Computational Approach for the Development of pH-Selective PD-1/PD-L1 Signaling Pathway Inhibition in Fight with Cancer

 ,
,
Abstract
:Simple Summary
Abstract
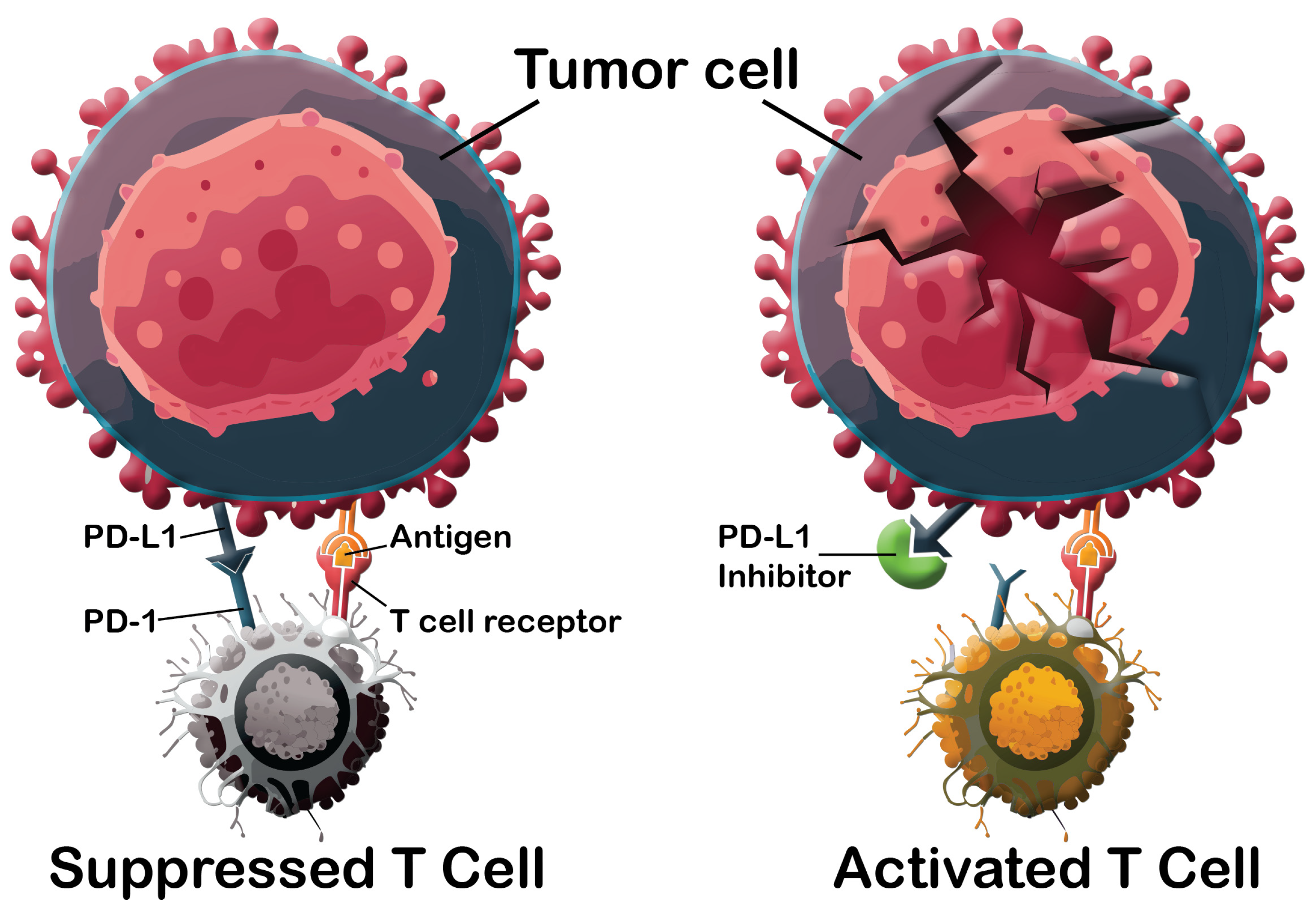
1. Introduction
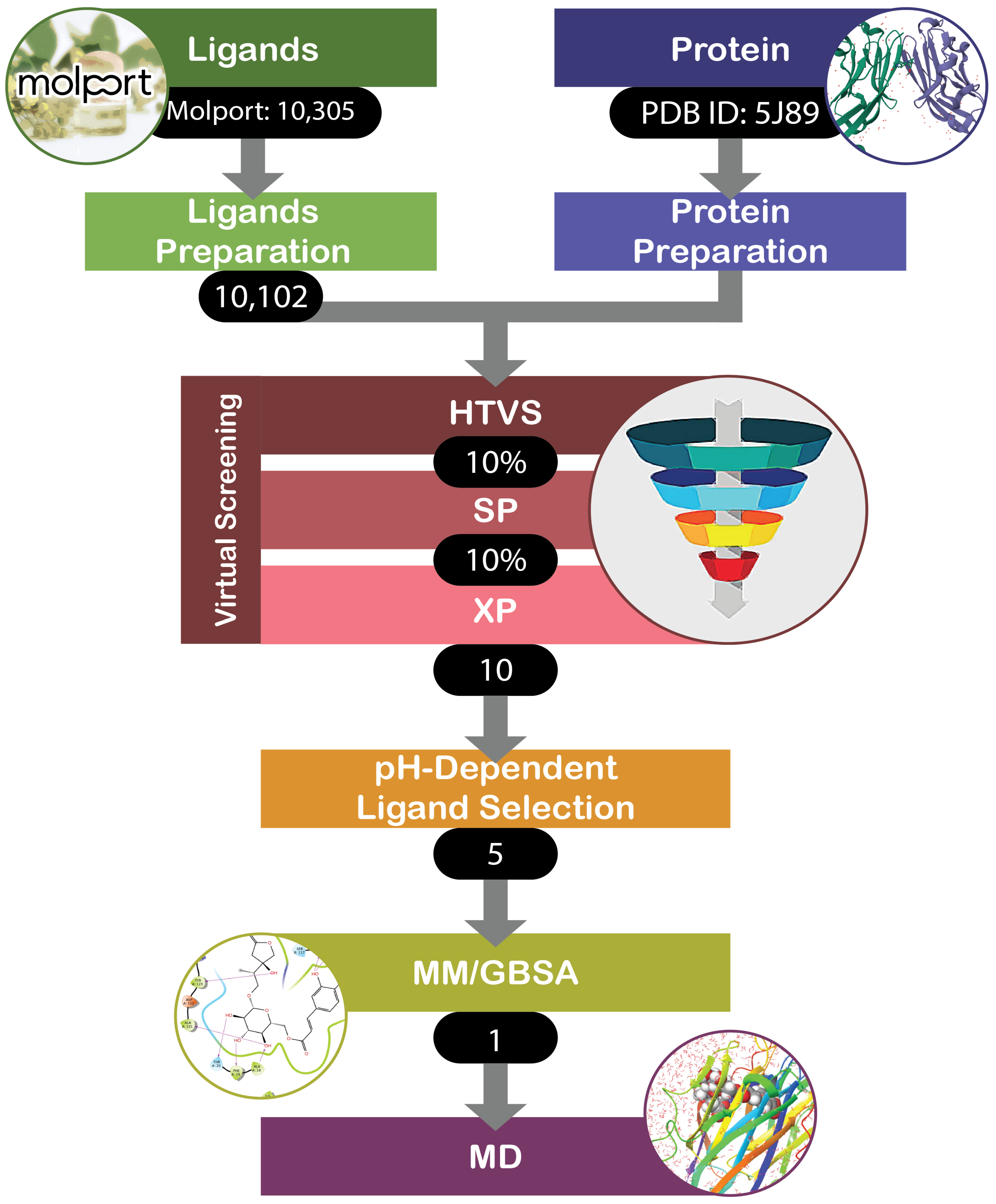
2. Materials and Methods
- O[C@@H]1[C@@H](COC(=O)c2cc(O)c(O)c(O)c2)O[C@@H](OC(=O)c2cc(O)c(O)c(O)c2)[C@H](O)[C@H]1OC(=O)c1cc(O)c(O)c(O)c1
- CC(C)(O[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O)C(OC(=O)C=Cc1ccc(O)cc1)C(=O)OCc1ccc(O[C@@H]2O[C@H](CO)[C@@H](O)[C@H](O)[C@H]2O)cc1
- Oc1cc(cc(O)c1O)C(=O)OC[C@H]1O[C@@H](OC(=O)c2cc(O)c(O)c(O)c2)[C@H](OC(=O)c2cc(O)c(O)c(O)c2)[C@@H](OC(=O)c2cc(O)c(O)c(O)c2)[C@@H]1OC(=O)c1cc(O)c(O)c(O)c1
- [#6]-[#6@@H]-1-[#8]-[#6@@H](-[#8]-[#6]-[#6@H]-2-[#8]-[#6@@H](-[#8]-[#6][#6]=[#6](/[#6])-[#6]-[#6][#6]=[#6](/[#6])-[#6]-[#6][#6]=[#6](/[#6])-[#6]-[#6][#6]=[#6]([#6])-[#6])-[#6@H](-[#8]-[#6@@H]-3-[#8]-[#6@@H](-[#6])-[#6@H](-[#8]-[#6](-[#6])=O)-[#6@@H](-[#8]-[#6](-[#6])=O)-[#6@H]-3-[#8]-[#6](-[#6])=O)-[#6@@H](-[#8])-[#6@@H]-2-[#8])-[#6@H](-[#8])-[#6@H](-[#8])-[#6@H]-1-[#8]
- CC(CO[C@@H]1O[C@H](COC(=O)C=Cc2ccc(O)c(O)c2)[C@@H](O)[C@H](O)[C@H]1O)C1(O)COC(=O)C1
- CC1=C(C=CC=C1C2=CC=CC=C2)COC3=NC(=C(C=C3)CNCCNC(=O)C)OC
- CC1=C(C=CC=C1OCC2=NN=C(O2)SCC3=CC=CC(=C3)CNC(CO)C(=O)O)C4=CC=CC=C4
3. Results
3.1. Benchmark and Positive Control
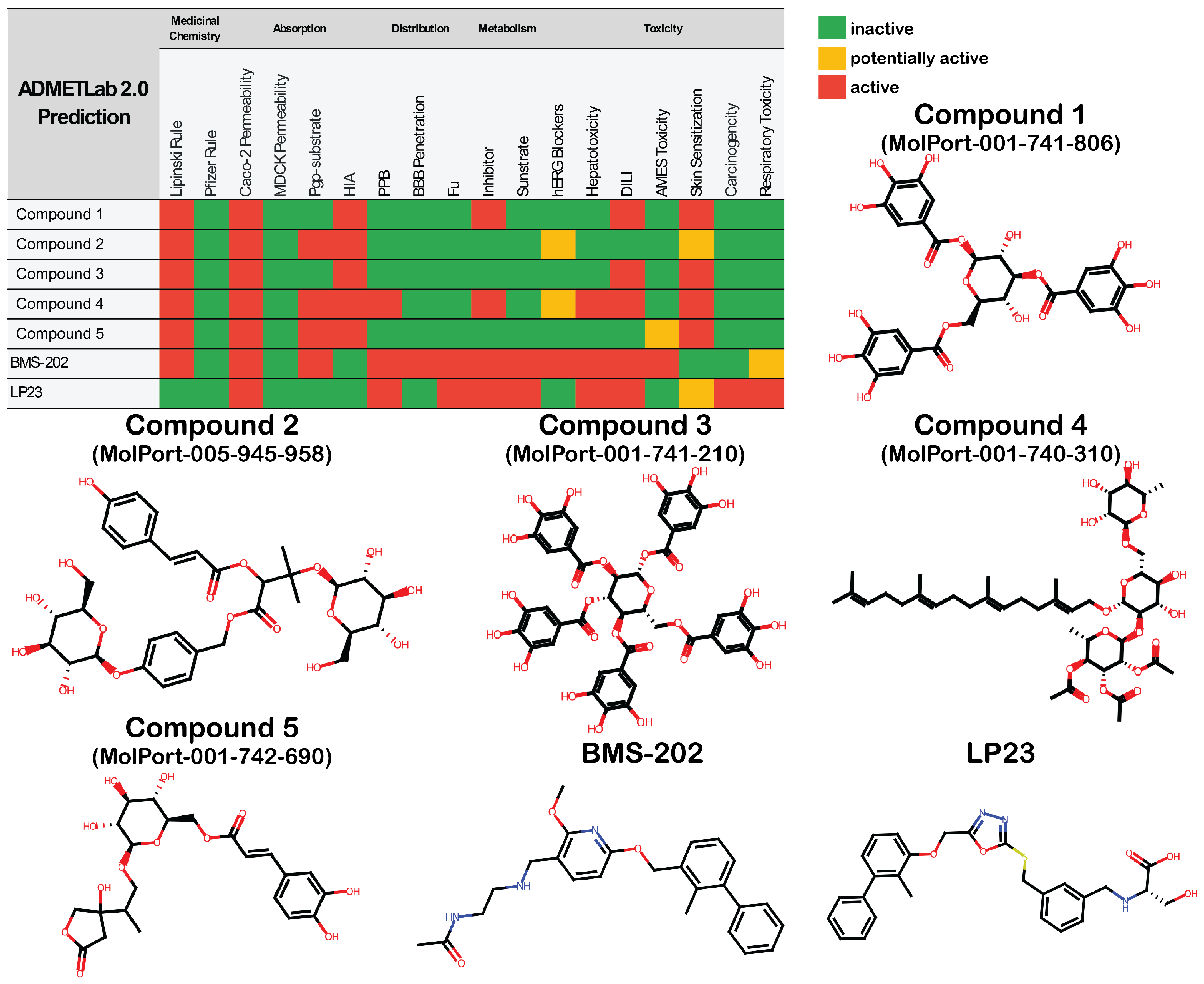
3.2. Library Screening and ADMET Profiling
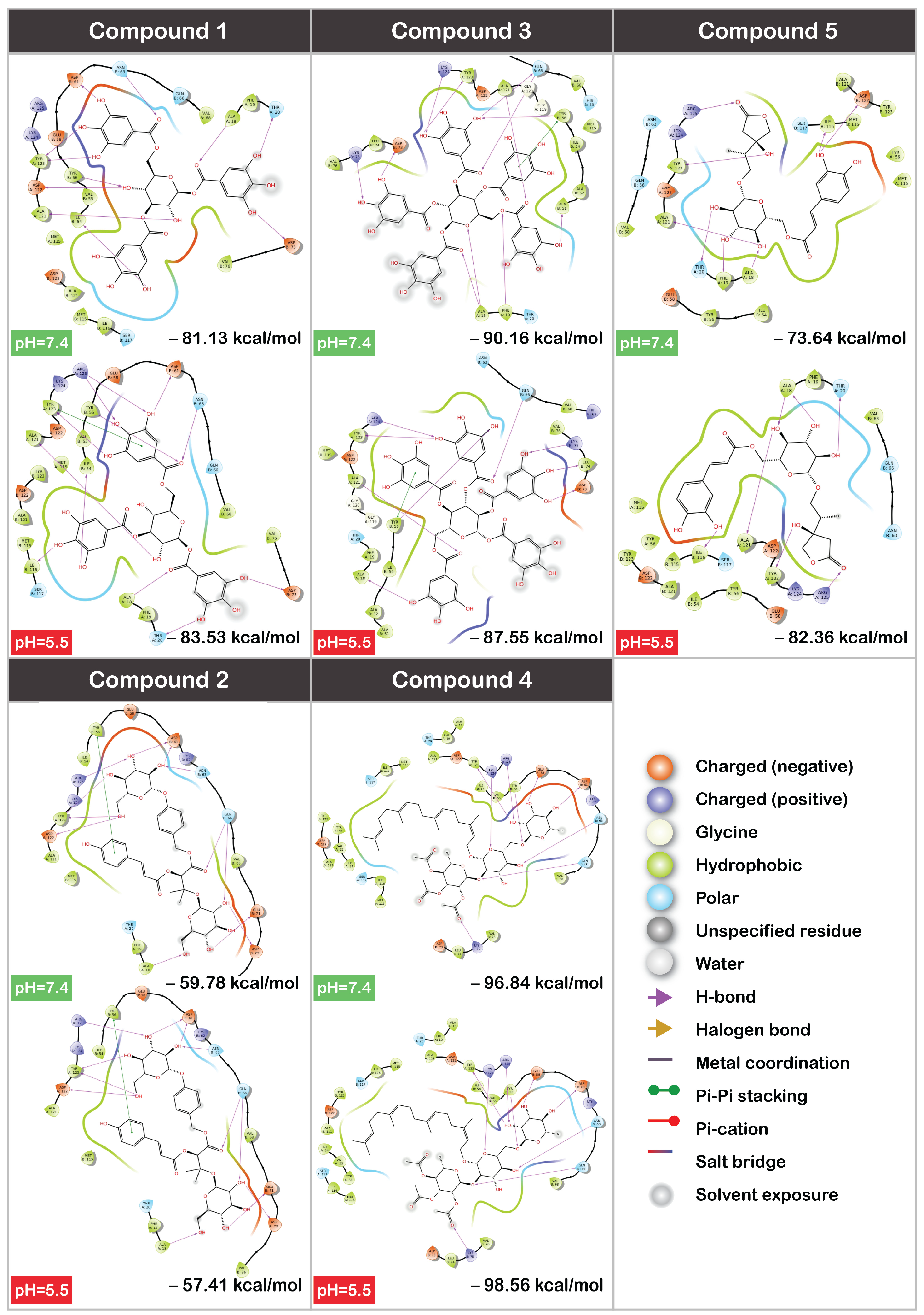
3.3. MM/GBSA
3.4. Molecular Dynamics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kennedy, S.R.; Loeb, L.A.; Herr, A.J. Somatic mutations in aging, cancer, and neurodegeneration. Mech. Ageing Dev. 2012, 133, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Ronellenfitsch, U. Surgical Oncology: Multidisciplinarity to Improve Cancer Treatment and Outcomes. Curr. Oncol. 2021, 28, 4471–4473. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes. Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef] [PubMed]
- Chaput, G.; Sumar, N. Endocrine therapies for breast and prostate cancers. Can. Fam. Physician 2022, 68, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and Radiation Therapy: Current Advances and Future Directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Starnes, C.O. Coley’s toxins in perspective. Nature 1992, 357, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’haese, J.G.; Schloesser, H.; et al. Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 268. [Google Scholar] [CrossRef]
- Yang, L.; Ning, Q.; Tang, S. Recent Advances and Next Breakthrough in Immunotherapy for Cancer Treatment. J. Immunol. Res. 2022, 2022, 8052212. [Google Scholar] [CrossRef]
- Mondal, M.; Guo, J.; He, P.; Zhou, D. Recent advances of oncolytic virus in cancer therapy. Hum. Vaccines Immunother. 2020, 16, 2389–2402. [Google Scholar] [CrossRef] [PubMed]
- Apolonio, J.S.; Gonçalves, V.L.d.S.; Santos, M.L.C.; Luz, M.S.; Souza, J.V.S.; Pinheiro, S.L.R.; de Souza, W.R.; Loureiro, M.S.; de Melo, F.F. Oncolytic virus therapy in cancer: A current review. World J. Virol. 2021, 10, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Propper, D.J.; Balkwill, F.R. Harnessing cytokines and chemokines for cancer therapy. Nat. Rev. Clin. Oncol. 2022, 19, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer vaccines: The next immunotherapy frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Rosenberg, S.A. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat. Rev. Cancer 2003, 3, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kwon, M.; Shin, E.-C. Immune checkpoint inhibitors for cancer treatment. Arch. Pharm. Res. 2016, 39, 1577–1587. [Google Scholar] [CrossRef]
- Cha, J.-H.; Chan, L.-C.; Li, C.-W.; Hsu, J.L.; Hung, M.-C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Makuku, R.; Khalili, N.; Razi, S.; Keshavarz-Fathi, M.; Rezaei, N. Current and Future Perspectives of PD-1/PDL-1 Blockade in Cancer Immunotherapy. J. Immunol. Res. 2021, 2021, 6661406. [Google Scholar] [CrossRef]
- Chamoto, K.; Hatae, R.; Honjo, T. Current issues and perspectives in PD-1 blockade cancer immunotherapy. Int. J. Clin. Oncol. 2020, 25, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Ning, Q.; Yang, L.; Mo, Z.; Tang, S. Mechanisms of immune escape in the cancer immune cycle. Int. Immunopharmacol. 2020, 86, 106700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dai, Z.; Wu, W.; Wang, Z.; Zhang, N.; Zhang, L.; Zeng, W.-J.; Liu, Z.; Cheng, Q. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 184. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, E. Nivolumab success in untreated metastatic melanoma. Lancet Oncol. 2015, 16, e9. [Google Scholar] [CrossRef] [PubMed]
- Bagcchi, S. Pembrolizumab for treatment of refractory melanoma. Lancet Oncol. 2014, 15, e419. [Google Scholar] [CrossRef]
- Westin, J.R.; Chu, F.; Zhang, M.; E Fayad, L.; Kwak, L.W.; Fowler, N.; Romaguera, J.; Hagemeister, F.; Fanale, M.; Samaniego, F.; et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: A single group, open-label, phase 2 trial. Lancet Oncol. 2014, 15, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef]
- Liu, J.; Cheng, Y.; Yuan, L.; Liu, T.; Ruan, Y.; Ren, Y.; Li, L.; Jiang, S.; Xiao, Y.; Chen, J. Discovery and Crystallography Study of Novel Biphenyl Ether and Oxadiazole Thioether (Non-Arylmethylamine)-Based Small-Molecule PD-1/PD-L1 Inhibitors as Immunotherapeutic Agents. J. Med. Chem. 2023, 66, 13172–13188. [Google Scholar] [CrossRef]
- Sun, C.; Yin, M.; Cheng, Y.; Kuang, Z.; Liu, X.; Wang, G.; Wang, X.; Yuan, K.; Min, W.; Dong, J.; et al. Novel Small-Molecule PD-L1 Inhibitor Induces PD-L1 Internalization and Optimizes the Immune Microenvironment. J. Med. Chem. 2023, 66, 2064–2083. [Google Scholar] [CrossRef]
- Cai, S.; Wang, K.; Qi, Z.; Ye, K.; Zhou, X.; Jiang, S.; Zhang, K.; Zhang, X.; Wang, T. Design, synthesis, and evaluation of PD-1/PD-L1 small-molecule inhibitors bearing a rigid indane scaffold. Eur. J. Med. Chem. 2023, 256, 115468. [Google Scholar] [CrossRef]
- Sasikumar, P.G.; Ramachandra, M. Small Molecule Agents Targeting PD-1 Checkpoint Pathway for Cancer Immunotherapy: Mechanisms of Action and Other Considerations for Their Advanced Development. Front. Immunol. 2022, 13, 752065. [Google Scholar] [CrossRef]
- Guo, Y.; Jin, Y.; Wang, B.; Liu, B. Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway through PD-L1 Dimerization. Int. J. Mol. Sci. 2021, 22, 4766. [Google Scholar] [CrossRef]
- Almahmoud, S.; Zhong, H.A. Molecular Modeling Studies on the Binding Mode of the PD-1/PD-L1 Complex Inhibitors. Int. J. Mol. Sci. 2019, 20, 4654. [Google Scholar] [CrossRef]
- Wang, F.; Ye, W.; Wang, S.; He, Y.; Zhong, H.; Wang, Y.; Zhu, Y.; Han, J.; Bing, Z.; Ji, S.; et al. Discovery of a new inhibitor targeting PD-L1 for cancer immunotherapy. Neoplasia 2021, 23, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Udhwani, T.; Mukherjee, S.; Sharma, K.; Sweta, J.; Khandekar, N.; Nayarisseri, A.; Singh, S.K. Design of PD-L1 inhibitors for lung cancer. Bioinformation 2019, 15, 139–150. [Google Scholar] [CrossRef]
- Choorakottayil Pushkaran, A.; Kumaran, K.; Ann Maria, T.; Biswas, R.; Mohan, C.G. Identification of a PD1/PD-L1 inhibitor by structure-based pharmacophore modelling, virtual screening, molecular docking and biological evaluation. Mol. Inform. 2023, 42, 2200254. [Google Scholar] [CrossRef]
- Kuang, Z.; Heng, Y.; Huang, S.; Shi, T.; Chen, L.; Xu, L.; Mei, H. Partial Least-Squares Discriminant Analysis and Ensemble-Based Flexible Docking of PD-1/PD-L1 Inhibitors: A Pilot Study. ACS Omega 2020, 5, 26914–26923. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhong, A.; Wang, Q.; Zheng, T. Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking, ADMET, and Molecular Dynamics (MD) Simulation of Potential Inhibitors of PD-L1 from the Library of Marine Natural Products. Mar. Drugs 2021, 20, 29. [Google Scholar] [CrossRef] [PubMed]
- Boussadia, Z.; Zanetti, C.; Parolini, I. Role of microenvironmental acidity and tumor exosomes in cancer immunomodulation. Transl. Cancer Res. 2020, 9, 5775–5786. [Google Scholar] [CrossRef]
- Mori, D.; Tsujikawa, T.; Sugiyama, Y.; Kotani, S.; Fuse, S.; Ohmura, G.; Arai, A.; Kawaguchi, T.; Hirano, S.; Mazda, O.; et al. Extracellular acidity in tumor tissue upregulates programmed cell death ligand 1 expression on tumor cells via proton-sensing G protein-coupled receptors. Int. J. Cancer 2021, 149, 2116–2124. [Google Scholar] [CrossRef]
- Korenchan, D.E.; Flavell, R.R. Spatiotemporal pH Heterogeneity as a Promoter of Cancer Progression and Therapeutic Resistance. Cancers 2019, 11, 1026. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06: 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43.
- Meador, W.E.; Kapusta, K.; Owolabi, I.; Autry, S.A.; Saloni, J.; Kolodziejczyk, W.; Hammer, N.I.; Flynt, A.S.; Hill, G.A.; Delcamp, J.H. Ultra-Bright Near-Infrared Sulfonate-Indolizine Cyanine- and Squaraine-Albumin Chaperones: Record Quantum Yields and Applications. ChemPhotoChem 2022, 6, e202200127. [Google Scholar] [CrossRef]
- Mulakala, C.; Viswanadhan, V.N. Could MM-GBSA be accurate enough for calculation of absolute protein/ligand binding free energies? J. Mol. Graph. Model. 2013, 46, 41–51. [Google Scholar] [CrossRef]
- Naliwajski, M.R.; Wileńska, B.; Misicka, A.; Pietrosiuk, A.; Sykłowska-Baranek, K. HPLC-PDA-ESI-HRMS-Based Profiling of Secondary Metabolites of Rindera graeca Anatomical and Hairy Roots Treated with Drought and Cold Stress. Cells 2022, 11, 931. [Google Scholar] [CrossRef]
- Alves, V.M.; Yasgar, A.; Wellnitz, J.; Rai, G.; Rath, M.; Braga, R.C.; Capuzzi, S.J.; Simeonov, A.; Muratov, E.N.; Zakharov, A.V.; et al. Lies and Liabilities: Computational Assessment of High-Throughput Screening Hits to Identify Artifact Compounds. J. Med. Chem. 2023, 66, 12828–12839. [Google Scholar] [CrossRef] [PubMed]
- Gorshkov, K.; Sima, N.; Sun, W.; Lu, B.; Huang, W.; Travers, J.; Klumpp-Thomas, C.; Michael, S.G.; Xu, T.; Huang, R.; et al. Quantitative Chemotherapeutic Profiling of Gynecologic Cancer Cell Lines Using Approved Drugs and Bioactive Compounds. Transl. Oncol. 2019, 12, 441–452. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Docking Score | Glide Ligand Efficiency | XP GScore | Glide GScore | Glide Evdw | Glide Ecoul | Glide Energy | Glide Einternal | XP HBond |
|---|---|---|---|---|---|---|---|---|---|
| pH = 7.4 | |||||||||
| MolPort-039-339-177 | −14.413 | −0.257 | −14.413 | −14.413 | −46.675 | −32.076 | −78.751 | 19.539 | −8.955 |
| MolPort-001-740-898 1 | −14.256 | −0.324 | −14.256 | −14.256 | −59.246 | −16.306 | −75.552 | 18.202 | −5.5 |
| MolPort-001-741-409 1 | −14.029 | −0.319 | −14.029 | −14.029 | −51.465 | −24.336 | −75.802 | 11.204 | −6.634 |
| MolPort-027-853-642 1 | −13.175 | −0.388 | −13.175 | −13.175 | −58.105 | −16.254 | −74.359 | 0 | −4.637 |
| MolPort-042-675-462 1 | −13.101 | −0.397 | −13.101 | −13.101 | −57.753 | −17.173 | −74.926 | 0 | −4.487 |
| MolPort-006-668-633 | −12.796 | −0.284 | −12.796 | −12.796 | −65.269 | −18.75 | −84.018 | 17.747 | −3.987 |
| MolPort-019-936-738 1 | −12.7 | −0.302 | −12.7 | −12.7 | −37.706 | −21.373 | −59.079 | 0 | −5.4 |
| MolPort-019-937-075 | −12.664 | −0.422 | −12.664 | −12.664 | −48.802 | −19.247 | −68.049 | 10.551 | −5.187 |
| MolPort-001-741-410 | −12.601 | −0.286 | −12.601 | −12.601 | −55.576 | −23.382 | −78.959 | 14.896 | −5 |
| MolPort-044-637-514 | −12.574 | −0.322 | −12.574 | −12.574 | −46.353 | −23.009 | −69.362 | 17.401 | −5.133 |
| pH = 5.5 | |||||||||
| MolPort-001-741-806 | −14.696 | −0.327 | −14.696 | −14.696 | −51.996 | −25.228 | −77.224 | 13.831 | −7.669 |
| MolPort-001-740-898 1 | −14.433 | −0.328 | −14.433 | −14.433 | −54.059 | −18.633 | −72.692 | 13.624 | −5.871 |
| MolPort-042-675-462 1 | −13.97 | −0.423 | −13.97 | −13.97 | −41.551 | −19.12 | −60.672 | 0 | −5.731 |
| MolPort-027-853-642 1 | −13.85 | −0.407 | −13.85 | −13.85 | −57.316 | −19.191 | −76.507 | 16.901 | −4.977 |
| MolPort-001-741-409 1 | −13.809 | −0.314 | −13.809 | −13.809 | −56.672 | −22.95 | −79.622 | 12.204 | −6.214 |
| MolPort-005-945-958 | −13.165 | −0.263 | −13.165 | −13.165 | −38.363 | −40.886 | −79.248 | 16.063 | −8.488 |
| MolPort-001-741-210 | −13.144 | −0.196 | −13.144 | −13.144 | −58.858 | −15.814 | −74.672 | 16.758 | −7.016 |
| MolPort-001-740-310 | −12.987 | −0.213 | −12.987 | −12.987 | −77.865 | −14.571 | −92.436 | 33.752 | −3.641 |
| MolPort-001-742-690 | −12.914 | −0.38 | −12.914 | −12.914 | −51.647 | −17.114 | −68.762 | 19.835 | −5.107 |
| MolPort-019-936-738 1 | −12.759 | −0.304 | −12.759 | −12.759 | −37.839 | −21.105 | −58.944 | 0 | −5.479 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDowell, R.C.; Booth, J.D.; McGowan, A.; Kolodziejczyk, W.; Hill, G.A.; Banerjee, S.; Feng, M.; Kapusta, K. Computational Approach for the Development of pH-Selective PD-1/PD-L1 Signaling Pathway Inhibition in Fight with Cancer. Cancers 2024, 16, 2295. https://doi.org/10.3390/cancers16132295
McDowell RC, Booth JD, McGowan A, Kolodziejczyk W, Hill GA, Banerjee S, Feng M, Kapusta K. Computational Approach for the Development of pH-Selective PD-1/PD-L1 Signaling Pathway Inhibition in Fight with Cancer. Cancers. 2024; 16(13):2295. https://doi.org/10.3390/cancers16132295
Chicago/Turabian StyleMcDowell, Roderick C., Jordhan D. Booth, Allyson McGowan, Wojciech Kolodziejczyk, Glake A. Hill, Santanu Banerjee, Manliang Feng, and Karina Kapusta. 2024. "Computational Approach for the Development of pH-Selective PD-1/PD-L1 Signaling Pathway Inhibition in Fight with Cancer" Cancers 16, no. 13: 2295. https://doi.org/10.3390/cancers16132295