
CRISPR-Cas9 Knockout Screens Identify DNA Damage Response Pathways and BTK as Essential for Cisplatin Response in Diffuse Large B-Cell Lymphoma

 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culturing
2.2. CRISPR Knockout Library Screening
2.3. Clinical Cohorts
2.4. Drug Response Assays
2.5. Generation of Single Gene Knockouts
2.6. DNA Damage and Cell Cycle Analysis
2.7. Biostatistical Analysis
3. Results
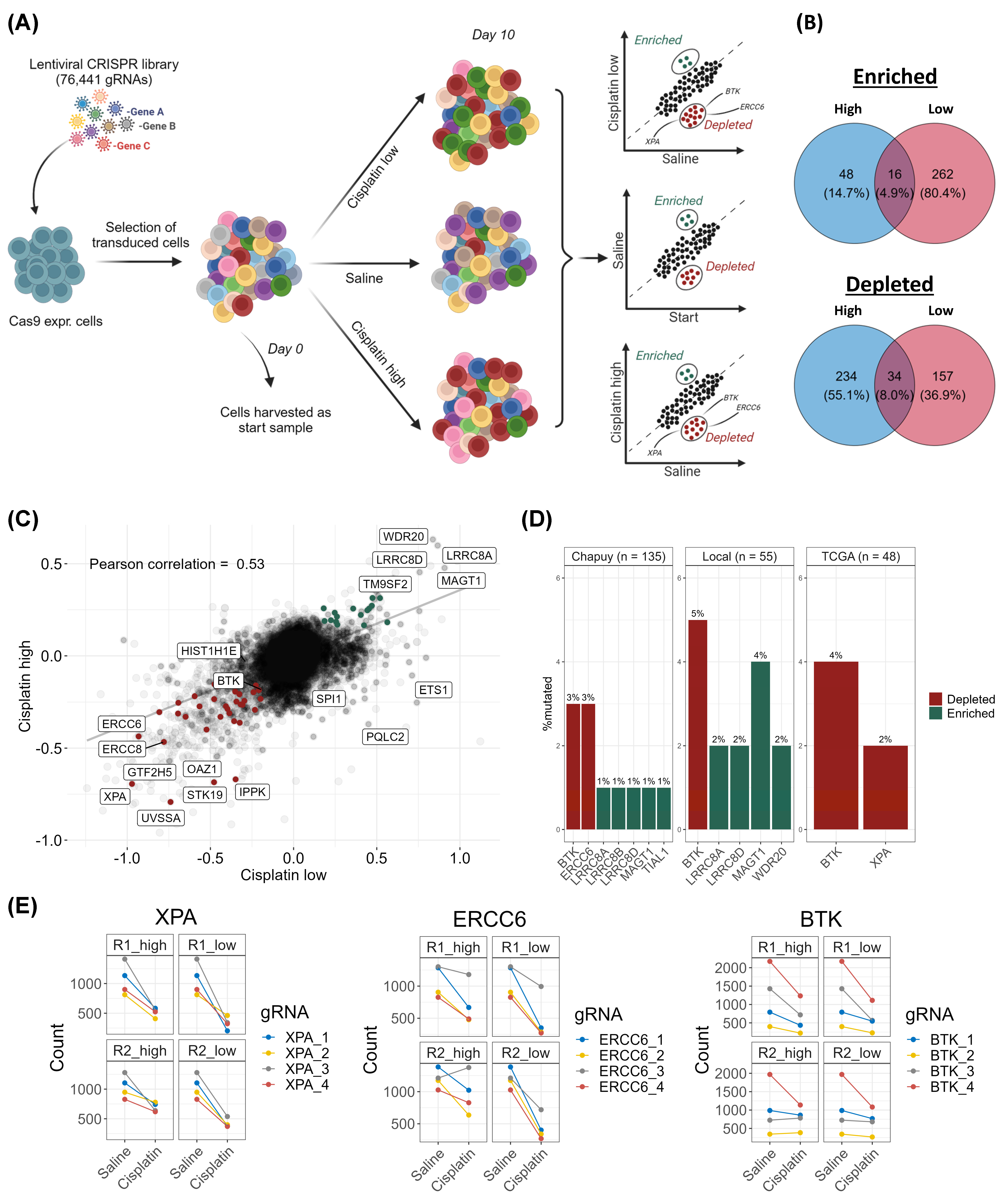
3.1. CRISPR-Cas9 Screens Identify DNA Damage Response Genes as Essential for Response to Cisplatin in DLBCL
3.2. Examination of Mutation Frequencies in Clinical Cohorts and Gene Selection for Validation
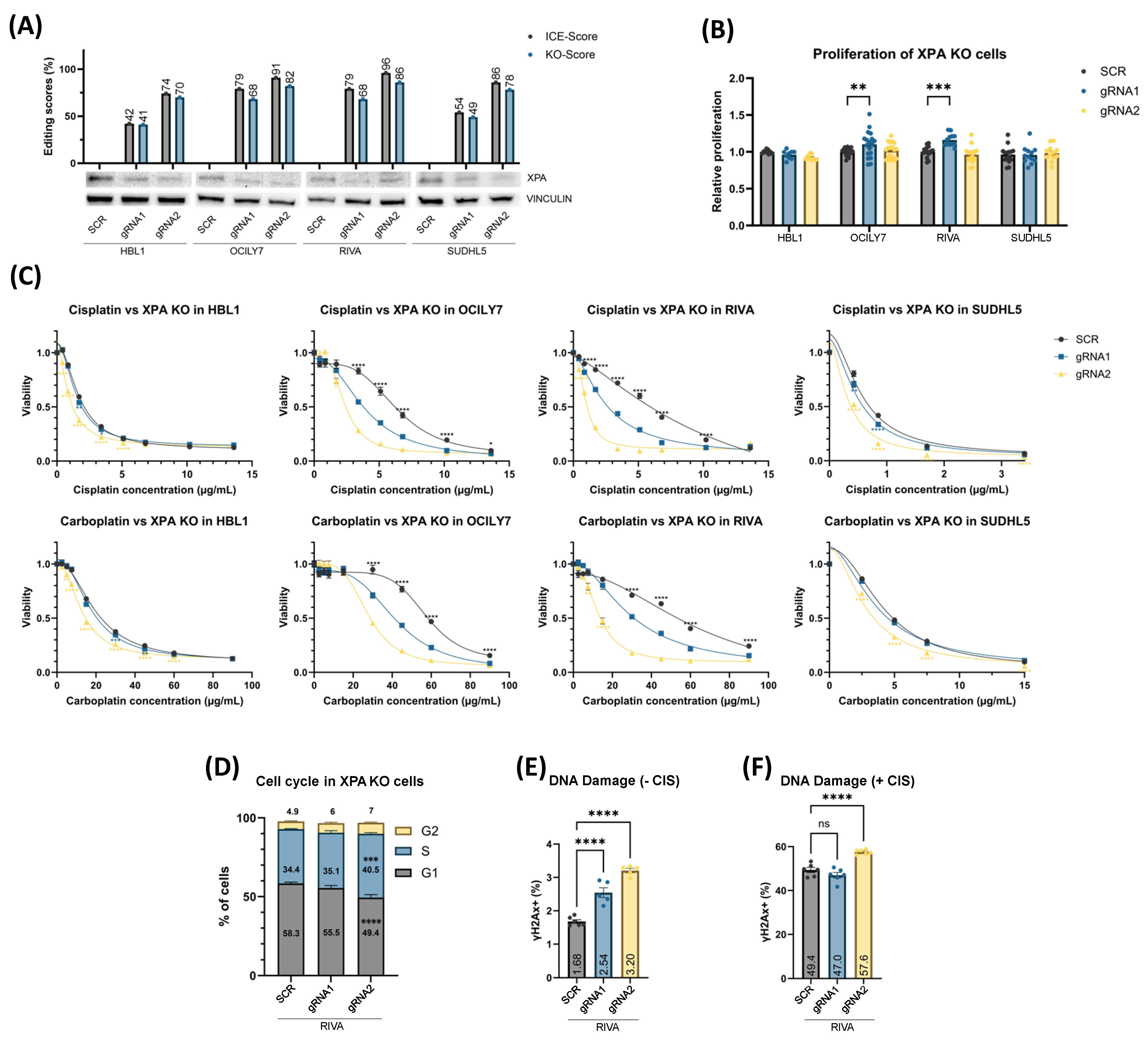
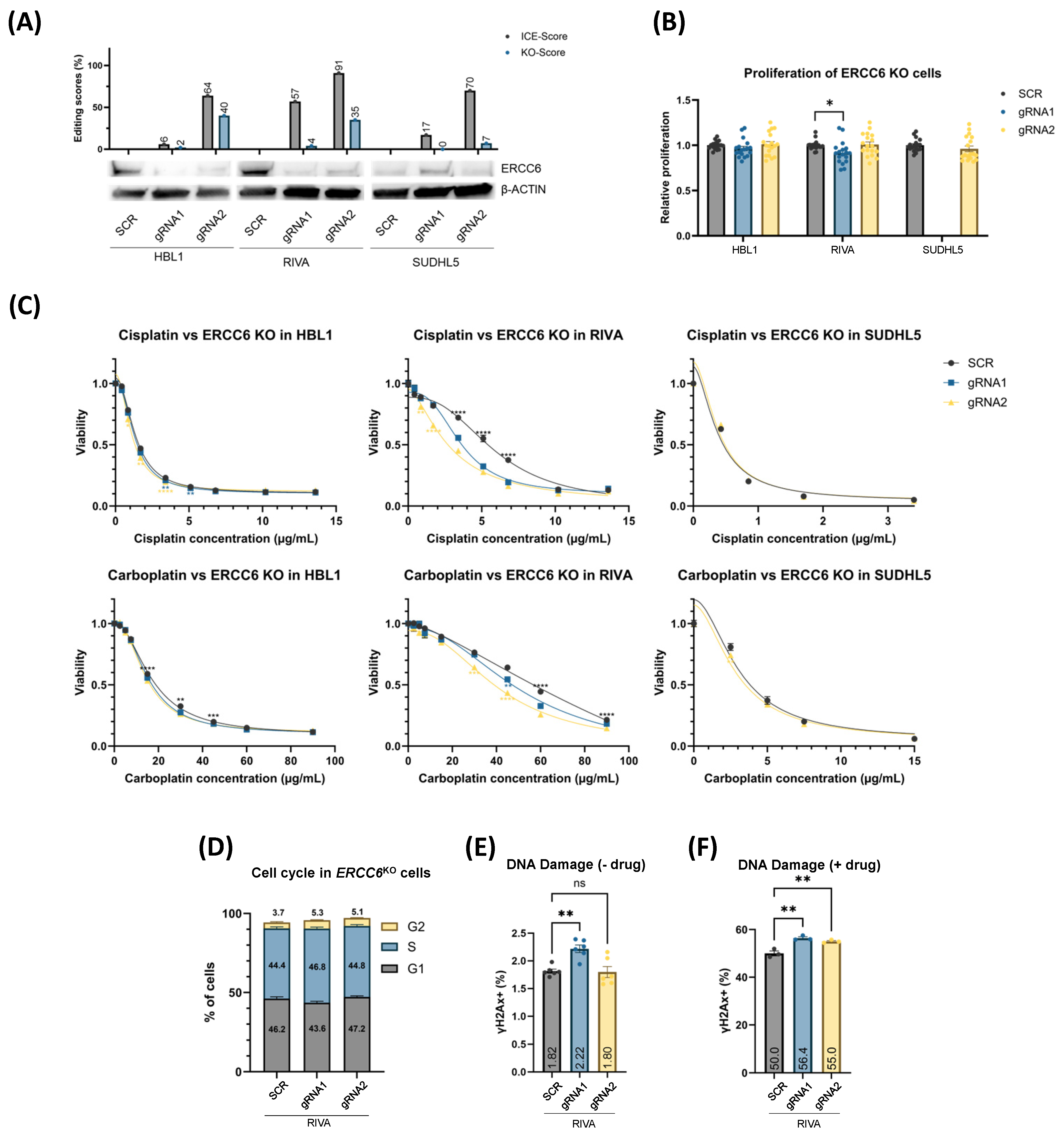
3.3. XPAKO and ERCC6KO Confers Sensitivity to Platinum Drugs Irrespective of Proliferation Rate
3.4. XPAKO and ERCC6KO Affect DNA Damage Response and Cell Cycle Distribution
3.5. BTKKO and Chemical Inhibition, Using Ibrutinib, Sensitize DLBCL Cells to Platinum Drugs
3.6. Mutations in DNA Damage Response Genes in Diagnostic DLBCL Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DLBCL | Diffuse large B-cell lymphoma |
| DDR | DNA damage response |
| DSBs | Double-strand breaks |
| gRNA | Guide RNA |
| HR | Homologous recombination |
| MMR | Mismatch repair |
| NER | Nucleotide excision repair |
| R-DHAP | Rituximab with dexamethasone, high-dose cytarabine, and cisplatin |
| R-GDP | Rituximab with gemcitabine, dexamethasone, and cisplatin |
| R-ICE | Rituximab with ifosfamide, carboplatin, and etoposide |
| RRA | Robust ranking aggregation |
| RR-DLBCL | Refractory/relapsed DLBCL |
References
- Sarkozy, C.; Sehn, L.H. Management of relapsed/refractory DLBCL. Best Pract. Res. Clin. Haematol. 2018, 31, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Salles, G. Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2021, 384, 842–858. [Google Scholar] [CrossRef]
- Abramson, J.S.; Solomon, S.R.; Arnason, J.; Johnston, P.B.; Glass, B.; Bachanova, V.; Ibrahimi, S.; Mielke, S.; Mutsaers, P.; Hernandez-Ilizaliturri, F.; et al. Lisocabtagene maraleucel as second-line therapy for large B-cell lymphoma: Primary analysis of the phase 3 TRANSFORM study. Blood 2023, 141, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Westin, J.R.; Oluwole, O.O.; Kersten, M.J.; Miklos, D.B.; Perales, M.-A.; Ghobadi, A.; Rapoport, A.P.; Sureda, A.; Jacobson, C.A.; Farooq, U.; et al. Survival with Axicabtagene Ciloleucel in Large B-Cell Lymphoma. N. Engl. J. Med. 2023, 389, 148–157. [Google Scholar] [CrossRef] [PubMed]
- García-Sancho, A.M.; Cabero, A.; Gutiérrez, N.C. Treatment of Relapsed or Refractory Diffuse Large B-Cell Lymphoma: New Approved Options. J. Clin. Med. 2024, 13, 70. [Google Scholar] [CrossRef] [PubMed]
- Gajra, A.; Zalenski, A.; Sannareddy, A.; Jeune-Smith, Y.; Kapinos, K.; Kansagra, A. Barriers to Chimeric Antigen Receptor T-Cell (CAR-T) Therapies in Clinical Practice. Pharmaceut. Med. 2022, 36, 163–171. [Google Scholar] [CrossRef]
- Gisselbrecht, C.; Glass, B.; Mounier, N.; Singh Gill, D.; Linch, D.C.; Trneny, M.; Bosly, A.; Ketterer, N.; Shpilberg, O.; Hagberg, H.; et al. Salvage Regimens With Autologous Transplantation for Relapsed Large B-Cell Lymphoma in the Rituximab Era. J. Clin. Oncol. 2010, 28, 4184–4190. [Google Scholar] [CrossRef]
- Crump, M.; Kuruvilla, J.; Couban, S.; MacDonald, D.A.; Kukreti, V.; Kouroukis, C.T.; Rubinger, M.; Buckstein, R.; Imrie, K.R.; Federico, M.; et al. Randomized comparison of gemcitabine, dexamethasone, and cisplatin versus dexamethasone, cytarabine, and cisplatin chemotherapy before autologous stem-cell transplantation for relapsed and refractory aggressive lymphomas: NCIC-CTG LY.12. J. Clin. Oncol. 2014, 32, 3490–3496. [Google Scholar] [CrossRef]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Van Den Neste, E.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef]
- Maurer, M.J.; Jakobsen, L.H.; Mwangi, R.; Schmitz, N.; Farooq, U.; Flowers, C.R.; de Nully Brown, P.; Thompson, C.A.; Frederiksen, H.; Cunningham, D.; et al. Relapsed/Refractory International Prognostic Index (R/R-IPI): An international prognostic calculator for relapsed/refractory diffuse large B-cell lymphoma. Am. J. Hematol. 2021, 96, 599–605. [Google Scholar] [CrossRef]
- Zhang, Z.; Xie, Z.; Sun, G.; Yang, P.; Li, J.; Yang, H.; Xiao, S.; Liu, Y.; Qiu, H.; Qin, L.; et al. Reversing drug resistance of cisplatin by hsp90 inhibitors in human ovarian cancer cells. Int. J. Clin. Exp. Med. 2015, 8, 6687–6701. [Google Scholar] [PubMed]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Issa, I.I.; Haraldsdóttir, H.; Hald, J.L.; Schmitz, A.; Due, H.; Dybkær, K. Hsp90 inhibition sensitizes DLBCL cells to cisplatin. Cancer Chemother. Pharmacol. 2022, 89, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M. DNA damage response and hematological malignancy. Int. J. Hematol. 2017, 106, 345–356. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.C.R.R.; Silva, M.M.M.; Quinet, A.; Cabral-Neto, J.B.J.; Menck, C.C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Ulibarri, J.; Liu, K.J.; Mao, P. Role of Nucleotide Excision Repair in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 9248. [Google Scholar] [CrossRef] [PubMed]
- Fadda, E. Role of the XPA protein in the NER pathway: A perspective on the function of structural disorder in macromolecular assembly. Comput. Struct. Biotechnol. J. 2016, 14, 78–85. [Google Scholar] [CrossRef]
- Falgreen, S.; Dybkær, K.; Young, K.H.; Xu-Monette, Z.Y.; El-Galaly, T.C.; Laursen, M.B.; Bødker, J.S.; Kjeldsen, M.K.; Schmitz, A.; Nyegaard, M.; et al. Predicting response to multidrug regimens in cancer patients using cell line experiments and regularised regression models. BMC Cancer 2015, 15, 235. [Google Scholar] [CrossRef]
- Sanson, K.R.; Hanna, R.E.; Hegde, M.; Donovan, K.F.; Strand, C.; Sullender, M.E.; Vaimberg, E.W.; Goodale, A.; Root, D.E.; Piccioni, F.; et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 2018, 9, 5416. [Google Scholar] [CrossRef] [PubMed]
- Schmierer, B.; Botla, S.K.; Zhang, J.; Turunen, M.; Kivioja, T.; Taipale, J. CRISPR/Cas9 screening using unique molecular identifiers. Mol. Syst. Biol. 2017, 13, 945. [Google Scholar] [CrossRef] [PubMed]
- Bødker, J.S.; Sønderkær, M.; Vesteghem, C.; Schmitz, A.; Brøndum, R.F.; Sommer, M.; Rytter, A.S.; Nielsen, M.M.; Madsen, J.; Jensen, P.; et al. Development of a Precision Medicine Workflow in Hematological Cancers, Aalborg University Hospital, Denmark. Cancers 2020, 12, 312. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst. 2018, 6, 271–281.e7. [Google Scholar] [CrossRef]
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.-B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef] [PubMed]
- Conant, D.; Hsiau, T.; Rossi, N.; Oki, J.; Maures, T.; Waite, K.; Yang, J.; Joshi, S.; Kelso, R.; Holden, K.; et al. Inference of CRISPR Edits from Sanger Trace Data. Cris. J. 2022, 5, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, M.; Zhang, W.; Xiao, T.; Chen, C.H.; Wu, A.; Wu, F.; Traugh, N.; Wang, X.; Li, Z.; et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat. Protoc. 2019, 14, 756–780. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 554. [Google Scholar] [CrossRef]
- Himmelstein, K.J.; Patton, T.F.; Belt, R.J.; Taylor, S.; Repta, A.J.; Sternson, L.A. Clinical kinetics on intact cisplatin and some related species. Clin. Pharmacol. Ther. 1981, 29, 658–664. [Google Scholar] [CrossRef]
- Dempster, J.M.; Rossen, J.; Kazachkova, M.; Pan, J.; Kugener, G.; Root, D.E.; Tsherniak, A. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2019. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Widmer, C.A.; Klebic, I.; Domanitskaya, N.; Decollogny, M.; Howald, D.; Siffert, M.; Essers, P.; Nowicka, Z.; Stokar-Regenscheit, N.; van de Ven, M.; et al. Loss of the Volume-regulated Anion Channel Components LRRC8A and LRRC8D Limits Platinum Drug Efficacy. Cancer Res. Commun. 2022, 2, 1266–1281. [Google Scholar] [CrossRef] [PubMed]
- Di Noia, J.M.; Neuberger, M.S. Molecular Mechanisms of Antibody Somatic Hypermutation. Annu. Rev. Biochem. 2007, 76, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Giallourakis, C.; Mostoslavsky, R.; Alt, F.W. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu. Rev. Immunol. 2006, 24, 541–570. [Google Scholar] [CrossRef]
- Bröckelmann, P.J.; de Jong, M.R.W.; Jachimowicz, R.D. Targeting DNA Repair, Cell Cycle, and Tumor Microenvironment in B Cell Lymphoma. Cells 2020, 9, 2287. [Google Scholar] [CrossRef] [PubMed]
- de Miranda, N.F.; Peng, R.; Georgiou, K.; Wu, C.; Sörqvist, E.F.; Berglund, M.; Chen, L.; Gao, Z.; Lagerstedt, K.; Lisboa, S.; et al. DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J. Exp. Med. 2013, 210, 1729–1742. [Google Scholar] [CrossRef]
- Carrassa, L.; Colombo, I.; Damia, G.; Bertoni, F. Targeting the DNA damage response for patients with lymphoma: Preclinical and clinical evidences. Cancer Treat. Rev. 2020, 90, 102090. [Google Scholar] [CrossRef]
- Szalat, R.; Samur, M.K.; Fulciniti, M.; Lopez, M.; Nanjappa, P.; Cleynen, A.; Wen, K.; Kumar, S.; Perini, T.; Calkins, A.S.; et al. Nucleotide excision repair is a potential therapeutic target in multiple myeloma. Leukemia 2018, 32, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Y.; Ji, H.; Pan, J.; He, P.; Zhou, X.; Zhang, H.; Zhou, Z.; Chen, Z. Downregulated XPA promotes carcinogenesis of bladder cancer via impairment of DNA repair. Tumor Biol. 2017, 39, 101042831769167. [Google Scholar] [CrossRef]
- Dai, W.; Wu, A.; Li, Y.; Yu, G.; Yan, X. XPA Enhances Temozolomide Resistance of Glioblastoma Cells by Promoting Nucleotide Excision Repair. Cell Transplant. 2022, 31, 09636897221092778. [Google Scholar] [CrossRef] [PubMed]
- Pulzová, L.B.; Ward, T.A.; Chovanec, M. XPA: DNA repair protein of significant clinical importance. Int. J. Mol. Sci. 2020, 21, 2182. [Google Scholar] [CrossRef] [PubMed]
- Goodspeed, A.; Jean, A.; Costello, J.C. A Whole-genome CRISPR Screen Identifies a Role of MSH2 in Cisplatin-mediated Cell Death in Muscle-invasive Bladder Cancer. Eur. Urol. 2019, 75, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Wu, S.; Fan, Y. Upregulation of exonuclease 1 caused by homology-dependent repair confers cisplatin resistance to gastric cancer cells. Can. J. Physiol. Pharmacol. 2022, 100, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Hu, J.; Han, H.; Hua, Y.; Zhou, L.; Shuai, W.; Du, W.; Kuang, C.; Chen, S.; Huang, W.; et al. High expression of XPA confers poor prognosis for nasopharyngeal carcinoma patients treated with platinum-based chemoradiotherapy. Oncotarget 2015, 6, 28478–28490. [Google Scholar] [CrossRef] [PubMed]
- Cierna, Z.; Miskovska, V.; Roska, J.; Jurkovicova, D.; Pulzova, L.B.; Sestakova, Z.; Hurbanova, L.; Machalekova, K.; Chovanec, M.; Rejlekova, K.; et al. Increased levels of XPA might be the basis of cisplatin resistance in germ cell tumours. BMC Cancer 2020, 20, 17. [Google Scholar] [CrossRef]
- Young, R.M.; Shaffer, A.L.; Phelan, J.D.; Staudt, L.M. B-Cell Receptor Signaling in Diffuse Large B-Cell lymphoma. Semin. Hematol. 2015, 52, 77–85. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Elsen, M.B.; Davis, R.E.; Ma, C.L.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Zucha, M.A.; Wu, A.T.H.; Lee, W.-H.; Wang, L.-S.; Lin, W.-W.; Yuan, C.-C.; Yeh, C.-T. Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib suppresses stem-like traits in ovarian cancer. Oncotarget 2015, 6, 13255–13268. [Google Scholar] [CrossRef]
- Liu, S.-C.; Wu, Y.-C.; Huang, C.-M.; Hsieh, M.-S.; Huang, T.-Y.; Huang, C.-S.; Hsu, T.-N.; Huang, M.-S.; Lee, W.-H.; Yeh, C.-T.; et al. Inhibition of Bruton’s tyrosine kinase as a therapeutic strategy for chemoresistant oral squamous cell carcinoma and potential suppression of cancer stemness. Oncogenesis 2021, 10, 20. [Google Scholar] [CrossRef]
- Palmer, A.C.; Chidley, C.; Sorger, P.K. A curative combination cancer therapy achieves high fractional cell killing through low cross-resistance and drug additivity. eLife 2019, 8, e50036. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Wright, G.W.; Huang, D.W.; Hodkinson, B.; Balasubramanian, S.; Fan, Y.; Vermeulen, J.; Shreeve, M.; Staudt, L.M. Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell 2021, 39, 1643–1653. [Google Scholar] [CrossRef] [PubMed]
- Cuccuini, W.; Briere, J.; Mounier, N.; Voelker, H.-U.; Rosenwald, A.; Sundstrom, C.; Cogliatti, S.; Hirchaud, E.; Ysebaert, L.; Bron, D.; et al. MYC + diffuse large B-cell lymphoma is not salvaged by classical R-ICE or R-DHAP followed by BEAM plus autologous stem cell transplantation. Blood 2012, 119, 4619–4624. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Papenhausen, P.; Shao, H. The Role of c-MYC in B-Cell Lymphomas: Diagnostic and Molecular Aspects. Genes 2017, 8, 116. [Google Scholar] [CrossRef]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Issa, I.I.; Due, H.; Brøndum, R.F.; Veeravakaran, V.; Haraldsdóttir, H.; Sylvester, C.; Brogaard, A.; Dhanjal, S.; Schmierer, B.; Dybkær, K. CRISPR-Cas9 Knockout Screens Identify DNA Damage Response Pathways and BTK as Essential for Cisplatin Response in Diffuse Large B-Cell Lymphoma. Cancers 2024, 16, 2437. https://doi.org/10.3390/cancers16132437
Issa II, Due H, Brøndum RF, Veeravakaran V, Haraldsdóttir H, Sylvester C, Brogaard A, Dhanjal S, Schmierer B, Dybkær K. CRISPR-Cas9 Knockout Screens Identify DNA Damage Response Pathways and BTK as Essential for Cisplatin Response in Diffuse Large B-Cell Lymphoma. Cancers. 2024; 16(13):2437. https://doi.org/10.3390/cancers16132437
Chicago/Turabian StyleIssa, Issa Ismail, Hanne Due, Rasmus Froberg Brøndum, Vidthdyan Veeravakaran, Hulda Haraldsdóttir, Cathrine Sylvester, Asta Brogaard, Soniya Dhanjal, Bernhard Schmierer, and Karen Dybkær. 2024. "CRISPR-Cas9 Knockout Screens Identify DNA Damage Response Pathways and BTK as Essential for Cisplatin Response in Diffuse Large B-Cell Lymphoma" Cancers 16, no. 13: 2437. https://doi.org/10.3390/cancers16132437
APA StyleIssa, I. I., Due, H., Brøndum, R. F., Veeravakaran, V., Haraldsdóttir, H., Sylvester, C., Brogaard, A., Dhanjal, S., Schmierer, B., & Dybkær, K. (2024). CRISPR-Cas9 Knockout Screens Identify DNA Damage Response Pathways and BTK as Essential for Cisplatin Response in Diffuse Large B-Cell Lymphoma. Cancers, 16(13), 2437. https://doi.org/10.3390/cancers16132437