Acad. Sci. USA 2008, 105, 20752–20757. [Google Scholar] [CrossRef] [Green Version]
- Syljuåsen, R.G.; Sørensen, C.S.; Hansen, L.T.; Fugger, K.; Lundin, C.; Johansson, F.; Helleday, T.; Sehested, M.; Lukas, J.; Bartek, J. Inhibition of Human Chk1 Causes Increased Initiation of DNA Replication, Phosphorylation of ATR Targets, and DNA Breakage. Mol. Cell. Biol. 2005, 25, 3553–3562. [Google Scholar] [CrossRef] [Green Version]
- Ando, K.; Nakamura, Y.; Nagase, H.; Nakagawara, A.; Koshinaga, T.; Wada, S.; Makishima, M. Co-Inhibition of the DNA damage response and CHK1 enhances apoptosis of neuroblastoma Cells. Int. J. Mol. Sci. 2019, 20, 3700. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.H.; Oh, D. ATM in DNA repair in cancer. Pharmacol. Ther. 2019, 203, 107391. [Google Scholar] [CrossRef] [PubMed]
- Lara-Gonzalez, P.; Westhorpe, F.G.; Taylor, S.S. The Spindle Assembly Checkpoint. Curr. Biol. 2012, 22, R966–R980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef]
- Brill, E.; Yokoyama, T.; Nair, J.; Yu, M.; Ahn, Y.R.; Lee, J.M. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 2017, 8, 111026–111040. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Yan, H.; Guo, W.; Tang, M.; Zhao, X.; Tong, A.; Peng, Y.; Li, Q.; Yuan, Z. ATM inhibition induces synthetic lethality and enhances sensitivity of PTEN-deficient breast cancer cells to cisplatin. Exp. Cell Res. 2018, 366, 24–33. [Google Scholar] [CrossRef]
- Abida, W.; Bang, Y.J.; Carter, L.; Azaro, A.; Krebs, M.; Im, S.; Chen, Y.; Buil-Bruna, N.; Li, Y.; Eaton, D.; et al. Abstract A094: Phase I modular study of AZD0156, a first-in-class oral selective inhibitor of ataxia telangiectasia mutated protein kinase (ATM), in combination with olaparib (AToM Study, Module 1). Mol. Cancer Ther. 2018, 17 (Suppl. 1), A094. [Google Scholar] [CrossRef]
- Berg, K.C.G.; Eide, P.W.; Eilertsen, I.A.; Johannessen, B.; Bruun, J.; Danielsen, S.A.; Bjørnslett, M.; Meza-Zepeda, L.A.; Eknæs, M.; Lind, G.E.; et al. Multi-omics of 34 colorectal cancer cell lines—a resource for biomedical studies. Mol. Cancer 2017, 16, 116. [Google Scholar] [CrossRef]
- Wan, Y.; Zhang, Y.; Wang, G.; Mwangi, P.M.; Cai, H.; Li, R. Recombinant KRAS G12D protein vaccines elicit significant anti-tumor effects in mouse CT26 tumor models. Front. Oncol. 2020, 10, 1326. [Google Scholar] [CrossRef] [PubMed]
- Nagro, C.J.D.; Choi, J.; Xiao, Y.; Rangell, L.; Mohan, S.; Pandita, A.; Zha, J.; Jackson, P.K.; Brien, T.O. Chk1 inhibition in p53-deficient cell lines drives rapid chromosome fragmentation followed by caspase-independent cell death. Cell Cycle 2014, 13, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [Green Version]
- Batey, M.A.; Zhao, Y.; Kyle, S.; Richardson, C.; Slade, A.; Martin, N.M.; Lau, A.; Newell, D.R.; Curtin, N.J. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer Ther. 2013, 12, 959–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tozaki, Y.; Aoki, H.; Kato, R.; Toriuchi, K.; Arame, S.; Inoue, Y.; Hayashi, H.; Kubota, E.; Kataoka, H.; Aoyama, M. The Combination of ATM and Chk1 Inhibitors Induces Synthetic Lethality in Colorectal Cancer Cells. Cancers 2023, 15, 735. https://doi.org/10.3390/cancers15030735
Tozaki Y, Aoki H, Kato R, Toriuchi K, Arame S, Inoue Y, Hayashi H, Kubota E, Kataoka H, Aoyama M. The Combination of ATM and Chk1 Inhibitors Induces Synthetic Lethality in Colorectal Cancer Cells. Cancers. 2023; 15(3):735. https://doi.org/10.3390/cancers15030735
Chicago/Turabian StyleTozaki, Yuri, Hiromasa Aoki, Rina Kato, Kohki Toriuchi, Saki Arame, Yasumichi Inoue, Hidetoshi Hayashi, Eiji Kubota, Hiromi Kataoka, and Mineyoshi Aoyama. 2023. "The Combination of ATM and Chk1 Inhibitors Induces Synthetic Lethality in Colorectal Cancer Cells" Cancers 15, no. 3: 735. https://doi.org/10.3390/cancers15030735
APA StyleTozaki, Y., Aoki, H., Kato, R., Toriuchi, K., Arame, S., Inoue, Y., Hayashi, H., Kubota, E., Kataoka, H., & Aoyama, M. (2023). The Combination of ATM and Chk1 Inhibitors Induces Synthetic Lethality in Colorectal Cancer Cells. Cancers, 15(3), 735. https://doi.org/10.3390/cancers15030735