


Phosphodiesterase Inhibition to Sensitize Non-Small-Cell Lung Cancer to Pemetrexed: A Double-Edged Strategy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Drugs and Reagents
2.2. Cells and Cell Culture
2.3. Western Blotting
2.4. Growth Assays
2.5. Cell Viability and Apoptosis Assays
2.6. Cell Proliferation Assay
2.7. cAMP and cGMP Measurements
2.8. PKA and PKG Activity Assays
2.9. Apo-Tox-Glo Triplex Assay and Drug Interaction Analysis
2.10. Statistical Analysis
3. Results
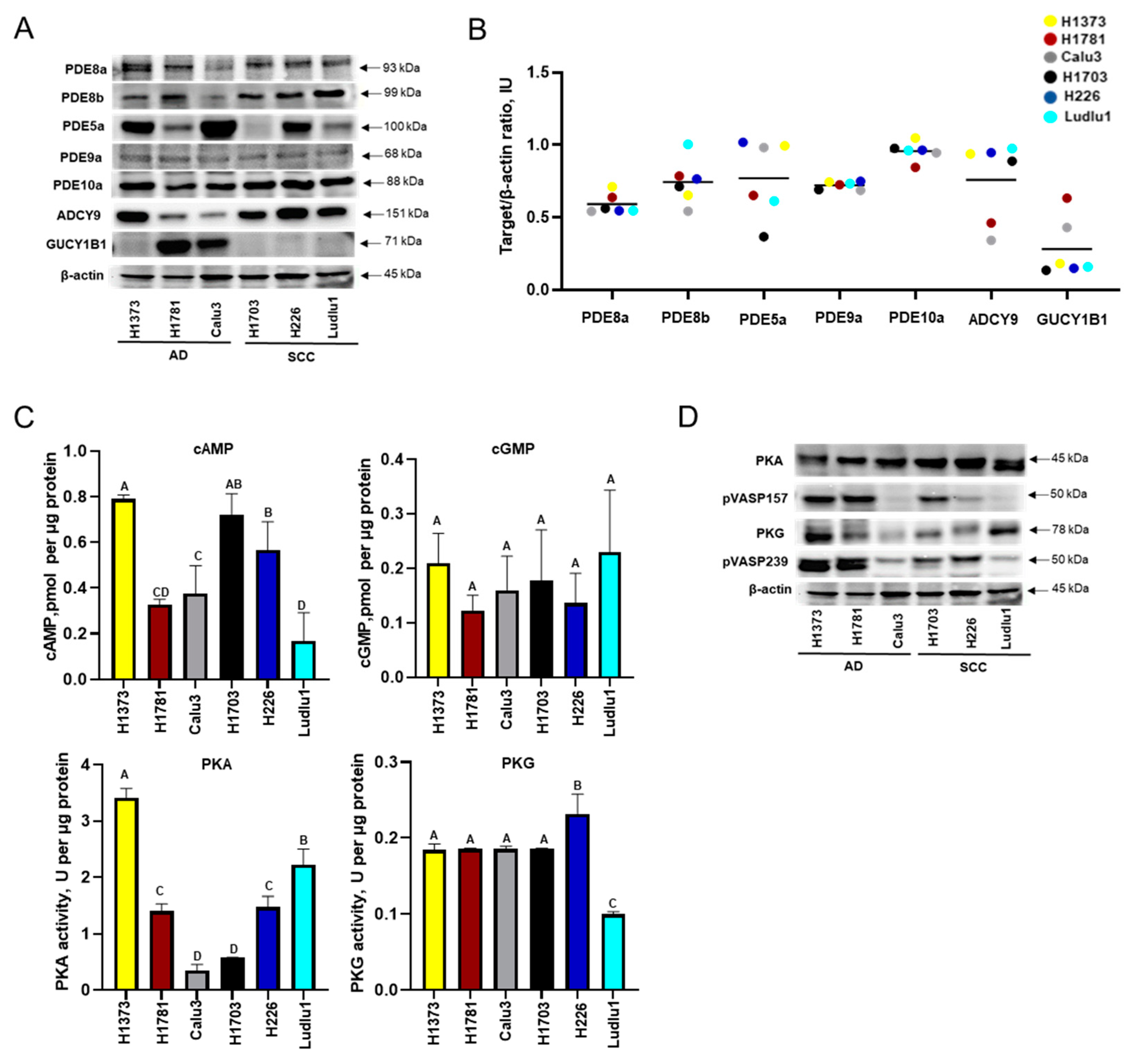
3.1. Baseline cAMP and cGMP Signaling in NSCLC Cell Lines
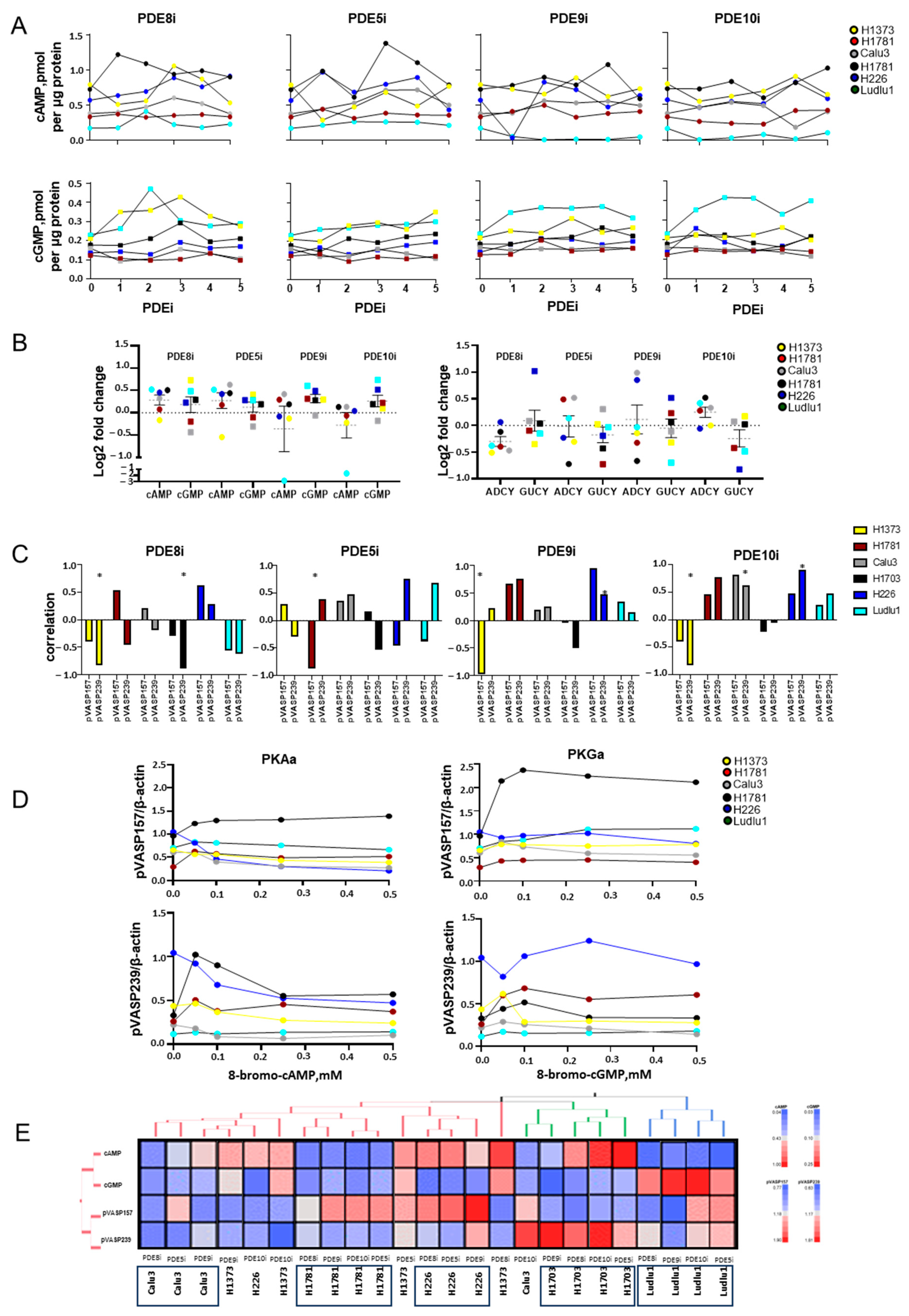
3.2. PDEi Modulates Cell Growth and Cyclic Nucleotide Signaling in NSCLC
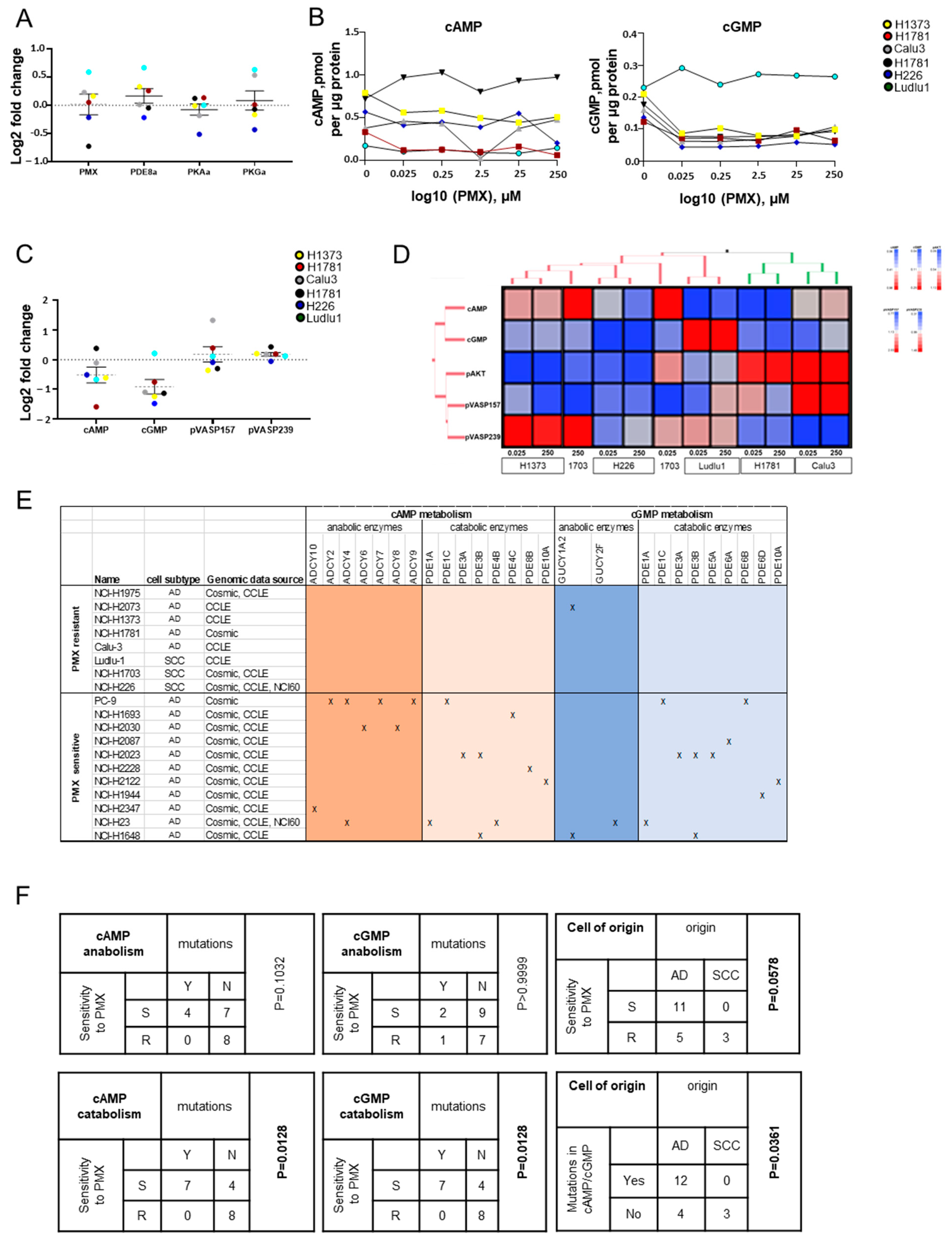
3.3. PMX Anti-NSCLC Activity Associates with Modulation of cAMP/cGMP Cellular Content and Downstream Signaling
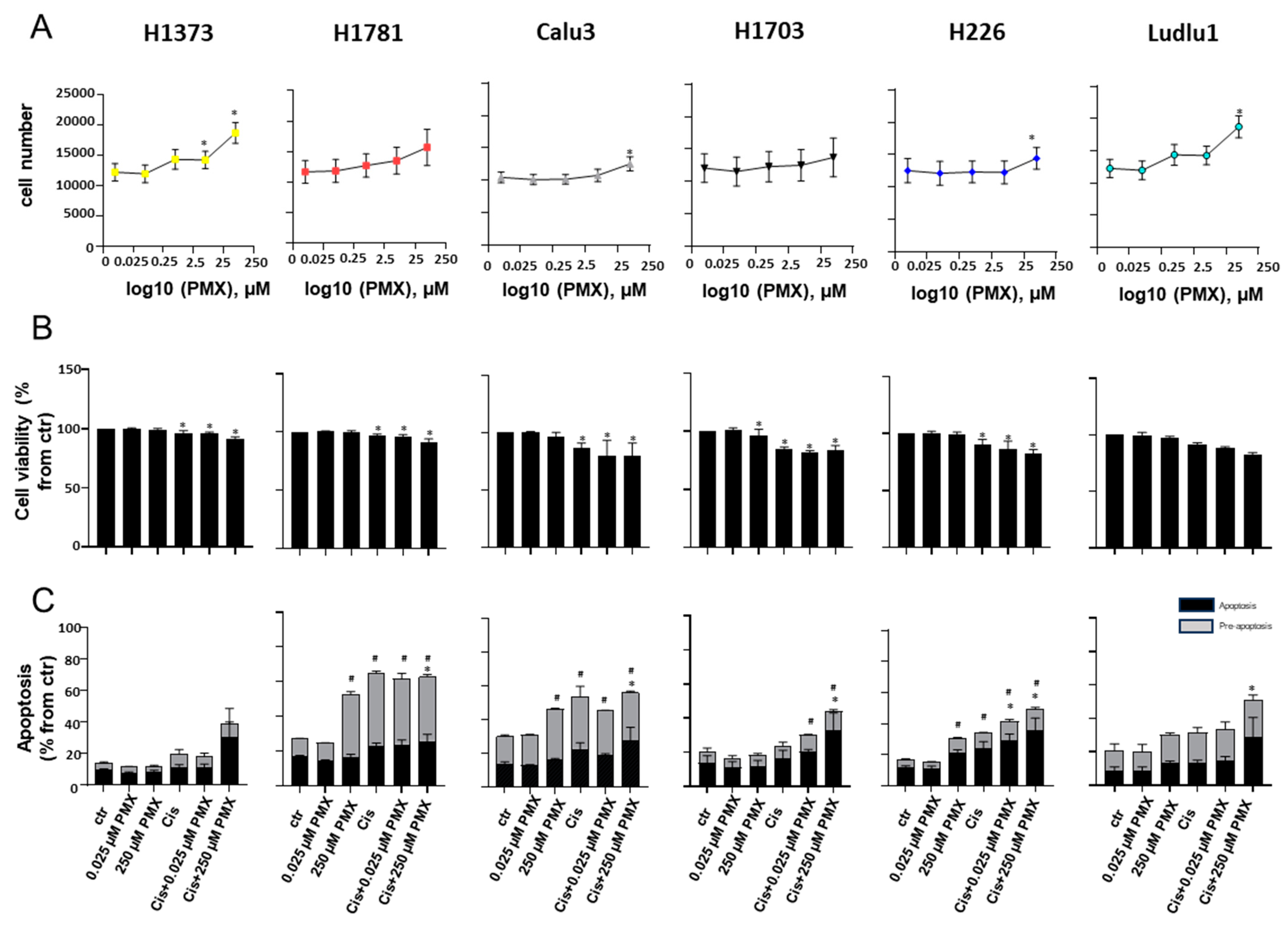
3.4. Biochemical PDE Inhibition Sensitizes NSCLC to Low-Dose PMX Killing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | protein kinase B |
| cAMP | cyclic adenosine monophosphate |
| cGMP | cyclic guanosine monophosphate |
| AD | lung cancer adenocarcinoma |
| SCC | lung cancer squamous-cell carcinoma |
| NSCLC | non-small-cell lung cancer |
| PDE | phosphodiesterase |
| PDEi | phosphodiesterase inhibitor |
| PKA | protein kinase A |
| PKG | protein kinase G |
| PMX | pemetrexed |
| TCGA | The Cancer Genome Atlas |
| WB | Western blotting |
References
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Gadgeel, S. Combination pembrolizumab plus chemotherapy: A new standard of care for patients with advanced non-small-cell lung cancer. Lung Cancer 2019, 10, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Mendes, C.; Serpa, J. Metabolic Remodelling: An Accomplice for New Therapeutic Strategies to Fight Lung Cancer. Antioxidants 2019, 8, 603. [Google Scholar] [CrossRef]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef]
- Adjei, A.A. Pharmacology and Mechanism of Action of Pemetrexed. Clin. Lung Cancer 2004, 5, S51–S55. [Google Scholar] [CrossRef]
- Wu, D.M.; Zhang, P.; Xu, G.C.; Tong, A.P.; Zhou, C.; Lang, J.Y.; Wang, C.T. Pemetrexed induces G1 phase arrest and apoptosis through inhibiting Akt activation in human non small lung cancer cell line A549. Asian Pac. J. Cancer Prev. APJCP 2015, 16, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Liu, J.; She, L.; Chen, J.; Zhu, T.; Yin, J.; Li, X.; Li, X.; Zhou, H.; Liu, Z. A perspective profile of ADCY1 in cAMP signaling with drug-resistance in lung cancer. J. Cancer 2019, 10, 6848–6857. [Google Scholar] [CrossRef]
- Hsien Lai, S.; Zervoudakis, G.; Chou, J.; Gurney, M.E.; Quesnelle, K.M. PDE4 subtypes in cancer. Oncogene 2020, 39, 3791–3802. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The role of cyclic nucleotide signaling pathways in cancer: Targets for prevention and treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef]
- Zhu, B.; Lindsey, A.; Li, N.; Lee, K.; Ramirez-Alcantara, V.; Canzoneri, J.C.; Fajardo, A.; da Silva, L.M.; Thomas, M.; Piazza, J.T.; et al. Phosphodiesterase 10A is overexpressed in lung tumor cells and inhibitors selectively suppress growth by blocking β-catenin and MAPK signaling. Oncotarget 2017, 8, 69264–69280. [Google Scholar] [CrossRef]
- Tuttle, T.R.; Mierzwa, M.L.; Wells, S.I.; Fox, S.R.; Ben-Jonathan, N. The cyclic GMP/protein kinase G pathway as a therapeutic target in head and neck squamous cell carcinoma. Cancer Lett. 2016, 370, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Gao, L.N.; Cui, Y.L.; Zhang, Y.; Zhou, X. The cyclic AMP signaling pathway: Exploring targets for successful drug discovery (Review). Mol. Med. Rep. 2016, 13, 3715–3723. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Kotera, J. Overview of PDEs and Their Regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Movsesian, M.A. cAMP and cGMP Signaling Cross-Talk. Circ. Res. 2007, 100, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Ivanina Foureau, A.V.; Sha, W.; Foureau, D.M.; Symanowski, J.T.; Farhangfar, C.J.; Mileham, K.F. Landscape and clinical impact of metabolic alterations in non-squamous non-small cell lung cancer. Transl. Lung Cancer Res. 2022, 11, 2464–2476. [Google Scholar] [CrossRef]
- Burvall, K.M.; Palmberg, L.; Larsson, K. The tyrosine kinase inhibitor genistein increases basal cAMP and potentiates forskolin-induced cAMP accumulation in A549 human airway epithelial cells. Mol. Cell. Biochem. 2002, 240, 131–133. [Google Scholar] [CrossRef]
- Ramezani, S.; Vousooghi, N.; Kapourchali, F.R.; Hadjighasem, M.; Hayat, P.; Amini, N.; Joghataei, M.T. Rolipram potentiates bevacizumab-induced cell death in human glioblastoma stem-like cells. Life Sci. 2017, 173, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Abusnina, A.; Keravis, T.; Yougbaré, I.; Bronner, C.; Lugnier, C. Anti-proliferative effect of curcumin on melanoma cells is mediated by PDE1A inhibition that regulates the epigenetic integrator UHRF1. Mol. Nutr. Food Res. 2011, 55, 1677–1689. [Google Scholar] [CrossRef]
- Sponziello, M.; Verrienti, A.; Rosignolo, F.; De Rose, R.F.; Pecce, V.; Maggisano, V.; Durante, C.; Bulotta, S.; Damante, G.; Giacomelli, L.; et al. PDE5 expression in human thyroid tumors and effects of PDE5 inhibitors on growth and migration of cancer cells. Endocrine 2015, 50, 434–441. [Google Scholar] [CrossRef]
- Mukherjee, P.; Bagchi, A.; Banerjee, A.; Roy, H.; Bhattacharya, A.; Biswas, A.; Chatterji, U. PDE4 inhibitor eliminates breast cancer stem cells via noncanonical activation of mTOR. J. Cell. Biochem. 2022, 123, 1980–1996. [Google Scholar] [CrossRef] [PubMed]
- Nagai, H.; Yasuda, H.; Hatachi, Y.; Xue, D.; Sasaki, T.; Yamaya, M.; Sakamori, Y.; Togashi, Y.; Masago, K.; Ito, I.; et al. Nitric oxide (NO) enhances pemetrexed cytotoxicity via NOcGMP signaling in lung adenocarcinoma cells in vitro and in vivo. Int. J. Oncol. 2012, 41, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Cross, T.G.; Scheel-Toellner, D.; Henriquez, N.V.; Deacon, E.; Salmon, M.; Lord, J.M. Serine/threonine protein kinases and apoptosis. Exp. Cell Res. 2000, 256, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, P.; Ronci, M.; Giuliani, P.; Caciagli, F.; Ciccarelli, R.; Caruso, V.; Beggiato, S.; Zuccarini, M. Pros and Cons of Pharmacological Manipulation of cGMP-PDEs in the Prevention and Treatment of Breast Cancer. Int. J. Mol. Sci. 2022, 23, 262. [Google Scholar] [CrossRef] [PubMed]
- Foureau, D.M.; Bhutani, M.; Robinson, M.; Guo, F.; Pham, D.; Buelow, B.; Steuerwald, N.; Rigby, K.; Tjaden, E.; Leonidas, M.; et al. Ex vivo efficacy of BCMA-bispecific antibody TNB-383B in relapsed/refractory multiple myeloma. EJHaem 2020, 1, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Gandra, D.; Mulama, D.H.; Foureau, D.M.; McKinney, K.Q.; Kim, E.; Smith, K.; Haw, J.; Nagulapally, A.; Saulnier Sholler, G.L. DFMO inhibition of neuroblastoma tumorigenesis. Cancer Med. 2024, 13, e7207. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef]
- Oelze, M.; Mollnau, H.; Hoffmann, N.; Warnholtz, A.; Bodenschatz, M.; Smolenski, A.; Walter, U.; Skatchkov, M.; Meinertz, T.; Münzel, T. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ. Res. 2000, 87, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Köhler, D.; Straub, A.; Weissmüller, T.; Faigle, M.; Bender, S.; Lehmann, R.; Wendel, H.-P.; Kurz, J.; Walter, U.; Zacharowski, K.; et al. Phosphorylation of Vasodilator-Stimulated Phosphoprotein Prevents Platelet-Neutrophil Complex Formation and Dampens Myocardial Ischemia-Reperfusion Injury. Circulation 2011, 123, 2579–2590. [Google Scholar] [CrossRef]
- Chen, K.C.; Yang, T.Y.; Wu, C.C.; Cheng, C.C.; Hsu, S.L.; Hung, H.W.; Chen, J.W.; Chang, G.C. Pemetrexed induces S-phase arrest and apoptosis via a deregulated activation of Akt signaling pathway. PLoS ONE 2014, 9, e97888. [Google Scholar] [CrossRef]
- Mohiuddin, M.; Kasahara, K. Cisplatin and Pemetrexed Have Distinctive Growth-inhibitory Effects in Monotherapy and Combination Therapy on KRAS-dependent A549 Lung Cancer Cells. Cancer Genom. Proteom. 2021, 18, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.-C.; Chen, J.-C.; Tseng, P.-Y.; Hsieh, J.-M.; Chiang, C.-S.; Liu, L.-L.; Chien, C.-C.; Huang, I.-H.; Lin, Y.-W. Nitroglycerin-induced downregulation of AKT- and ERK1/2-mediated radiation-sensitive 52 expression to enhance pemetrexed-induced cytotoxicity in human lung cancer cells. Toxicol. Res. 2022, 11, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Shields, M.D. Discovery and Development of Predictive Biomarkers for the Personalization of Pemetrexed Therapy in Non-Small Cell Lung Cancer. Ph.D. Thesis, The University of Texas Southwestern Medical Center, Dallas, TX, USA, 2012. [Google Scholar]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Liu, W.; Schiöth, H.B. Recent developments of phosphodiesterase inhibitors: Clinical trials, emerging indications and novel molecules. Front. Pharmacol. 2022, 13, 1057083. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.A.; Ward, A.; Chen, X.; Maxuitenko, Y.; Coley, A.; Aboelella, N.S.; Buchsbaum, D.J.; Boyd, M.R.; Keeton, A.B.; Zhou, G. PDE5 and PDE10 inhibition activates cGMP/PKG signaling to block Wnt/β-catenin transcription, cancer cell growth, and tumor immunity. Drug Discov. Today 2020, 25, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, T.M.; Cornwell, T.L.; Taylor, A.E. cGMP-dependent protein kinase mediates the reduction of Ca2+ by cAMP in vascular smooth muscle cells. Am. J. Physiol. 1990, 258, C399–C407. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Kryman, J.P.; El-Mowafy, A.M.; Han, G.; Carrier, G.O. cAMP-Dependent Vasodilators Cross-Activate the cGMP-Dependent Protein Kinase to Stimulate BKCa Channel Activity in Coronary Artery Smooth Muscle Cells. Circ. Res. 2000, 86, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-J.; Bond, M.; Sala-Newby, G.B.; Newby, A.C. Altered S-Phase Kinase-Associated Protein-2 Levels Are a Major Mediator of Cyclic Nucleotide–Induced Inhibition of Vascular Smooth Muscle Cell Proliferation. Circ. Res. 2006, 98, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Eckly-Michel, A.; Martin, V.; Lugnier, C. Involvement of cyclic nucleotide-dependent protein kinases in cyclic AMP-mediated vasorelaxation. Br. J. Pharmacol. 1997, 122, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Haushalter, K.J.; Casteel, D.E.; Raffeiner, A.; Stefan, E.; Patel, H.H.; Taylor, S.S. Phosphorylation of protein kinase A (PKA) regulatory subunit RIα by protein kinase G (PKG) primes PKA for catalytic activity in cells. J. Biol. Chem. 2018, 293, 4411–4421. [Google Scholar] [CrossRef]
- Li, T.; Ling, Y.-H.; Goldman, I.D.; Perez-Soler, R. Schedule-Dependent Cytotoxic Synergism of Pemetrexed and Erlotinib in Human Non–Small Cell Lung Cancer Cells. Clin. Cancer Res. 2007, 13, 3413–3422. [Google Scholar] [CrossRef]
- Smith, P.G.; Wang, F.; Wilkinson, K.N.; Savage, K.J.; Klein, U.; Neuberg, D.S.; Bollag, G.; Shipp, M.A.; Aguiar, R.C.T. The phosphodiesterase PDE4B limits cAMP-associated PI3K/AKT–dependent apoptosis in diffuse large B-cell lymphoma. Blood 2005, 105, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Wang, Y.; Ali, A.K.; Ashraf, M. Long-acting phosphodiesterase-5 inhibitor, tadalafil, induces sustained cardioprotection against lethal ischemic injury. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H387–H391. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ponuwei, G.A.; Moore, C.E.; Willars, G.B.; Tee, A.R.; Herbert, T.P. cAMP inhibits mammalian target of rapamycin complex-1 and -2 (mTORC1 and 2) by promoting complex dissociation and inhibiting mTOR kinase activity. Cell. Signal. 2011, 23, 1927–1935. [Google Scholar] [CrossRef] [PubMed]
- Bischof, M.; Abdollahi, A.; Gong, P.; Stoffregen, C.; Lipson, K.E.; Debus, J.ü.; Weber, K.J.; Huber, P.E. Triple combination of irradiation, chemotherapy (pemetrexed), and VEGFR inhibition (SU5416) in human endothelial and tumor cells. Int. J. Radiat. Oncol. Biol. Phys. 2004, 60, 1220–1232. [Google Scholar] [CrossRef] [PubMed]
- Tekle, C.; Giovannetti, E.; Sigmond, J.; Graff, J.R.; Smid, K.; Peters, G.J. Molecular pathways involved in the synergistic interaction of the PKCβ inhibitor enzastaurin with the antifolate pemetrexed in non-small cell lung cancer cells. Br. J. Cancer 2008, 99, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Vemavarapu, L.; Thompson, W.J.; Strada, S.J. Suppression of cyclic GMP-specific phosphodiesterase 5 promotes apoptosis and inhibits growth in HT29 cells. J. Cell. Biochem. 2005, 94, 336–350. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.H.; Jung, J.M.; Hong, S.H. 8-Chloro-cyclic AMP-induced growth inhibition and apoptosis is mediated by p38 mitogen-activated protein kinase activation in HL60 cells. Cancer Res. 2005, 65, 4896–4901. [Google Scholar] [CrossRef] [PubMed]
- Massimi, M.; Cardarelli, S.; Galli, F.; Giardi, M.F.; Ragusa, F.; Panera, N.; Cinque, B.; Cifone, M.G.; Biagioni, S.; Giorgi, M. Increase of Intracellular Cyclic AMP by PDE4 Inhibitors Affects HepG2 Cell Cycle Progression and Survival. J. Cell. Biochem. 2017, 118, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Naderi, E.H.; Findley, H.W.; Ruud, E.; Blomhoff, H.K.; Naderi, S. Activation of cAMP signaling inhibits DNA damage–induced apoptosis in BCP-ALL cells through abrogation of p53 accumulation. Blood 2009, 114, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Lan, Y.W.; Tsai, Y.T.; Chen, Y.C.; Staniczek, T.; Tsou, Y.A.; Yen, C.C.; Chen, C.M. Additive Antiproliferative and Antiangiogenic Effects of Metformin and Pemetrexed in a Non-Small-Cell Lung Cancer Xenograft Model. Front. Cell Dev. Biol. 2021, 9, 688062. [Google Scholar] [CrossRef]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Dent, P. PDE5 inhibitors enhance the lethality of [pemetrexed + sorafenib]. Oncotarget 2017, 8, 13464–13475. [Google Scholar] [CrossRef] [PubMed]
- Tanino, R.; Tsubata, Y.; Harashima, N.; Harada, M.; Isobe, T. Novel drug-resistance mechanisms of pemetrexed-treated non-small cell lung cancer. Oncotarget 2018, 9, 16807–16821. [Google Scholar] [CrossRef] [PubMed]
- Hang, T.; Yang, L.; Zhang, X.; Li, J.; Long, F.; Zhu, N.; Li, Y.; Xia, J.; Zhang, Y.; Zhang, P.; et al. Peroxisome proliferator-activated receptor γ improves pemetrexed therapeutic efficacy in non-squamous non-small cell lung cancer. Am. J. Transl. Res. 2021, 13, 2296–2307. [Google Scholar] [PubMed]
- Paz-Ares, L.G.; de Marinis, F.; Dediu, M.; Thomas, M.; Pujol, J.L.; Bidoli, P.; Molinier, O.; Sahoo, T.P.; Laack, E.; Reck, M.; et al. PARAMOUNT: Final overall survival results of the phase III study of maintenance pemetrexed versus placebo immediately after induction treatment with pemetrexed plus cisplatin for advanced nonsquamous non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 2895–2902. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chi, Y.; Cao, G.; Zhao, J.; An, T.; Wu, M.; Wang, Y.; Zhuo, M.; Yang, X.; Jia, B.; et al. Efficacy and safety of pemetrexed maintenance chemotherapy for advanced non-small cell lung cancer in a real-world setting. J. Thorac. Dis. 2021, 13, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-Y.; Park, C.-K.; Choi, Y.-D.; Oh, I.-J.; Kim, Y.-C. Predictive factors for long-term responders of pemetrexed maintenance treatment in non-small cell lung cancer. Thorac. Cancer 2019, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Kim, J.S.; Govindan, R. Pemetrexed maintenance with or without pembrolizumab in non-squamous non-small cell lung cancer: A cross-trial comparison of KEYNOTE-189 versus PARAMOUNT, PRONOUNCE, and JVBL. Lung Cancer 2021, 151, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Dent, P. [pemetrexed + sildenafil], via autophagy-dependent HDAC downregulation, enhances the immunotherapy response of NSCLC cells. Cancer Biol. Ther. 2017, 18, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, C.; Jin, S.; Li, J.; Dai, J.; Zhang, Z.; Guo, R. Pemetrexed induces ROS generation and cellular senescence by attenuating TS-mediated thymidylate metabolism to reverse gefitinib resistance in NSCLC. J. Cell. Mol. Med. 2023, 27, 2032–2044. [Google Scholar] [CrossRef]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef]
- Ho, G.Y.; Woodward, N.; Coward, J.I. Cisplatin versus carboplatin: Comparative review of therapeutic management in solid malignancies. Crit. Rev. Oncol./Hematol. 2016, 102, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Brader, H.S.; Athappilly, G.K.; Loewenstein, J. Retinal Toxicity Associated With Excessive Sildenafil Ingestion. JAMA Ophthalmol. 2019, 137, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Al Ibrahim, A.H.; Ghallab, K.Q.; Alhumaid, F.I.; Almahfoudh, H.H.; Almadan, A.J.; Al Eid, M.A.; AlMishqab, M.H.; Alsaffar, M.F.; Aljamea, J.H. A Systematic Review of Sildenafil Mortality through the Years. Cureus 2022, 14, e32179. [Google Scholar] [CrossRef] [PubMed]
- Charnigo, R.J.; Beidler, D.; Rybin, D.; Pittman, D.D.; Tan, B.; Howard, J.; Michelson, A.D.; Frelinger, A.L.; Clarke, N. PF-04447943, a Phosphodiesterase 9A Inhibitor, in Stable Sickle Cell Disease Patients: A Phase Ib Randomized, Placebo-Controlled Study. Clin. Transl. Sci. 2019, 12, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.J.; Muirhead, G.J.; Harness, J.A. Pharmacokinetics of sildenafil after single oral doses in healthy male subjects: Absolute bioavailability, food effects and dose proportionality. Br. J. Clin. Pharmacol. 2002, 53 (Suppl. 1), 5s–12s. [Google Scholar] [CrossRef] [PubMed]
- Walling, D.P.; Banerjee, A.; Dawra, V.; Boyer, S.; Schmidt, C.J.; DeMartinis, N. Phosphodiesterase 10A Inhibitor Monotherapy Is Not an Effective Treatment of Acute Schizophrenia. J. Clin. Psychopharmacol. 2019, 39, 575–582. [Google Scholar] [CrossRef]
- Li, K.M.; Rivory, L.P.; Clarke, S.J. Pemetrexed pharmacokinetics and pharmacodynamics in a phase I/II study of doublet chemotherapy with vinorelbine: Implications for further optimization of pemetrexed schedules. Br. J. Cancer 2007, 97, 1071–1076. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanina Foureau, A.V.; Foureau, D.M.; McHale, C.C.; Guo, F.; Farhangfar, C.J.; Mileham, K.F. Phosphodiesterase Inhibition to Sensitize Non-Small-Cell Lung Cancer to Pemetrexed: A Double-Edged Strategy. Cancers 2024, 16, 2475. https://doi.org/10.3390/cancers16132475
Ivanina Foureau AV, Foureau DM, McHale CC, Guo F, Farhangfar CJ, Mileham KF. Phosphodiesterase Inhibition to Sensitize Non-Small-Cell Lung Cancer to Pemetrexed: A Double-Edged Strategy. Cancers. 2024; 16(13):2475. https://doi.org/10.3390/cancers16132475
Chicago/Turabian StyleIvanina Foureau, Anna V., David M. Foureau, Cody C. McHale, Fei Guo, Carol J. Farhangfar, and Kathryn F. Mileham. 2024. "Phosphodiesterase Inhibition to Sensitize Non-Small-Cell Lung Cancer to Pemetrexed: A Double-Edged Strategy" Cancers 16, no. 13: 2475. https://doi.org/10.3390/cancers16132475
APA StyleIvanina Foureau, A. V., Foureau, D. M., McHale, C. C., Guo, F., Farhangfar, C. J., & Mileham, K. F. (2024). Phosphodiesterase Inhibition to Sensitize Non-Small-Cell Lung Cancer to Pemetrexed: A Double-Edged Strategy. Cancers, 16(13), 2475. https://doi.org/10.3390/cancers16132475