Functional Investigation of IGF1R Mutations in Multiple Myeloma

 , , , , ,
, , , , , 
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. IGF1 Stimulation
2.3. Pharmacologic Inhibitors
2.4. siRNA Knockdown
2.5. CRISPR-Cas9 Screen
2.6. Generation of IGF1RWT and IGF1Rmut-Overexpressing HMCL Sublines
2.6.1. Mutagenesis PCR
2.6.2. Expression Cloning and Stable Transfection of HMCLs Using the Sleeping Beauty Transposon System
2.7. SDS-PAGE and Immunoblotting
2.8. Viability and Proliferation Assays
2.9. Apoptosis and Survival Assay (Annexin-V/PI)
3. Results
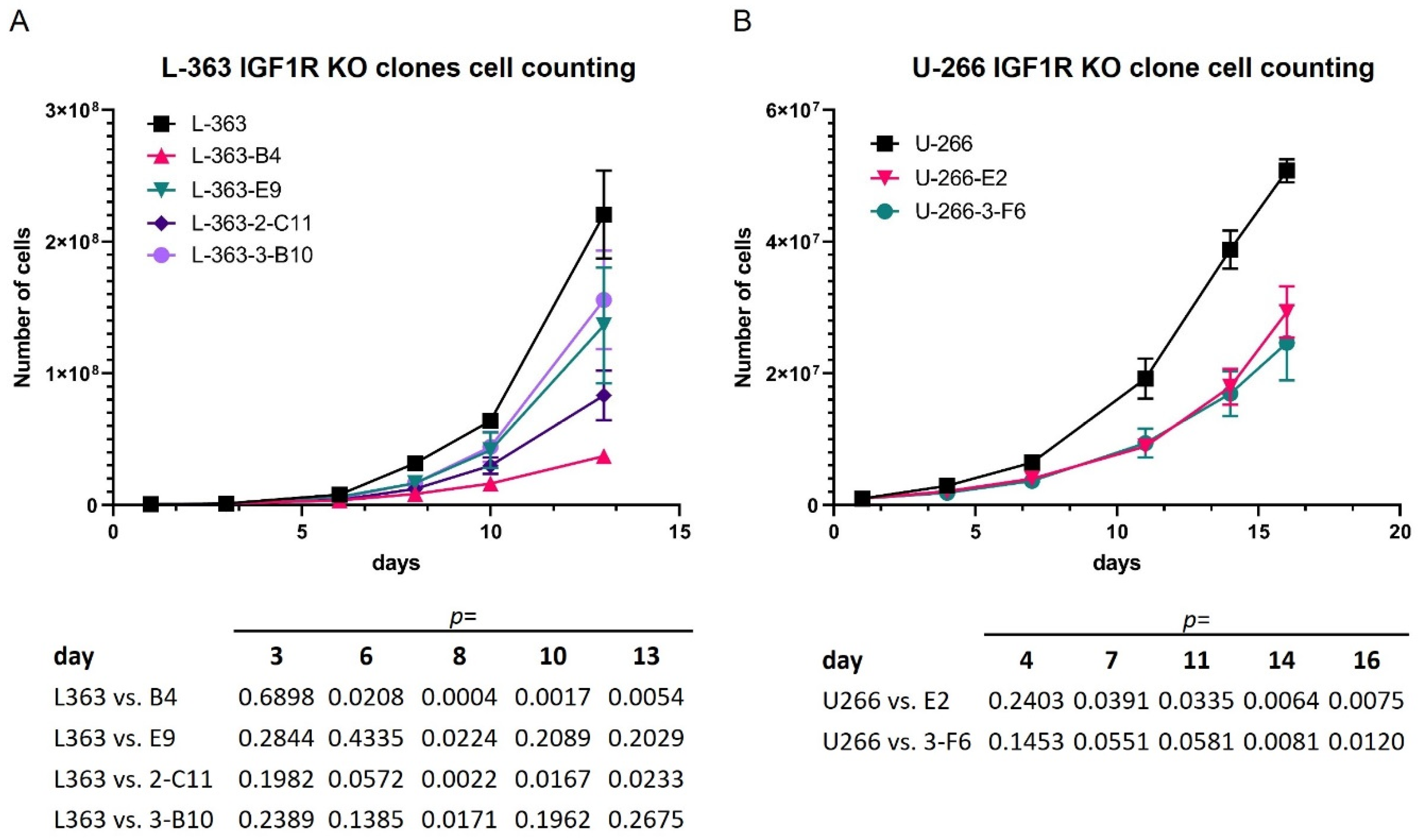
3.1. Stimulation as Well as Attenuation of IGF1R Signaling Influence the Activation of IGF1R Effectors and the Proliferation Rate in HMCLs
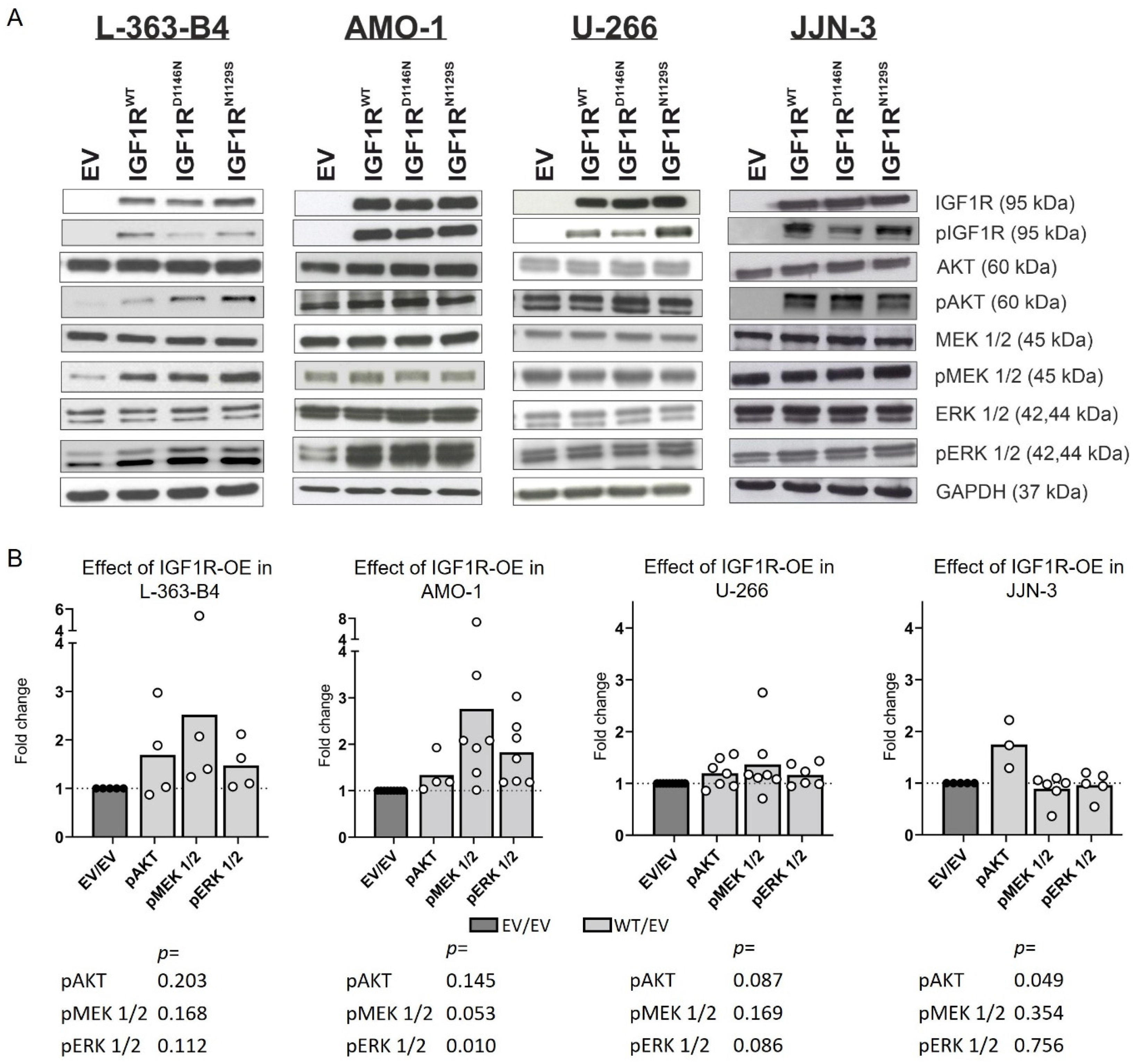
3.2. IGF1R Overexpression Impacts the Activation of MEK/ERK and AKT
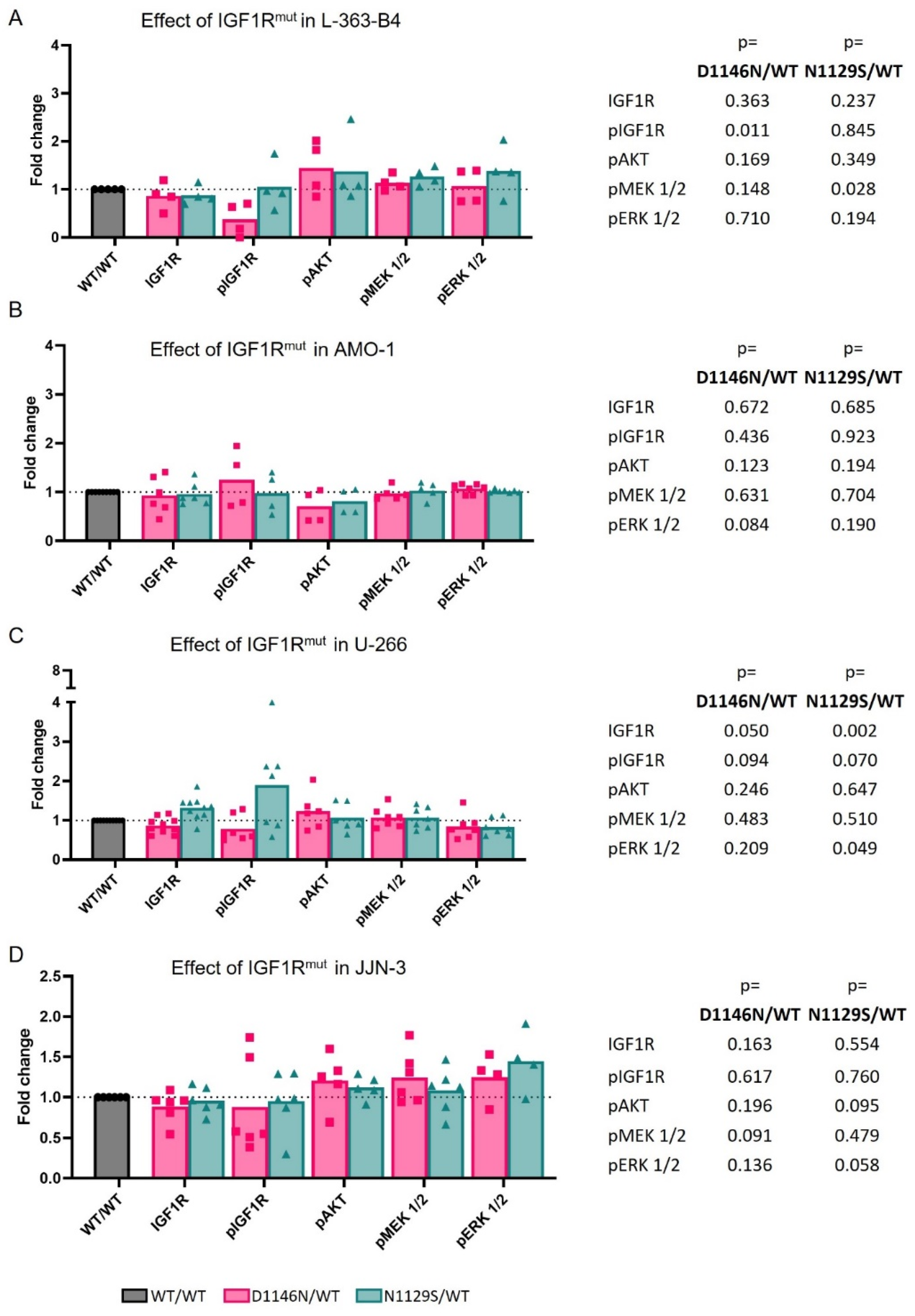
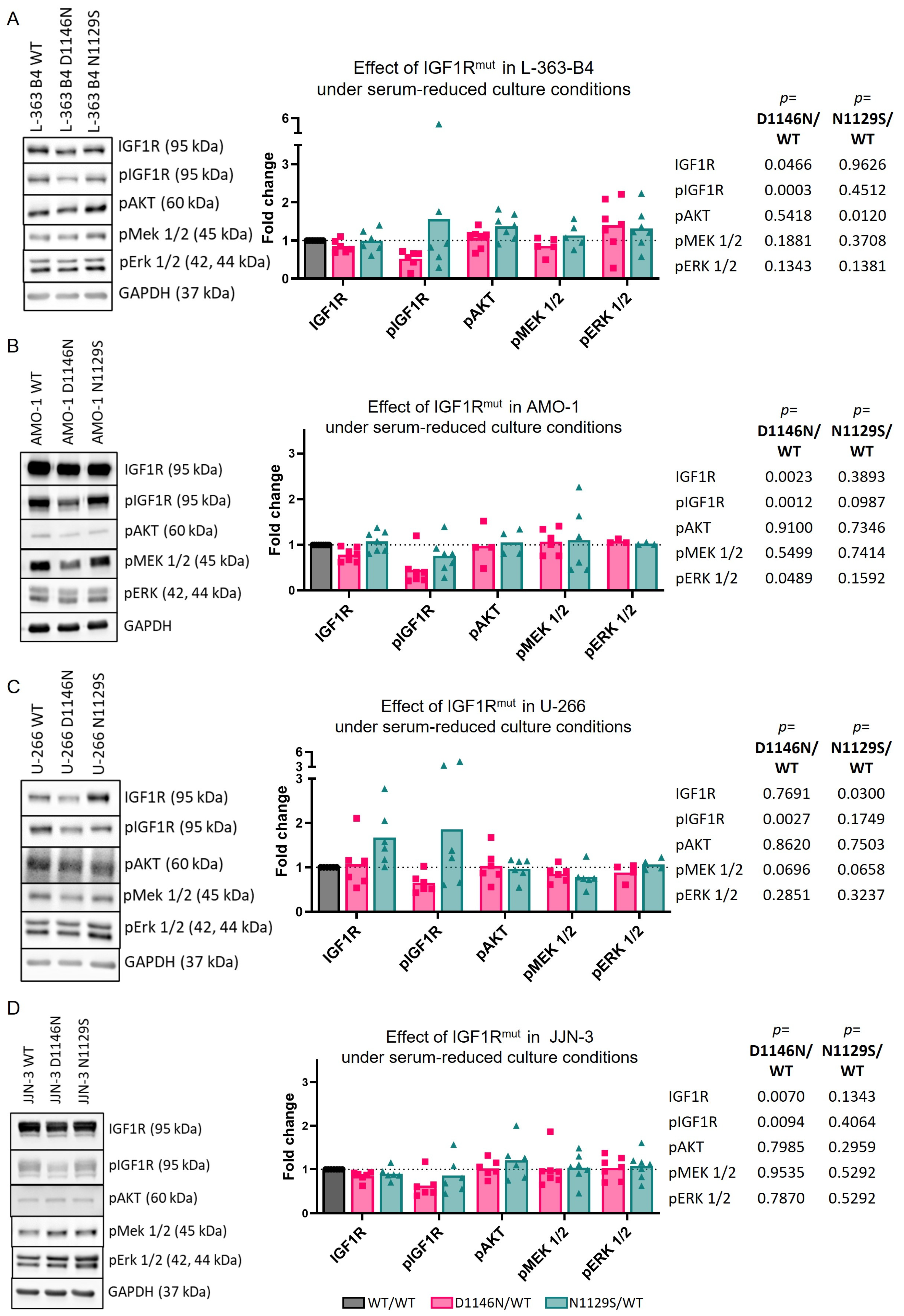
3.3. Mutations Can Impact IGF1R Activation and/or Downstream Signaling
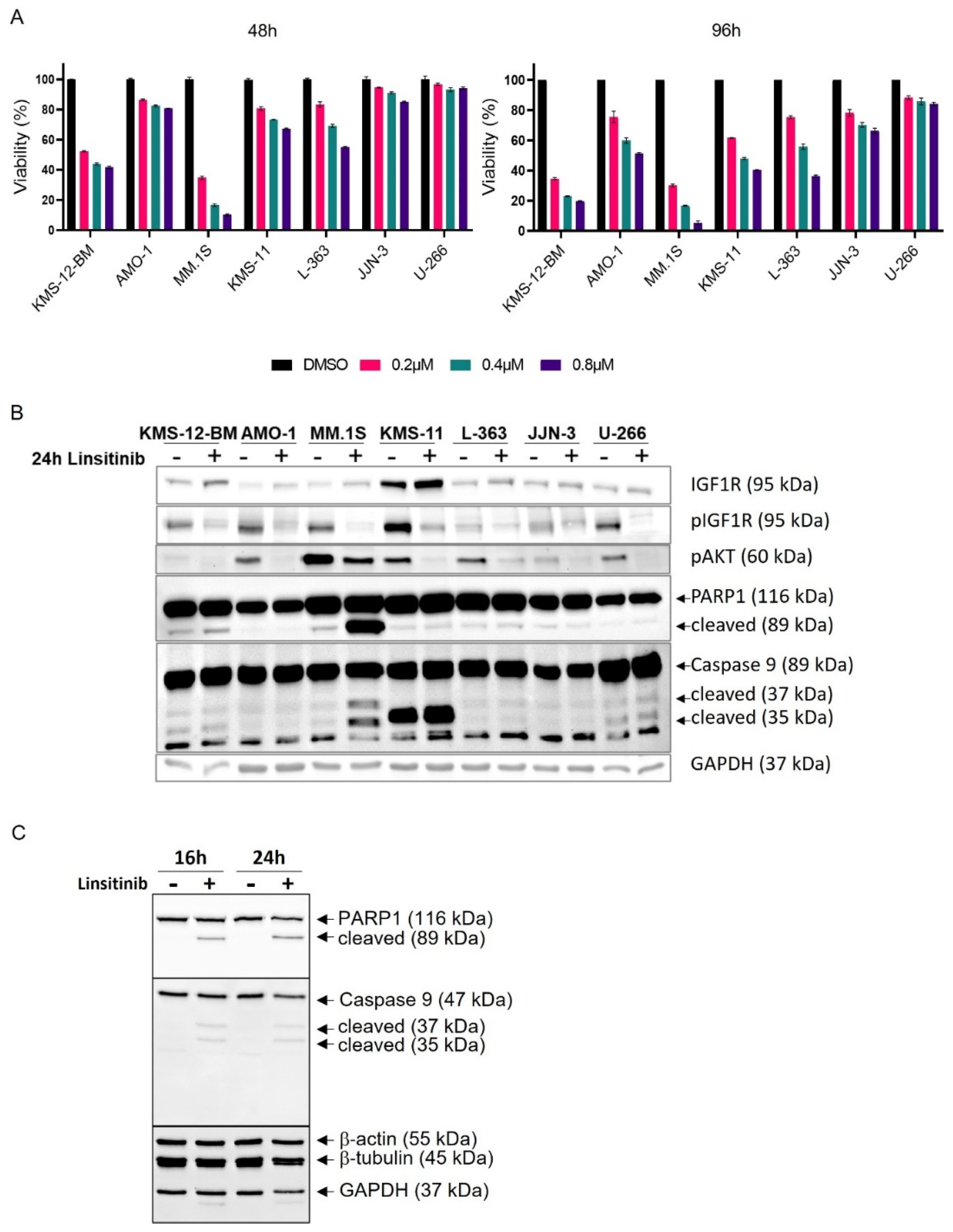
3.4. Linsitinib Affects AKT Signaling, Viability and Survival in All HMCLs
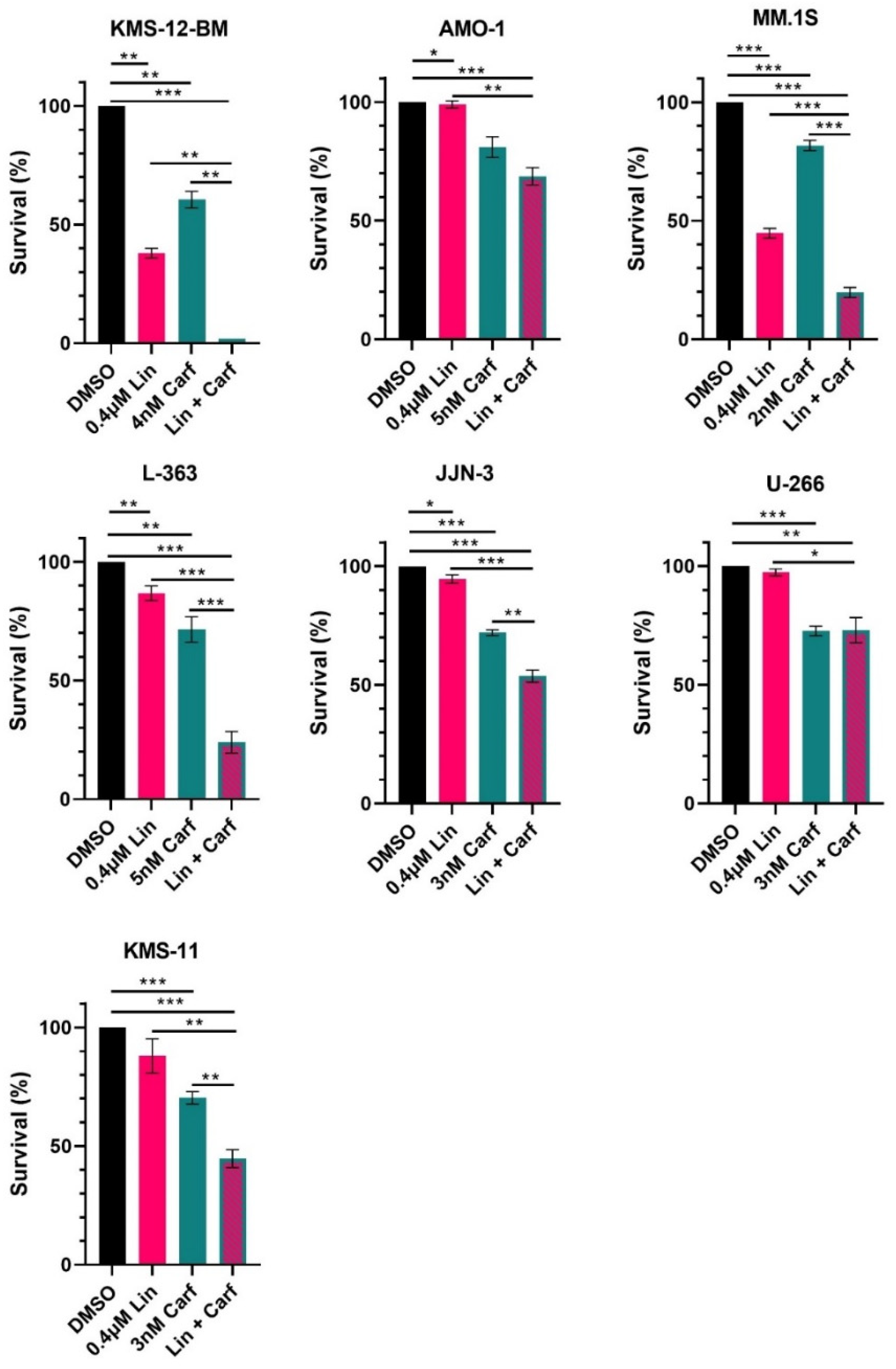
3.5. Combined Treatment with Linsitinib and Carfilzomib Is Effective in HMCL with and without IGF1R Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Insulin-like growth factor 1 receptor | IGF1R |
| Multiple myeloma | MM |
| Human multiple myeloma cell lines | HMCLs |
| Receptor tyrosine kinases | RTKs |
| Epidermal growth factor receptor | EGFR |
| Fibroblast growth factor receptor 3 | FGFR3 |
| erb-b2 receptor tyrosine kinase 2 | ERBB2 |
| Single nucleotide variants | SNVs |
| Single nucleotide polymorphisms | SNPs |
References
- Engelhardt, M.; Yong, K.; Bringhen, S.; Wasch, R. Carfilzomib combination treatment as first-line therapy in multiple myeloma: Where do we go from the carthadex (ktd)-trial update? Haematologica 2019, 104, 2128–2131. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Hulin, C.; Perrot, A.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Bene, M.C.; Zweegman, S.; Caillon, H.; Caillot, D.; et al. Maintenance with daratumumab or observation following treatment with bortezomib, thalidomide, and dexamethasone with or without daratumumab and autologous stem-cell transplant in patients with newly diagnosed multiple myeloma (cassiopeia): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 1378–1390. [Google Scholar] [PubMed]
- Rajkumar, S.V. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2022, 97, 1086–1107. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Nooka, A.K.; Larson, S.M. Moving toward a cure for myeloma. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Andrulis, M.; Lehners, N.; Capper, D.; Penzel, R.; Heining, C.; Huellein, J.; Zenz, T.; von Deimling, A.; Schirmacher, P.; Ho, A.D.; et al. Targeting the braf v600e mutation in multiple myeloma. Cancer Discov. 2013, 3, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Giesen, N.; Chatterjee, M.; Scheid, C.; Poos, A.M.; Besemer, B.; Miah, K.; Benner, A.; Becker, N.; Moehler, T.; Metzler, I.; et al. A phase 2 clinical trial of combined braf/mek inhibition for brafv600e-mutated multiple myeloma. Blood 2023, 141, 1685–1690. [Google Scholar] [CrossRef]
- Kaufman, J.L.; Gasparetto, C.; Schjesvold, F.H.; Moreau, P.; Touzeau, C.; Facon, T.; Boise, L.H.; Jiang, Y.; Yang, X.; Dunbar, F.; et al. Targeting bcl-2 with venetoclax and dexamethasone in patients with relapsed/refractory t(11;14) multiple myeloma. Am. J. Hematol. 2021, 96, 418–427. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, W.M.; Bergsagel, P.L. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J. Clin. Invest. 2012, 122, 3456–3463. [Google Scholar] [CrossRef]
- Leich, E.; Weissbach, S.; Klein, H.U.; Grieb, T.; Pischimarov, J.; Stühmer, T.; Chatterjee, M.; Steinbrunn, T.; Langer, C.; Eilers, M.; et al. Multiple myeloma is affected by multiple and heterogeneous somatic mutations in adhesion- and receptor tyrosine kinase signaling molecules. Blood Cancer J. 2013, 3, e102. [Google Scholar] [CrossRef]
- Chng, W.J.; Gonzalez-Paz, N.; Price-Troska, T.; Jacobus, S.; Rajkumar, S.V.; Oken, M.M.; Kyle, R.A.; Henderson, K.J.; Van Wier, S.; Greipp, P.; et al. Clinical and biological significance of ras mutations in multiple myeloma. Leukemia 2008, 22, 2280–2284. [Google Scholar] [CrossRef] [PubMed]
- Heuck, C.J.; Jethava, Y.; Khan, R.; van Rhee, F.; Zangari, M.; Chavan, S.; Robbins, K.; Miller, S.E.; Matin, A.; Mohan, M.; et al. Inhibiting mek in mapk pathway-activated myeloma. Leukemia 2016, 30, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, M.; Barbieri, M.; Todoerti, K.; Agnelli, L.; Marzorati, S.; Fabris, S.; Ciceri, G.; Galletti, S.; Milesi, G.; Manzoni, M.; et al. Molecular spectrum of braf, nras and kras gene mutations in plasma cell dyscrasias: Implication for mek-erk pathway activation. Oncotarget 2015, 6, 24205–24217. [Google Scholar] [CrossRef] [PubMed]
- Steinbrunn, T.; Stühmer, T.; Gattenlohner, S.; Rosenwald, A.; Mottok, A.; Unzicker, C.; Einsele, H.; Chatterjee, M.; Bargou, R.C. Mutated ras and constitutively activated akt delineate distinct oncogenic pathways, which independently contribute to multiple myeloma cell survival. Blood 2011, 117, 1998–2004. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Gardner, A.; Lichtenstein, A. The phosphatidylinositol 3-kinase/akt kinase pathway in multiple myeloma plasma cells: Roles in cytokine-dependent survival and proliferative responses. Cancer Res. 2000, 60, 6763–6770. [Google Scholar] [PubMed]
- Zollinger, A.; Stuhmer, T.; Chatterjee, M.; Gattenlohner, S.; Haralambieva, E.; Muller-Hermelink, H.K.; Andrulis, M.; Greiner, A.; Wesemeier, C.; Rath, J.C.; et al. Combined functional and molecular analysis of tumor cell signaling defines 2 distinct myeloma subgroups: Akt-dependent and akt-independent multiple myeloma. Blood 2008, 112, 3403–3411. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Nappi, A.; Berretta, M.; Romano, C.; Tafuto, S.; Cassata, A.; Casaretti, R.; Silvestro, L.; Divitiis, C.; Alessandrini, L.; Fiorica, F.; et al. Metastatic colorectal cancer: Role of target therapies and future perspectives. Curr. Cancer Drug Targets 2018, 18, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Regad, T. Targeting rtk signaling pathways in cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Schiavoni, G.; Forconi, F.; Santi, A.; Trentin, L.; Ambrosetti, A.; Cecchini, D.; Sozzi, E.; Francia di Celle, P.; Di Bello, C.; et al. Simple genetic diagnosis of hairy cell leukemia by sensitive detection of the braf-v600e mutation. Blood 2012, 119, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. Braf mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [PubMed]
- Keppler, S.; Weissbach, S.; Langer, C.; Knop, S.; Pischimarov, J.; Kull, M.; Stühmer, T.; Steinbrunn, T.; Bargou, R.; Einsele, H.; et al. Rare snps in receptor tyrosine kinases are negative outcome predictors in multiple myeloma. Oncotarget 2016, 7, 38762–38774. [Google Scholar] [CrossRef] [PubMed]
- Leich, E.; Schreder, M.; Pischimarov, J.; Stühmer, T.; Steinbrunn, T.; Rudelius, M.; Brunnert, D.; Chatterjee, M.; Langer, C.; Keppler, S.; et al. Novel molecular subgroups within the context of receptor tyrosine kinase and adhesion signalling in multiple myeloma. Blood Cancer J. 2021, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Gualberto, A.; Fonseca, R. Igf-1r is overexpressed in poor-prognostic subtypes of multiple myeloma. Leukemia 2006, 20, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Georgii-Hemming, P.; Wiklund, H.J.; Ljunggren, O.; Nilsson, K. Insulin-like growth factor i is a growth and survival factor in human multiple myeloma cell lines. Blood 1996, 88, 2250–2258. [Google Scholar] [CrossRef]
- Jernberg-Wiklund, H.; Nilsson, K. Targeting the igf-1r signaling and mechanisms for epigenetic gene silencing in human multiple myeloma. Ups. J. Med. Sci. 2012, 117, 166–177. [Google Scholar] [CrossRef]
- Khan, S.; LeBlanc, R.; Gyger, M.; White, D.; Kaufman, J.; Jazubowiak, A.; Gul, E.; Paul, H.; Le, L.W.; Lau, A.; et al. A phase-1 trial of linsitinib (osi-906) in combination with bortezomib and dexamethasone for the treatment of relapsed/refractory multiple myeloma. Leuk. Lymphoma 2021, 62, 1721–1729. [Google Scholar] [CrossRef]
- Wang, P.; Mak, V.C.; Cheung, L.W. Drugging igf-1r in cancer: New insights and emerging opportunities. Genes Dis. 2023, 10, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Descamps, G.; Pellat-Deceunynck, C.; Szpak, Y.; Bataille, R.; Robillard, N.; Amiot, M. The magnitude of akt/phosphatidylinositol 3′-kinase proliferating signaling is related to cd45 expression in human myeloma cells. J. Immunol. 2004, 173, 4953–4959. [Google Scholar] [CrossRef] [PubMed]
- Descamps, G.; Wuilleme-Toumi, S.; Trichet, V.; Venot, C.; Debussche, L.; Hercend, T.; Collette, M.; Robillard, N.; Bataille, R.; Amiot, M. Cd45neg but not cd45pos human myeloma cells are sensitive to the inhibition of igf-1 signaling by a murine anti-igf-1r monoclonal antibody, mave1642. J. Immunol. 2006, 177, 4218–4223. [Google Scholar] [CrossRef]
- Liang, S.B.; Yang, X.Z.; Trieu, Y.; Li, Z.; Zive, J.; Leung-Hagesteijn, C.; Wei, E.; Zozulya, S.; Coss, C.C.; Dalton, J.T.; et al. Molecular target characterization and antimyeloma activity of the novel, insulin-like growth factor 1 receptor inhibitor, gtx-134. Clin. Cancer Res. 2011, 17, 4693–4704. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Akiyama, M.; Hideshima, T.; Chauhan, D.; Joseph, M.; Libermann, T.A.; et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell 2004, 5, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Menu, E.; van Valckenborgh, E.; van Camp, B.; Vanderkerken, K. The role of the insulin-like growth factor 1 receptor axis in multiple myeloma. Arch. Physiol. Biochem. 2009, 115, 49–57. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Bene, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (cassiopeia): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Singh, P.; Alex, J.M.; Bast, F. Insulin receptor (ir) and insulin-like growth factor receptor 1 (igf-1r) signaling systems: Novel treatment strategies for cancer. Med. Oncol. 2014, 31, 805. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Berkova, Z.; Jones, R.J.; Woessner, R.; Bjorklund, C.C.; Ma, W.; Davis, R.E.; Lin, P.; Wang, H.; Madden, T.L.; et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood 2012, 120, 3260–3270. [Google Scholar] [CrossRef]
- Beauvais, D.M.; Jung, O.; Yang, Y.; Sanderson, R.D.; Rapraeger, A.C. Syndecan-1 (cd138) suppresses apoptosis in multiple myeloma by activating igf1 receptor: Prevention by synstatinigf1r inhibits tumor growth. Cancer Res. 2016, 76, 4981–4993. [Google Scholar] [CrossRef] [PubMed]
- de Matos Simoes, R.; Shirasaki, R.; Downey-Kopyscinski, S.L.; Matthews, G.M.; Barwick, B.G.; Gupta, V.A.; Dupere-Richer, D.; Yamano, S.; Hu, Y.; Sheffer, M.; et al. Genome-scale functional genomics identify genes preferentially essential for multiple myeloma cells compared to other neoplasias. Nat. Cancer 2023, 4, 754–773. [Google Scholar] [CrossRef] [PubMed]
- Ge, N.L.; Rudikoff, S. Insulin-like growth factor i is a dual effector of multiple myeloma cell growth. Blood 2000, 96, 2856–2861. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.J. The role of carfilzomib in relapsed/refractory multiple myeloma. Ther. Adv. Hematol. 2021, 12, 20406207211019612. [Google Scholar] [CrossRef] [PubMed]
- Keppler, S. Characterization of novel mutations in receptor-tyrosine kinases in multiple myeloma. In Charakterisierung Neuer Mutationen in Rezeptor-Tyrosin Kinasen im Multiplen Myelom; Universitätsbibliothek Würzburg: Würzburg, Germany, 2020. [Google Scholar]
- Weissbach, S.; Heredia-Guerrero, S.C.; Barnsteiner, S.; Grosshans, L.; Bodem, J.; Starz, H.; Langer, C.; Appenzeller, S.; Knop, S.; Steinbrunn, T.; et al. Exon-4 mutations in kras affect mek/erk and pi3k/akt signaling in human multiple myeloma cell lines. Cancers 2020, 12, 455. [Google Scholar] [CrossRef] [PubMed]
- Steinbrunn, T.; Chatterjee, M.; Bargou, R.C.; Stühmer, T. Efficient transient transfection of human multiple myeloma cells by electroporation—An appraisal. PLoS ONE 2014, 9, e97443. [Google Scholar] [CrossRef] [PubMed]
- Gouda, M.; Subbiah, V. Precision oncology for braf-mutant cancers with braf and mek inhibitors: From melanoma to tissue-agnostic therapy. ESMO Open 2023, 8, 100788. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Esposito Abate, R.; Lambiase, M.; Forgione, L.; Cardone, C.; Iannaccone, A.; Sacco, A.; Rachiglio, A.M.; Martinelli, E.; Rizzi, D.; et al. Ras testing of liquid biopsy correlates with the outcome of metastatic colorectal cancer patients treated with first-line folfiri plus cetuximab in the capri-goim trial. Ann. Oncol. 2018, 29, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Ciardullo, C.; Gaidano, G. Genetic aberrations of signaling pathways in lymphomagenesis: Revelations from next generation sequencing studies. Semin. Cancer Biol. 2013, 23, 422–430. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. Egfr mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer 2012, 12, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Steinbrunn, T.; Stühmer, T.; Sayehli, C.; Chatterjee, M.; Einsele, H.; Bargou, R.C. Combined targeting of mek/mapk and pi3k/akt signalling in multiple myeloma. Br. J. Haematol. 2012, 159, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Sehat, B.; Andersson, S.; Vasilcanu, R.; Girnita, L.; Larsson, O. Role of ubiquitination in igf-1 receptor signaling and degradation. PLoS ONE 2007, 2, e340. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| IGF1R Exon 18 F | 5′ CATAAACAACCCACGGTGCC 3′ |
| IGF1R Exon 18 R | 5′ AAGGAGTCCGTGCACTCAAG 3′ |
| IGF1R Exon 2 F | 5′ GACATCCGCAACGACTATCA 3′ |
| IGF1R Exon 2 R | 5′ TTCTCACACATCGGCTTCTC 3′ |
| Table | Catalogue Numbers# | Dilution | Company |
|---|---|---|---|
| IGF1R | #9750 | 1:1000 | Cell Signaling Technology (CST) (Danvers, MA, USA) |
| pIGF1R (Y1135) | #3918 | 1:1000 | CST |
| pPYK2 (Y402) | #3291 | 1:2000 | CST |
| AKT | #4691 | 1:2000 | CST |
| pAKT (S473) | #4058 | 1:2000 | CST |
| MEK1/2 | #9122 | 1:8000 | CST |
| pMEK1/2 (S217/221) | #9154 | 1:4000 | CST |
| ERK1/2 | #9102 | 1:4000 | CST |
| pERK1/2 (T202/Y204) | #9101 | 1:4000 | CST |
| CASP9 | #9508 | 1:1000 | CST |
| PARP | #9532 | 1:1000 | CST |
| beta-tubulin | #2146 | 1:7500 | CST |
| beta-actin | #4970 | 1:16,000 | CST |
| GAPDH | #5174 | 1:100,000 | CST |
| HRP-linked anti-rabbit | #7074 | 1:1000–1:3000 | CST |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heredia-Guerrero, S.C.; Evers, M.; Keppler, S.; Schwarzfischer, M.; Fuhr, V.; Rauert-Wunderlich, H.; Krügl, A.; Nedeva, T.; Grieb, T.; Pickert, J.; et al. Functional Investigation of IGF1R Mutations in Multiple Myeloma. Cancers 2024, 16, 2139. https://doi.org/10.3390/cancers16112139
Heredia-Guerrero SC, Evers M, Keppler S, Schwarzfischer M, Fuhr V, Rauert-Wunderlich H, Krügl A, Nedeva T, Grieb T, Pickert J, et al. Functional Investigation of IGF1R Mutations in Multiple Myeloma. Cancers. 2024; 16(11):2139. https://doi.org/10.3390/cancers16112139
Chicago/Turabian StyleHeredia-Guerrero, Sofia Catalina, Marietheres Evers, Sarah Keppler, Marlene Schwarzfischer, Viktoria Fuhr, Hilka Rauert-Wunderlich, Anne Krügl, Theodora Nedeva, Tina Grieb, Julia Pickert, and et al. 2024. "Functional Investigation of IGF1R Mutations in Multiple Myeloma" Cancers 16, no. 11: 2139. https://doi.org/10.3390/cancers16112139
APA StyleHeredia-Guerrero, S. C., Evers, M., Keppler, S., Schwarzfischer, M., Fuhr, V., Rauert-Wunderlich, H., Krügl, A., Nedeva, T., Grieb, T., Pickert, J., Koch, H., Steinbrunn, T., Bayrhof, O.-J., Bargou, R. C., Rosenwald, A., Stühmer, T., & Leich, E. (2024). Functional Investigation of IGF1R Mutations in Multiple Myeloma. Cancers, 16(11), 2139. https://doi.org/10.3390/cancers16112139