
The Versatile Attributes of MGMT: Its Repair Mechanism, Crosstalk with Other DNA Repair Pathways, and Its Role in Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
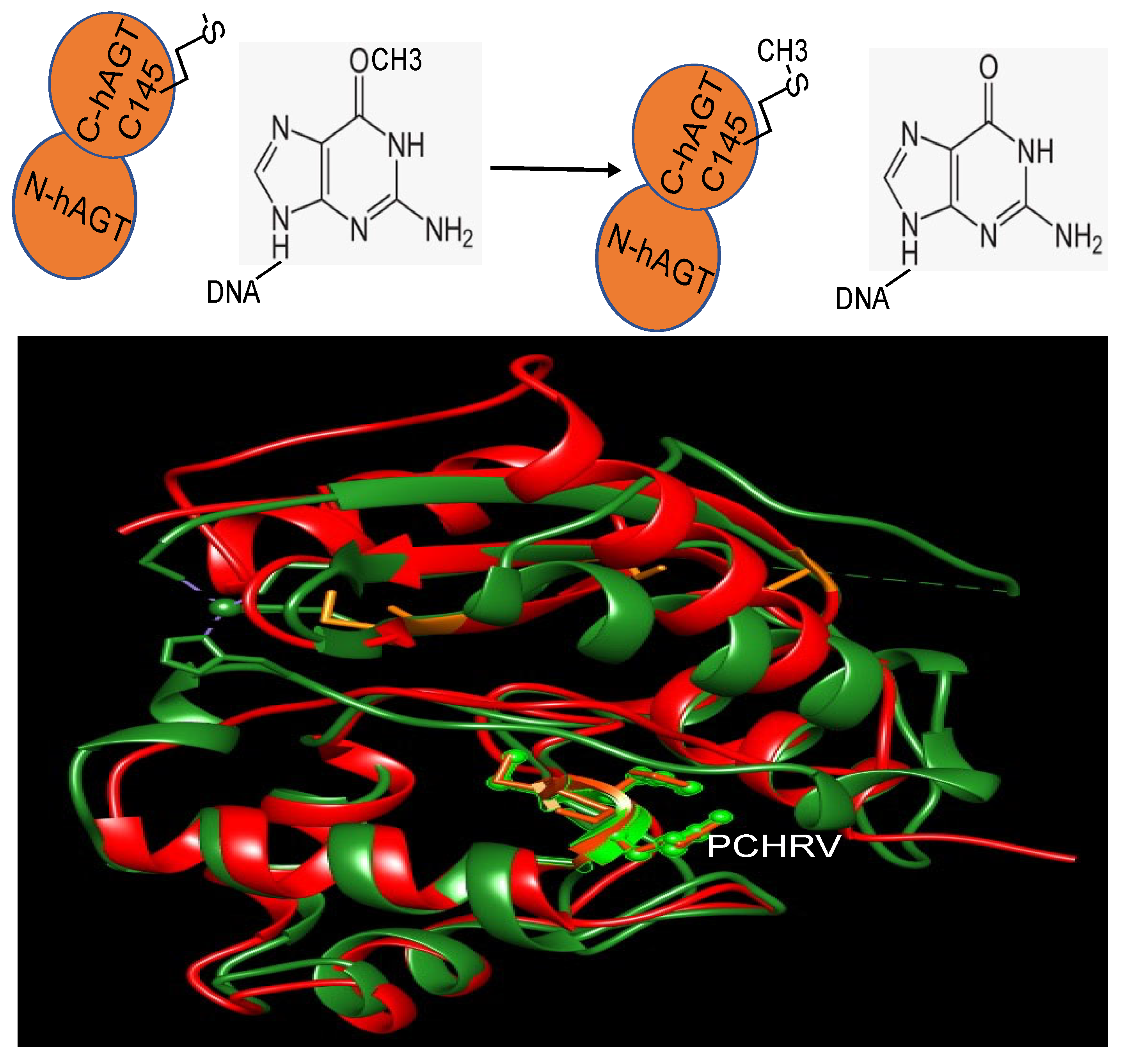
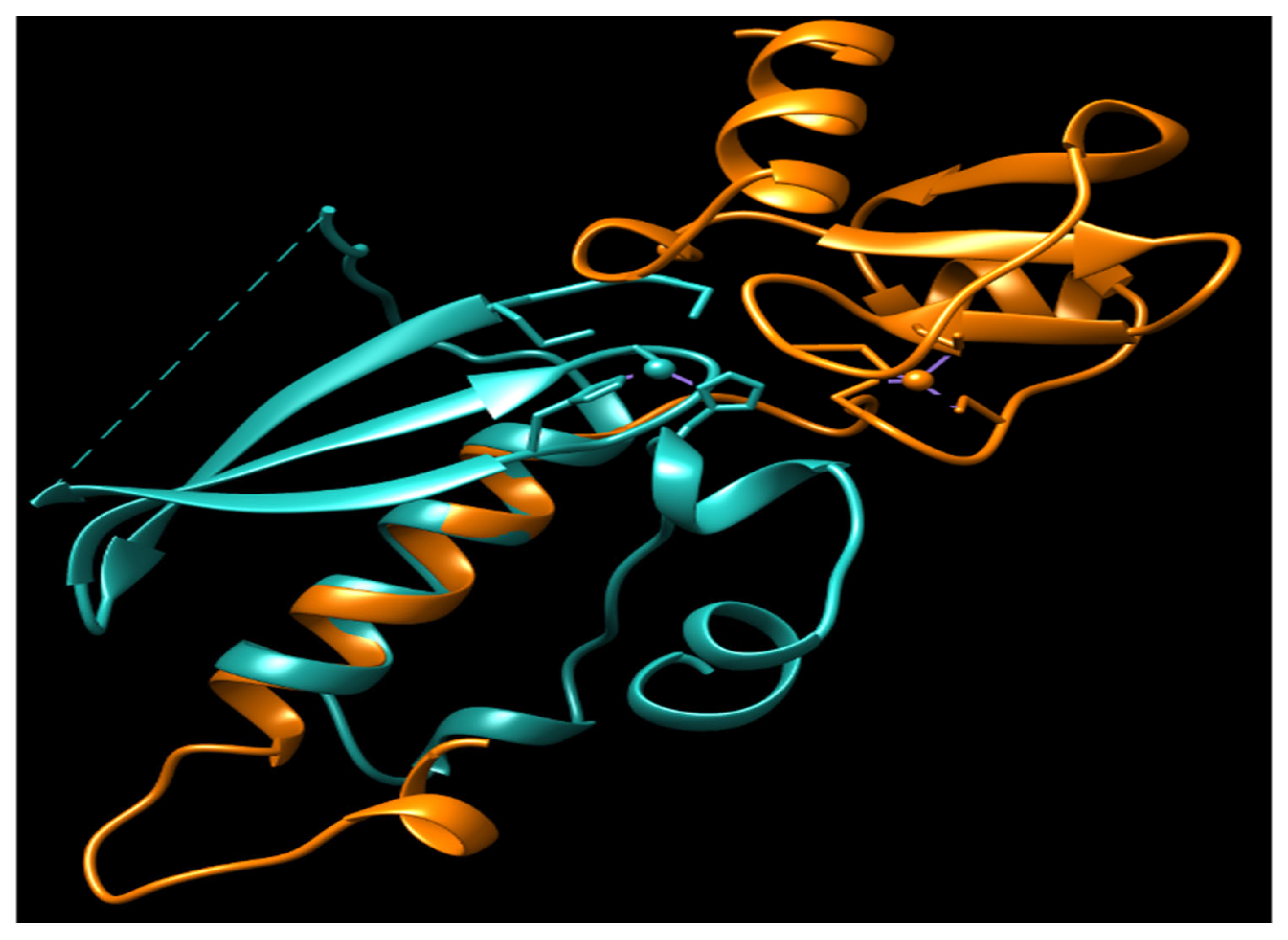
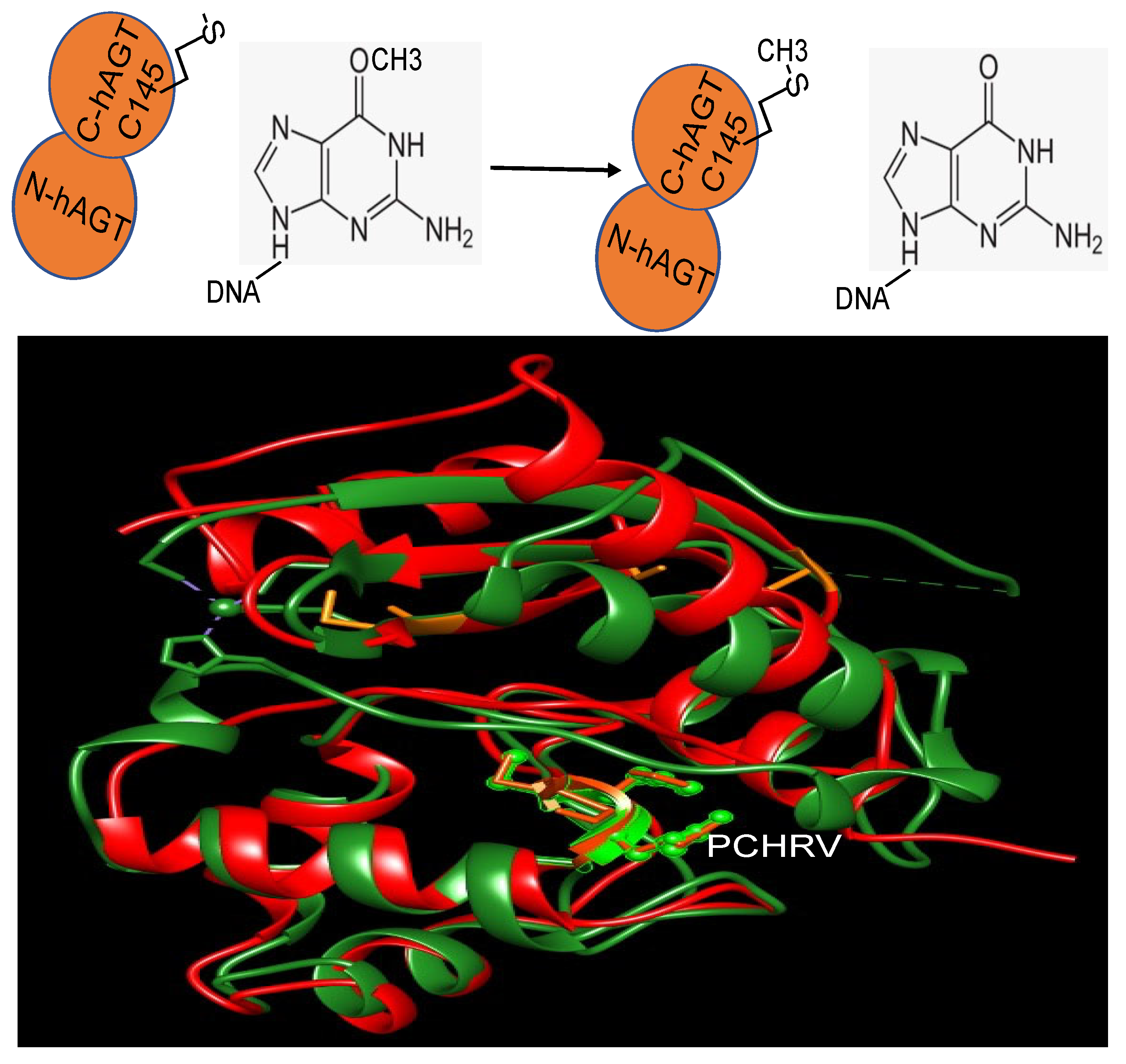
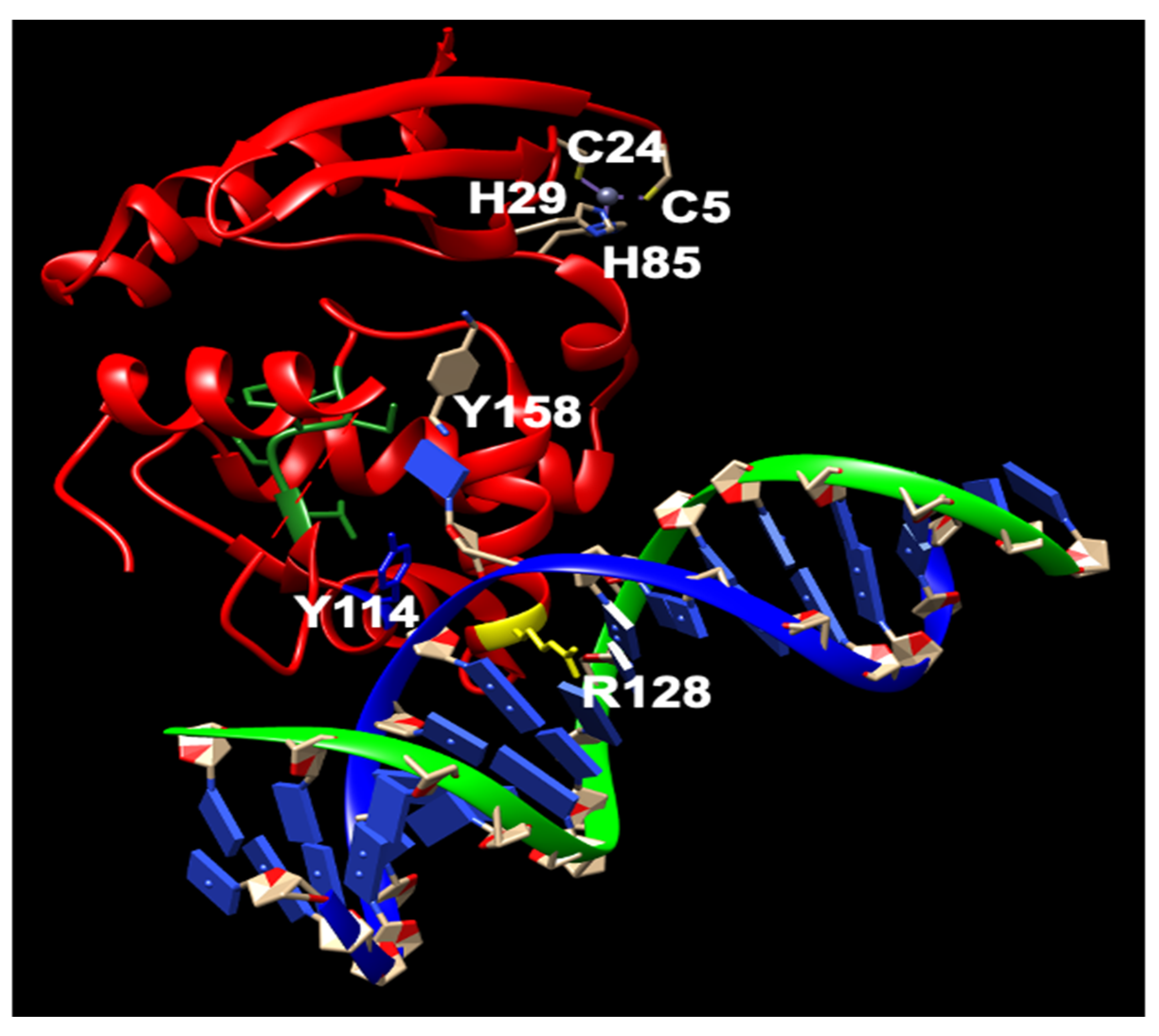
2. Repair Mechanism
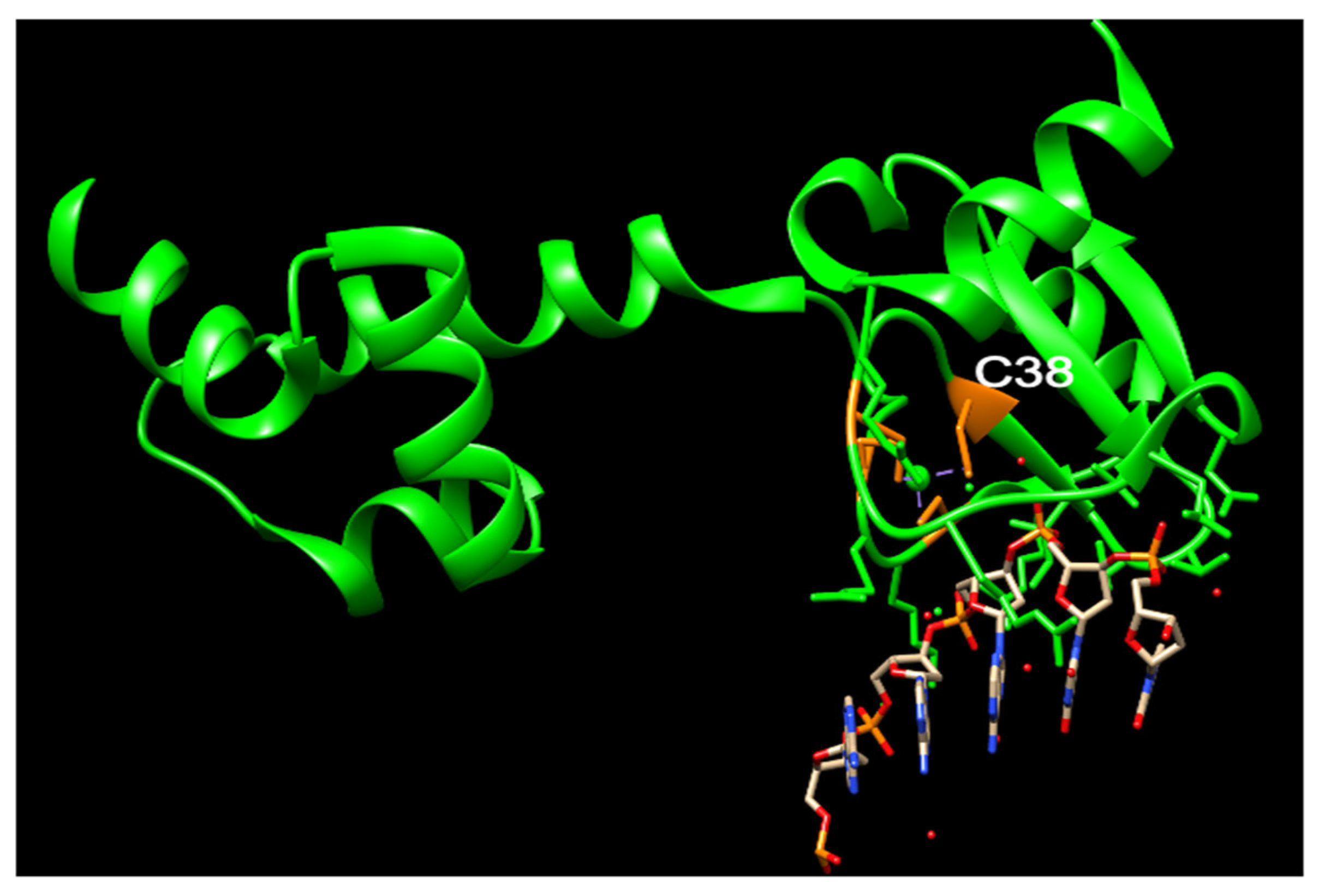
3. The Function of N-Ada and the Potential Role of N-hMGMT
4. The Fate of Alkylated MGMT
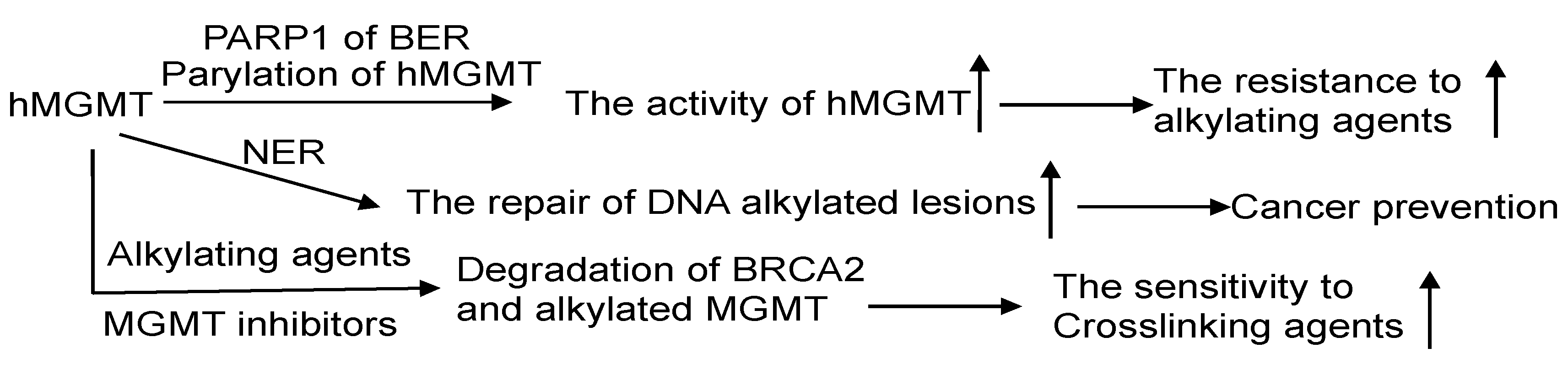
5. The Role of hMGMT in Cancer Prevention and Chemotherapy
6. The Development of Strategies That Target MGMT
6.1. Non Cancer-Selective MGMT Inhibitors
6.2. Cancer-Selective MGMT Inhibitors
6.3. Local Drug Delivery
6.4. Targeting the Expression of hMGMT
6.5. Others
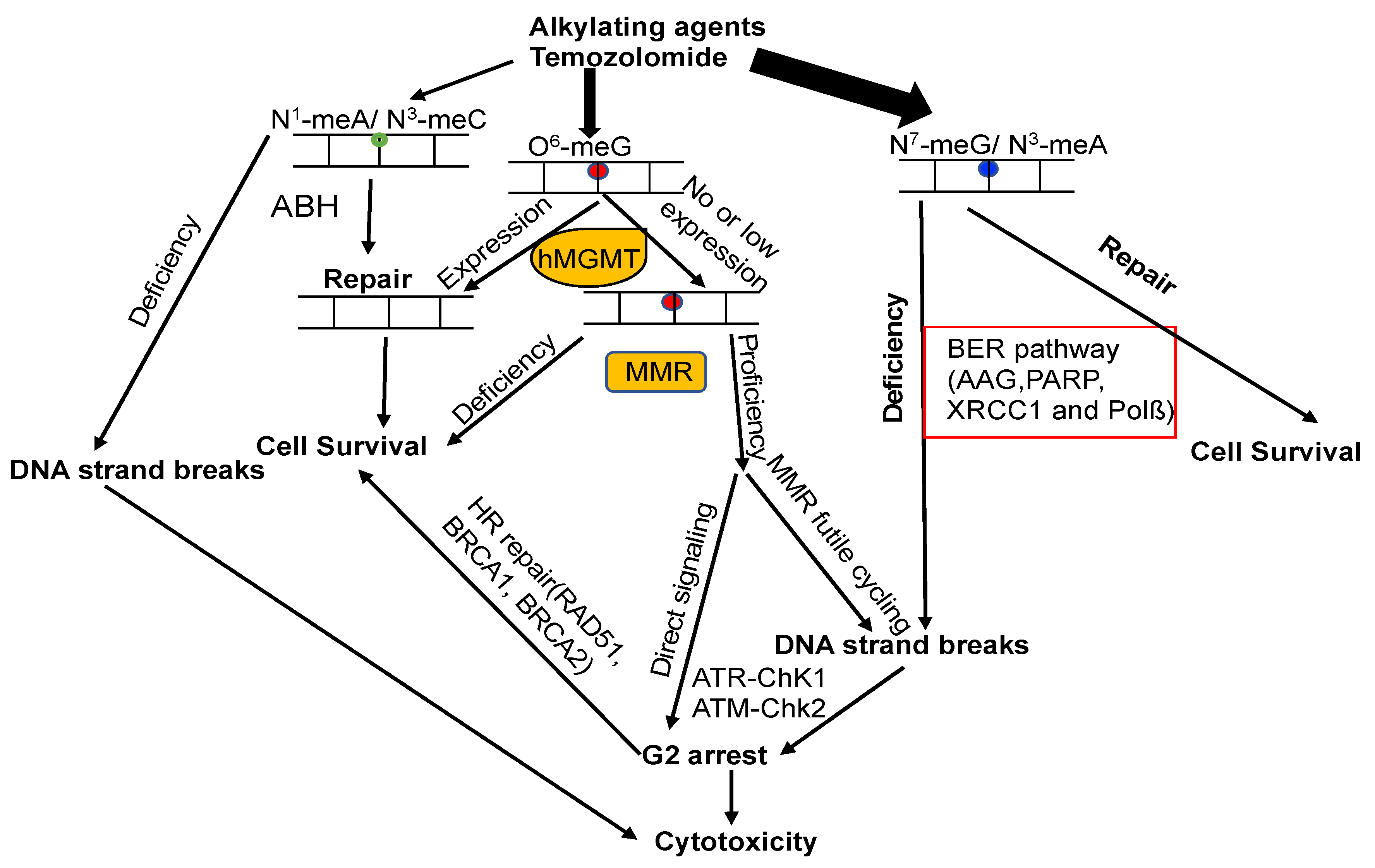
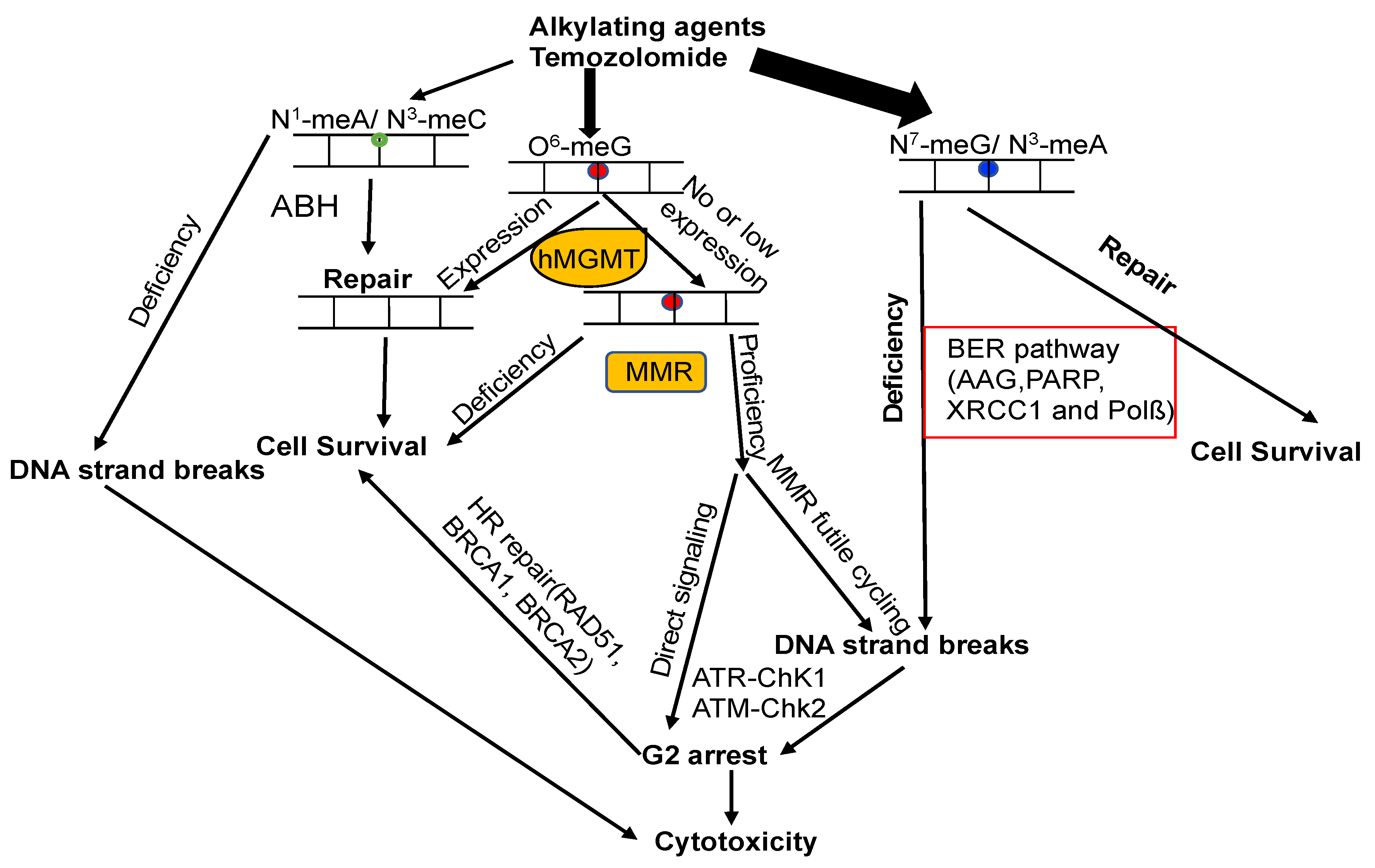
7. The Crosstalk of MGMT with Other DNA Repair Pathways
7.1. MGMT and BER
7.2. MGMT and Nucleotide Excision Repair (NER)
7.3. MGMT and MMR
7.4. MGMT and DSB Repair
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. Out of the shadows and into the light: The emergence of DNA repair. Trends Biochem. Sci. 1995, 20, 381. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Repair of O(6)-alkylguanine by alkyltransferases. Mutat. Res. 2000, 462, 83–100. [Google Scholar] [CrossRef]
- Pegg, A.E.; Dolan, M.E.; Moschel, R.C. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase. Prog. Nucleic Acid Res. Mol. Biol. 1995, 51, 167–223. [Google Scholar] [CrossRef]
- Mishina, Y.; Duguid, E.M.; He, C. Direct reversal of DNA alkylation damage. Chem. Rev. 2006, 106, 215–232. [Google Scholar] [CrossRef]
- Hecht, S.S. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat. Res. 1999, 424, 127–142. [Google Scholar] [CrossRef]
- Hurley, L.H. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 188–200. [Google Scholar] [CrossRef]
- Rajski, S.R.; Williams, R.M. DNA Cross-Linking Agents as Antitumor Drugs. Chem. Rev. 1998, 98, 2723–2796. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, B. Repairing DNA-methylation damage. Nat. Rev. Mol. Cell Biol. 2004, 5, 148–157. [Google Scholar] [CrossRef]
- Vaughan, P.; Lindahl, T.; Sedgwick, B. Induction of the adaptive response of Escherichia coli to alkylation damage by the environmental mutagen, methyl chloride. Mutat. Res. 1993, 293, 249–257. [Google Scholar] [CrossRef]
- Barrows, L.R.; Magee, P.N. Nonenzymatic methylation of DNA by S-adenosylmethionine in vitro. Carcinogenesis 1982, 3, 349–351. [Google Scholar] [CrossRef]
- Rydberg, B.; Lindahl, T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J. 1982, 1, 211–216. [Google Scholar] [CrossRef]
- Pegg, A.E. Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem. Res. Toxicol. 2011, 24, 618–639. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, M.D.; Pittman, D.L. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006, 19, 1580–1594. [Google Scholar] [CrossRef] [PubMed]
- Margison, G.P.; Povey, A.C.; Kaina, B.; Santibanez Koref, M.F. Variability and regulation of O6-alkylguanine-DNA alkyltransferase. Carcinogenesis 2003, 24, 625–635. [Google Scholar] [CrossRef]
- Beranek, D.T. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat. Res. 1990, 231, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Sedgwick, B.; Sekiguchi, M.; Nakabeppu, Y. Regulation and expression of the adaptive response to alkylating agents. Annu. Rev. Biochem. 1988, 57, 133–157. [Google Scholar] [CrossRef]
- Fornace, A.J., Jr.; Papathanasiou, M.A.; Hollander, M.C.; Yarosh, D.B. Expression of the O6-methylguanine-DNA methyltransferase gene MGMT in MER+ and MER- human tumor cells. Cancer Res. 1990, 50, 7908–7911. [Google Scholar] [PubMed]
- Bouras, E.; Karakioulaki, M.; Bougioukas, K.I.; Aivaliotis, M.; Tzimagiorgis, G.; Chourdakis, M. Gene promoter methylation and cancer: An umbrella review. Gene 2019, 710, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Kroes, R.A.; Erickson, L.C. The role of mRNA stability and transcription in O6-methylguanine DNA methyltransferase (MGMT) expression in Mer+ human tumor cells. Carcinogenesis 1995, 16, 2255–2257. [Google Scholar] [CrossRef]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Costello, J.F.; Futscher, B.W.; Tano, K.; Graunke, D.M.; Pieper, R.O. Graded methylation in the promoter and body of the O6-methylguanine DNA methyltransferase (MGMT) gene correlates with MGMT expression in human glioma cells. J. Biol. Chem. 1994, 269, 17228–17237. [Google Scholar] [CrossRef]
- Watts, G.S.; Pieper, R.O.; Costello, J.F.; Peng, Y.M.; Dalton, W.S.; Futscher, B.W. Methylation of discrete regions of the O6-methylguanine DNA methyltransferase (MGMT) CpG island is associated with heterochromatinization of the MGMT transcription start site and silencing of the gene. Mol. Cell. Biol. 1997, 17, 5612–5619. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zhang, L.; Wei, Q.; Shao, A. O(6)-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Front. Oncol. 2019, 9, 1547. [Google Scholar] [CrossRef]
- Juillerat, A.; Gronemeyer, T.; Keppler, A.; Gendreizig, S.; Pick, H.; Vogel, H.; Johnsson, K. Directed evolution of O6-alkylguanine-DNA alkyltransferase for efficient labeling of fusion proteins with small molecules in vivo. Chem. Biol. 2003, 10, 313–317. [Google Scholar] [CrossRef]
- Damoiseaux, R.; Keppler, A.; Johnsson, K. Synthesis and applications of chemical probes for human O6-alkylguanine-DNA alkyltransferase. Chembiochem 2001, 2, 285–287. [Google Scholar] [CrossRef]
- Keppler, A.; Gendreizig, S.; Gronemeyer, T.; Pick, H.; Vogel, H.; Johnsson, K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 2003, 21, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Keppler, A.; Kindermann, M.; Gendreizig, S.; Pick, H.; Vogel, H.; Johnsson, K. Labeling of fusion proteins of O6-alkylguanine-DNA alkyltransferase with small molecules in vivo and in vitro. Methods 2004, 32, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Keppler, A.; Pick, H.; Arrivoli, C.; Vogel, H.; Johnsson, K. Labeling of fusion proteins with synthetic fluorophores in live cells. Proc. Natl. Acad. Sci. USA 2004, 101, 9955–9959. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef]
- Gerson, S.L. MGMT: Its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer 2004, 4, 296–307. [Google Scholar] [CrossRef]
- Tubbs, J.L.; Pegg, A.E.; Tainer, J.A. DNA binding, nucleotide flipping, and the helix-turn-helix motif in base repair by O6-alkylguanine-DNA alkyltransferase and its implications for cancer chemotherapy. DNA Repair 2007, 6, 1100–1115. [Google Scholar] [CrossRef]
- Eker, A.P.; Quayle, C.; Chaves, I.; van der Horst, G.T. DNA repair in mammalian cells: Direct DNA damage reversal: Elegant solutions for nasty problems. Cell. Mol. Life Sci. 2009, 66, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Dalhus, B.; Laerdahl, J.K.; Backe, P.H.; Bjoras, M. DNA base repair--recognition and initiation of catalysis. FEMS Microbiol. Rev. 2009, 33, 1044–1078. [Google Scholar] [CrossRef]
- Sharma, S.; Salehi, F.; Scheithauer, B.W.; Rotondo, F.; Syro, L.V.; Kovacs, K. Role of MGMT in tumor development, progression, diagnosis, treatment and prognosis. Anticancer Res. 2009, 29, 3759–3768. [Google Scholar]
- Shrivastav, N.; Li, D.; Essigmann, J.M. Chemical biology of mutagenesis and DNA repair: Cellular responses to DNA alkylation. Carcinogenesis 2010, 31, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Fan, T.; Sun, G.; Wang, X.; Zhao, L.; Zhong, R. The dual role of DNA repair protein MGMT in cancer prevention and treatment. DNA Repair 2023, 123, 103449. [Google Scholar] [CrossRef]
- Kirstein, A.; Schmid, T.E.; Combs, S.E. The Role of miRNA for the Treatment of MGMT Unmethylated Glioblastoma Multiforme. Cancers 2020, 12, 1099. [Google Scholar] [CrossRef]
- Kaina, B.; Margison, G.P.; Christmann, M. Targeting O(6)-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell. Mol. Life Sci. 2010, 67, 3663–3681. [Google Scholar] [CrossRef]
- Lindahl, T.; Demple, B.; Robins, P. Suicide inactivation of the E. coli O6-methylguanine-DNA methyltransferase. EMBO J. 1982, 1, 1359–1363. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Lindahl, T. Repair of alkylated DNA in Escherichia coli. Methyl group transfer from O6-methylguanine to a protein cysteine residue. J. Biol. Chem. 1980, 255, 10569–10571. [Google Scholar] [CrossRef] [PubMed]
- Samson, L. The suicidal DNA repair methyltransferases of microbes. Mol. Microbiol. 1992, 6, 825–831. [Google Scholar] [CrossRef]
- Samson, L.; Cairns, J. A new pathway for DNA repair in Escherichia coli. Nature 1977, 267, 281–283. [Google Scholar] [CrossRef]
- Potter, P.M.; Wilkinson, M.C.; Fitton, J.; Carr, F.J.; Brennand, J.; Cooper, D.P.; Margison, G.P. Characterisation and nucleotide sequence of ogt, the O6-alkylguanine-DNA-alkyltransferase gene of E. coli. Nucleic Acids Res. 1987, 15, 9177–9193. [Google Scholar] [CrossRef]
- Rebeck, G.W.; Smith, C.M.; Goad, D.L.; Samson, L. Characterization of the major DNA repair methyltransferase activity in unadapted Escherichia coli and identification of a similar activity in Salmonella typhimurium. J. Bacteriol. 1989, 171, 4563–4568. [Google Scholar] [CrossRef] [PubMed]
- Koike, G.; Maki, H.; Takeya, H.; Hayakawa, H.; Sekiguchi, M. Purification, structure, and biochemical properties of human O6-methylguanine-DNA methyltransferase. J. Biol. Chem. 1990, 265, 14754–14762. [Google Scholar] [CrossRef]
- Tano, K.; Shiota, S.; Collier, J.; Foote, R.S.; Mitra, S. Isolation and structural characterization of a cDNA clone encoding the human DNA repair protein for O6-alkylguanine. Proc. Natl. Acad. Sci. USA 1990, 87, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Mijal, R.S.; Thomson, N.M.; Fleischer, N.L.; Pauly, G.T.; Moschel, R.C.; Kanugula, S.; Fang, Q.; Pegg, A.E.; Peterson, L.A. The repair of the tobacco specific nitrosamine derived adduct O6-[4-Oxo-4-(3-pyridyl)butyl]guanine by O6-alkylguanine-DNA alkyltransferase variants. Chem. Res. Toxicol. 2004, 17, 424–434. [Google Scholar] [CrossRef]
- Fang, Q.; Noronha, A.M.; Murphy, S.P.; Wilds, C.J.; Tubbs, J.L.; Tainer, J.A.; Chowdhury, G.; Guengerich, F.P.; Pegg, A.E. Repair of O6-G-alkyl-O6-G interstrand cross-links by human O6-alkylguanine-DNA alkyltransferase. Biochemistry 2008, 47, 10892–10903. [Google Scholar] [CrossRef]
- McManus, F.P.; Fang, Q.; Booth, J.D.; Noronha, A.M.; Pegg, A.E.; Wilds, C.J. Synthesis and characterization of an O(6)-2′-deoxyguanosine-alkyl-O(6)-2′-deoxyguanosine interstrand cross-link in a 5′-GNC motif and repair by human O(6)-alkylguanine-DNA alkyltransferase. Org. Biomol. Chem. 2010, 8, 4414–4426. [Google Scholar] [CrossRef]
- Samson, L.; Han, S.; Marquis, J.C.; Rasmussen, L.J. Mammalian DNA repair methyltransferases shield O4MeT from nucleotide excision repair. Carcinogenesis 1997, 18, 919–924. [Google Scholar] [CrossRef]
- Fang, Q.; Kanugula, S.; Tubbs, J.L.; Tainer, J.A.; Pegg, A.E. Repair of O4-alkylthymine by O6-alkylguanine-DNA alkyltransferases. J. Biol. Chem. 2010, 285, 8185–8195. [Google Scholar] [CrossRef] [PubMed]
- Wibley, J.E.; Pegg, A.E.; Moody, P.C. Crystal structure of the human O(6)-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 2000, 28, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.S.; Mol, C.D.; Arvai, A.S.; Kanugula, S.; Pegg, A.E.; Tainer, J.A. Active and alkylated human AGT structures: A novel zinc site, inhibitor and extrahelical base binding. EMBO J. 2000, 19, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.S.; Woo, T.T.; Luu, K.X.; Noll, D.M.; Clarke, N.D.; Pegg, A.E.; Tainer, J.A. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat. Struct. Mol. Biol. 2004, 11, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Duguid, E.M.; Rice, P.A.; He, C. The structure of the human AGT protein bound to DNA and its implications for damage detection. J. Mol. Biol. 2005, 350, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Kanugula, S.; Pegg, A.E. Function of domains of human O6-alkylguanine-DNA alkyltransferase. Biochemistry 2005, 44, 15396–15405. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.S.; Tainer, J.A. Conserved structural motifs governing the stoichiometric repair of alkylated DNA by O(6)-alkylguanine-DNA alkyltransferase. Mutat. Res. 2000, 460, 151–163. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Fang, Q.; Liu, L.; Hachey, D.L.; Pegg, A.E. O6-alkylguanine-DNA alkyltransferase: Low pKa and high reactivity of cysteine 145. Biochemistry 2003, 42, 10965–10970. [Google Scholar] [CrossRef]
- Hu, J.; Ma, A.; Dinner, A.R. A two-step nucleotide-flipping mechanism enables kinetic discrimination of DNA lesions by AGT. Proc. Natl. Acad. Sci. USA 2008, 105, 4615–4620. [Google Scholar] [CrossRef]
- Demple, B.; Sedgwick, B.; Robins, P.; Totty, N.; Waterfield, M.D.; Lindahl, T. Active site and complete sequence of the suicidal methyltransferase that counters alkylation mutagenesis. Proc. Natl. Acad. Sci. USA 1985, 82, 2688–2692. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, B.; Robins, P.; Totty, N.; Lindahl, T. Functional domains and methyl acceptor sites of the Escherichia coli ada protein. J. Biol. Chem. 1988, 263, 4430–4433. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Hus, J.C.; Sun, L.J.; Zhou, P.; Norman, D.P.; Dotsch, V.; Wei, H.; Gross, J.D.; Lane, W.S.; Wagner, G.; et al. A methylation-dependent electrostatic switch controls DNA repair and transcriptional activation by E. coli ada. Mol. Cell 2005, 20, 117–129. [Google Scholar] [CrossRef]
- Teo, I.; Sedgwick, B.; Kilpatrick, M.W.; McCarthy, T.V.; Lindahl, T. The intracellular signal for induction of resistance to alkylating agents in E. coli. Cell 1986, 45, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Akimaru, H.; Sakumi, K.; Yoshikai, T.; Anai, M.; Sekiguchi, M. Positive and negative regulation of transcription by a cleavage product of Ada protein. J. Mol. Biol. 1990, 216, 261–273. [Google Scholar] [CrossRef]
- Landini, P.; Busby, S.J. The Escherichia coli Ada protein can interact with two distinct determinants in the sigma70 subunit of RNA polymerase according to promoter architecture: Identification of the target of Ada activation at the alkA promoter. J. Bacteriol. 1999, 181, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Landini, P.; Volkert, M.R. Transcriptional activation of the Escherichia coli adaptive response gene aidB is mediated by binding of methylated Ada protein. Evidence for a new consensus sequence for Ada-binding sites. J. Biol. Chem. 1995, 270, 8285–8289. [Google Scholar] [CrossRef] [PubMed]
- Myers, L.C.; Terranova, M.P.; Ferentz, A.E.; Wagner, G.; Verdine, G.L. Repair of DNA methylphosphotriesters through a metalloactivated cysteine nucleophile. Science 1993, 261, 1164–1167. [Google Scholar] [CrossRef]
- Myers, L.C.; Jackow, F.; Verdine, G.L. Metal dependence of transcriptional switching in Escherichia coli Ada. J. Biol. Chem. 1995, 270, 6664–6670. [Google Scholar] [CrossRef]
- Myers, L.C.; Terranova, M.P.; Nash, H.M.; Markus, M.A.; Verdine, G.L. Zinc binding by the methylation signaling domain of the Escherichia coli Ada protein. Biochemistry 1992, 31, 4541–4547. [Google Scholar] [CrossRef]
- Moore, M.H.; Gulbis, J.M.; Dodson, E.J.; Demple, B.; Moody, P.C. Crystal structure of a suicidal DNA repair protein: The Ada O6-methylguanine-DNA methyltransferase from E. coli. EMBO J. 1994, 13, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Paalman, S.R.; Sung, C.; Clarke, N.D. Specificity of DNA repair methyltransferases determined by competitive inactivation with oligonucleotide substrates: Evidence that Escherichia coli Ada repairs O6-methylguanine and O4-methylthymine with similar efficiency. Biochemistry 1997, 36, 11118–11124. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E.; Boosalis, M.; Samson, L.; Moschel, R.C.; Byers, T.L.; Swenn, K.; Dolan, M.E. Mechanism of inactivation of human O6-alkylguanine-DNA alkyltransferase by O6-benzylguanine. Biochemistry 1993, 32, 11998–12006. [Google Scholar] [CrossRef] [PubMed]
- Pullman, A.; Pullman, B. Molecular electrostatic potential of the nucleic acids. Q. Rev. Biophys. 1981, 14, 289–380. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Laurent, B.; Hsu, C.H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Yang, C.G.; Yang, S.; Jian, X.; Yi, C.; Zhou, Z.; He, C. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett. 2008, 582, 3313–3319. [Google Scholar] [CrossRef] [PubMed]
- Aas, P.A.; Otterlei, M.; Falnes, P.O.; Vagbo, C.B.; Skorpen, F.; Akbari, M.; Sundheim, O.; Bjoras, M.; Slupphaug, G.; Seeberg, E.; et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 2003, 421, 859–863. [Google Scholar] [CrossRef]
- Ougland, R.; Zhang, C.M.; Liiv, A.; Johansen, R.F.; Seeberg, E.; Hou, Y.M.; Remme, J.; Falnes, P.O. AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol. Cell 2004, 16, 107–116. [Google Scholar] [CrossRef]
- Friedman, S.; Parsa, I. DNA adduct formation in rat, human and hamster pancreas treated with methylnitrosourea. Cancer Lett. 1985, 26, 269–276. [Google Scholar] [CrossRef]
- Falnes, P.O. RNA repair--the latest addition to the toolbox for macromolecular maintenance. RNA Biol. 2005, 2, 14–16. [Google Scholar] [CrossRef]
- Wang, T.; Pickard, A.J.; Gallo, J.M. Histone Methylation by Temozolomide; A Classic DNA Methylating Anticancer Drug. Anticancer Res. 2016, 36, 3289–3299. [Google Scholar] [PubMed]
- Yan, L.L.; Zaher, H.S. How do cells cope with RNA damage and its consequences? J. Biol. Chem. 2019, 294, 15158–15171. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E.; Wiest, L.; Mummert, C.; Stine, L.; Moschel, R.C.; Dolan, M.E. Use of antibodies to human O6-alkylguanine-DNA alkyltransferase to study the content of this protein in cells treated with O6-benzylguanine or N-methyl-N′-nitro-N-nitrosoguanidine. Carcinogenesis 1991, 12, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Srivenugopal, K.S.; Yuan, X.H.; Friedman, H.S.; Ali-Osman, F. Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemistry 1996, 35, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Xu-Welliver, M.; Pegg, A.E. Degradation of the alkylated form of the DNA repair protein, O(6)-alkylguanine-DNA alkyltransferase. Carcinogenesis 2002, 23, 823–830. [Google Scholar] [CrossRef]
- Edara, S.; Kanugula, S.; Pegg, A.E. Expression of the inactive C145A mutant human O6-alkylguanine-DNA alkyltransferase in E. coli increases cell killing and mutations by N-methyl-N′-nitro-N-nitrosoguanidine. Carcinogenesis 1999, 20, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Rasimas, J.J.; Dalessio, P.A.; Ropson, I.J.; Pegg, A.E.; Fried, M.G. Active-site alkylation destabilizes human O6-alkylguanine DNA alkyltransferase. Protein Sci. Publ. Protein Soc. 2004, 13, 301–305. [Google Scholar] [CrossRef]
- Rasimas, J.J.; Pegg, A.E.; Fried, M.G. DNA-binding mechanism of O6-alkylguanine-DNA alkyltransferase. Effects of protein and DNA alkylation on complex stability. J. Biol. Chem. 2003, 278, 7973–7980. [Google Scholar] [CrossRef]
- Ali, R.B.; Teo, A.K.; Oh, H.K.; Chuang, L.S.; Ayi, T.C.; Li, B.F. Implication of localization of human DNA repair enzyme O6-methylguanine-DNA methyltransferase at active transcription sites in transcription-repair coupling of the mutagenic O6-methylguanine lesion. Mol. Cell. Biol. 1998, 18, 1660–1669. [Google Scholar] [CrossRef]
- Teo, A.K.; Oh, H.K.; Ali, R.B.; Li, B.F. The modified human DNA repair enzyme O(6)-methylguanine-DNA methyltransferase is a negative regulator of estrogen receptor-mediated transcription upon alkylation DNA damage. Mol. Cell. Biol. 2001, 21, 7105–7114. [Google Scholar] [CrossRef]
- Wu, X.; Luo, Q.; Zhao, P.; Chang, W.; Wang, Y.; Shu, T.; Ding, F.; Li, B.; Liu, Z. MGMT-activated DUB3 stabilizes MCL1 and drives chemoresistance in ovarian cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 2961–2966. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, M.; Kim, J.; Matsunaga, N.; Kaji, H.; Egawa, T.; Makino, K.; Koyanagi, S.; Ohdo, S. Glucocorticoid-dependent expression of O(6)-methylguanine-DNA methyltransferase gene modulates dacarbazine-induced hepatotoxicity in mice. J. Pharmacol. Exp. Ther. 2010, 333, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E.; Perry, W.; Bennett, R.A. Effect of partial hepatectomy on removal of O6-methylguanine from alkylated DNA by rat liver extracts. Biochem. J. 1981, 197, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, A.; Bailey, N.I.; Konduri, S.; Bobustuc, G.C.; Ali-Osman, F.; Yusuf, M.A.; Punganuru, S.R.; Madala, H.R.; Basak, D.; Mostofa, A.; et al. New insights into estrogenic regulation of O(6)-methylguanine DNA-methyltransferase (MGMT) in human breast cancer cells: Co-degradation of ER-alpha and MGMT proteins by fulvestrant or O(6)-benzylguanine indicates fresh avenues for therapy. J. Biomed. Res. 2016, 30, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Franceschi, E.; Tosoni, A.; Bartolini, S.; Bacci, A.; Agati, R.; Ghimenton, C.; Turazzi, S.; Talacchi, A.; Skrap, M.; et al. O(6)-methylguanine DNA-methyltransferase methylation status can change between first surgery for newly diagnosed glioblastoma and second surgery for recurrence: Clinical implications. Neuro Oncol. 2010, 12, 283–288. [Google Scholar] [CrossRef]
- Wiewrodt, D.; Nagel, G.; Dreimuller, N.; Hundsberger, T.; Perneczky, A.; Kaina, B. MGMT in primary and recurrent human glioblastomas after radiation and chemotherapy and comparison with p53 status and clinical outcome. Int. J. Cancer 2008, 122, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Mullapudi, S.R.; Ali-Osman, F.; Shou, J.; Srivenugopal, K.S. DNA repair protein O6-alkylguanine-DNA alkyltransferase is phosphorylated by two distinct and novel protein kinases in human brain tumour cells. Biochem. J. 2000, 351 Pt 2, 393–402. [Google Scholar] [CrossRef]
- Cropper, J.D.; Alimbetov, D.S.; Brown, K.T.G.; Likhotvorik, R.I.; Robles, A.J.; Guerra, J.T.; He, B.; Chen, Y.; Kwon, Y.; Kurmasheva, R.T. PARP1-MGMT complex underpins pathway crosstalk in O(6)-methylguanine repair. J. Hematol. Oncol. 2022, 15, 146. [Google Scholar] [CrossRef]
- Wu, S.; Li, X.; Gao, F.; de Groot, J.F.; Koul, D.; Yung, W.K.A. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro Oncol. 2021, 23, 920–931. [Google Scholar] [CrossRef]
- Srivenugopal, K.S.; Mullapudi, S.R.; Shou, J.; Hazra, T.K.; Ali-Osman, F. Protein phosphorylation is a regulatory mechanism for O6-alkylguanine-DNA alkyltransferase in human brain tumor cells. Cancer Res. 2000, 60, 282–287. [Google Scholar]
- Choi, J.Y.; Chowdhury, G.; Zang, H.; Angel, K.C.; Vu, C.C.; Peterson, L.A.; Guengerich, F.P. Translesion synthesis across O6-alkylguanine DNA adducts by recombinant human DNA polymerases. J. Biol. Chem. 2006, 281, 38244–38256. [Google Scholar] [CrossRef]
- Loechler, E.L.; Green, C.L.; Essigmann, J.M. In vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc. Natl. Acad. Sci. USA 1984, 81, 6271–6275. [Google Scholar] [CrossRef]
- Pence, M.G.; Choi, J.Y.; Egli, M.; Guengerich, F.P. Structural basis for proficient incorporation of dTTP opposite O6-methylguanine by human DNA polymerase iota. J. Biol. Chem. 2010, 285, 40666–40672. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.J.; Forsberg, L.J.; Beese, L.S. The structural basis for the mutagenicity of O(6)-methyl-guanine lesions. Proc. Natl. Acad. Sci. USA 2006, 103, 19701–19706. [Google Scholar] [CrossRef]
- Woodside, A.M.; Guengerich, F.P. Effect of the O6 substituent on misincorporation kinetics catalyzed by DNA polymerases at O(6)-methylguanine and O(6)-benzylguanine. Biochemistry 2002, 41, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Pongor, L.; Su, Y.T.; Xi, L.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Kaina, B. O6-methylguanine-DNA methyltransferase in the defense against N-nitroso compounds and colorectal cancer. Carcinogenesis 2013, 34, 2435–2442. [Google Scholar] [CrossRef]
- Zaidi, N.H.; Pretlow, T.P.; O’Riordan, M.A.; Dumenco, L.L.; Allay, E.; Gerson, S.L. Transgenic expression of human MGMT protects against azoxymethane-induced aberrant crypt foci and G to A mutations in the K-ras oncogene of mouse colon. Carcinogenesis 1995, 16, 451–456. [Google Scholar] [CrossRef]
- Wali, R.K.; Skarosi, S.; Hart, J.; Zhang, Y.; Dolan, M.E.; Moschel, R.C.; Nguyen, L.; Mustafi, R.; Brasitus, T.A.; Bissonnette, M. Inhibition of O(6)-methylguanine-DNA methyltransferase increases azoxymethane-induced colonic tumors in rats. Carcinogenesis 1999, 20, 2355–2360. [Google Scholar] [CrossRef]
- Wirtz, S.; Nagel, G.; Eshkind, L.; Neurath, M.F.; Samson, L.D.; Kaina, B. Both base excision repair and O6-methylguanine-DNA methyltransferase protect against methylation-induced colon carcinogenesis. Carcinogenesis 2010, 31, 2111–2117. [Google Scholar] [CrossRef]
- Bugni, J.M.; Meira, L.B.; Samson, L.D. Alkylation-induced colon tumorigenesis in mice deficient in the Mgmt and Msh6 proteins. Oncogene 2009, 28, 734–741. [Google Scholar] [CrossRef]
- Hecht, S.S.; Hatsukami, D.K. Smokeless tobacco and cigarette smoking: Chemical mechanisms and cancer prevention. Nat. Rev. Cancer 2022, 22, 143–155. [Google Scholar] [CrossRef]
- Schrader, E.; Hirsch-Ernst, K.I.; Richter, E.; Foth, H. Metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in isolated rat lung and liver. Naunyn-Schmiedebergs Arch. Pharmacol. 1998, 357, 336–343. [Google Scholar] [CrossRef]
- Liu, L.; Qin, X.; Gerson, S.L. Reduced lung tumorigenesis in human methylguanine DNA--methyltransferase transgenic mice achieved by expression of transgene within the target cell. Carcinogenesis 1999, 20, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Staretz, M.E.; Foiles, P.G.; Miglietta, L.M.; Hecht, S.S. Evidence for an important role of DNA pyridyloxobutylation in rat lung carcinogenesis by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: Effects of dose and phenethyl isothiocyanate. Cancer Res. 1997, 57, 259–266. [Google Scholar] [PubMed]
- Dumenco, L.L.; Allay, E.; Norton, K.; Gerson, S.L. The prevention of thymic lymphomas in transgenic mice by human O6-alkylguanine-DNA alkyltransferase. Science 1993, 259, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Sakumi, K.; Shiraishi, A.; Shimizu, S.; Tsuzuki, T.; Ishikawa, T.; Sekiguchi, M. Methylnitrosourea-induced tumorigenesis in MGMT gene knockout mice. Cancer Res. 1997, 57, 2415–2418. [Google Scholar]
- Hanigan, M.H.; Kemp, C.J.; Ginsler, J.J.; Drinkwater, N.R. Rapid growth of preneoplastic lesions in hepatocarcinogen-sensitive C3H/HeJ male mice relative to C57BL/6J male mice. Carcinogenesis 1988, 9, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuru, Y.; Matsukuma, S.; Nemoto, N.; Sugano, H.; Sekiguchi, M.; Ishikawa, T. O6-methylguanine-DNA methyltransferase protects against nitrosamine-induced hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 1993, 90, 6468–6472. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Zhang, S.; Matsukuma, S.; Zarkovic, M.; Shimizu, S.; Ishikawa, T.; Nakatsuru, Y. Protection against malignant progression of spontaneously developing liver tumors in transgenic mice expressing O(6)-methylguanine-DNA methyltransferase. Jpn. J. Cancer Res. 2000, 91, 1085–1089. [Google Scholar] [CrossRef]
- Becker, K.; Gregel, C.M.; Kaina, B. The DNA repair protein O6-methylguanine-DNA methyltransferase protects against skin tumor formation induced by antineoplastic chloroethylnitrosourea. Cancer Res. 1997, 57, 3335–3338. [Google Scholar]
- Becker, K.; Dosch, J.; Gregel, C.M.; Martin, B.A.; Kaina, B. Targeted expression of human O(6)-methylguanine-DNA methyltransferase (MGMT) in transgenic mice protects against tumor initiation in two-stage skin carcinogenesis. Cancer Res. 1996, 56, 3244–3249. [Google Scholar] [PubMed]
- Becker, K.; Gregel, C.; Fricke, C.; Komitowski, D.; Dosch, J.; Kaina, B. DNA repair protein MGMT protects against N-methyl-N-nitrosourea-induced conversion of benign into malignant tumors. Carcinogenesis 2003, 24, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Bobola, M.S.; Blank, A.; Berger, M.S.; Silber, J.R. O6-methylguanine-DNA methyltransferase deficiency in developing brain: Implications for brain tumorigenesis. DNA Repair 2007, 6, 1127–1133. [Google Scholar] [CrossRef]
- Kisby, G.E.; Olivas, A.; Park, T.; Churchwell, M.; Doerge, D.; Samson, L.D.; Gerson, S.L.; Turker, M.S. DNA repair modulates the vulnerability of the developing brain to alkylating agents. DNA Repair 2009, 8, 400–412. [Google Scholar] [CrossRef]
- Silber, J.R.; Blank, A.; Bobola, M.S.; Mueller, B.A.; Kolstoe, D.D.; Ojemann, G.A.; Berger, M.S. Lack of the DNA repair protein O6-methylguanine-DNA methyltransferase in histologically normal brain adjacent to primary human brain tumors. Proc. Natl. Acad. Sci. USA 1996, 93, 6941–6946. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair 2019, 78, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Godard, S.; Dietrich, P.Y.; Regli, L.; Ostermann, S.; Otten, P.; Van Melle, G.; de Tribolet, N.; Stupp, R. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 1871–1874. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef]
- Aoki, K.; Natsume, A. Overview of DNA methylation in adult diffuse gliomas. Brain Tumor Pathol. 2019, 36, 84–91. [Google Scholar] [CrossRef]
- Reifenberger, G.; Hentschel, B.; Felsberg, J.; Schackert, G.; Simon, M.; Schnell, O.; Westphal, M.; Wick, W.; Pietsch, T.; Loeffler, M.; et al. Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. Int. J. Cancer 2012, 131, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Yin, Z.; Wang, G.; Zeng, S. Clinical and Prognostic Significance of O(6)-Methylguanine-DNA Methyltransferase Promoter Methylation in Patients with Melanoma: A Systematic Meta-Analysis. Ann. Dermatol. 2018, 30, 129–135. [Google Scholar] [CrossRef]
- Pandith, A.A.; Qasim, I.; Zahoor, W.; Shah, P.; Bhat, A.R.; Sanadhya, D.; Shah, Z.A.; Naikoo, N.A. Concordant association validates MGMT methylation and protein expression as favorable prognostic factors in glioma patients on alkylating chemotherapy (Temozolomide). Sci. Rep. 2018, 8, 6704. [Google Scholar] [CrossRef] [PubMed]
- Dahlrot, R.H.; Larsen, P.; Boldt, H.B.; Kreutzfeldt, M.S.; Hansen, S.; Hjelmborg, J.B.; Kristensen, B.W. Posttreatment Effect of MGMT Methylation Level on Glioblastoma Survival. J. Neuropathol. Exp. Neurol. 2019, 78, 633–640. [Google Scholar] [CrossRef]
- Malmstrom, A.; Gronberg, B.H.; Marosi, C.; Stupp, R.; Frappaz, D.; Schultz, H.; Abacioglu, U.; Tavelin, B.; Lhermitte, B.; Hegi, M.E.; et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: The Nordic randomised, phase 3 trial. Lancet Oncol. 2012, 13, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Platten, M.; Meisner, C.; Felsberg, J.; Tabatabai, G.; Simon, M.; Nikkhah, G.; Papsdorf, K.; Steinbach, J.P.; Sabel, M.; et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: The NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012, 13, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Jaeckle, K.A.; Eyre, H.J.; Townsend, J.J.; Schulman, S.; Knudson, H.M.; Belanich, M.; Yarosh, D.B.; Bearman, S.I.; Giroux, D.J.; Schold, S.C. Correlation of tumor O6 methylguanine-DNA methyltransferase levels with survival of malignant astrocytoma patients treated with bis-chloroethylnitrosourea: A Southwest Oncology Group study. J. Clin. Oncol. 1998, 16, 3310–3315. [Google Scholar] [CrossRef]
- Van den Bent, M.J.; Dubbink, H.J.; Sanson, M.; van der Lee-Haarloo, C.R.; Hegi, M.; Jeuken, J.W.; Ibdaih, A.; Brandes, A.A.; Taphoorn, M.J.; Frenay, M.; et al. MGMT promoter methylation is prognostic but not predictive for outcome to adjuvant PCV chemotherapy in anaplastic oligodendroglial tumors: A report from EORTC Brain Tumor Group Study 26951. J. Clin. Oncol. 2009, 27, 5881–5886. [Google Scholar] [CrossRef]
- Wick, W.; Hartmann, C.; Engel, C.; Stoffels, M.; Felsberg, J.; Stockhammer, F.; Sabel, M.C.; Koeppen, S.; Ketter, R.; Meyermann, R.; et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J. Clin. Oncol. 2009, 27, 5874–5880. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, L.; Hu, J.; Wang, S.; Liu, Y.; Yang, M.; Zhang, J.; Tang, B. Inhibition of MGMT-mediated autophagy suppression decreases cisplatin chemosensitivity in gastric cancer. Biomed. Pharmacother. 2020, 125, 109896. [Google Scholar] [CrossRef]
- Gefen, N.; Brkic, G.; Galron, D.; Priel, E.; Ozer, J.; Benharroch, D.; Gopas, J. Acquired resistance to 6-thioguanine in melanoma cells involves the repair enzyme O6-methylguanine-DNA methyltransferase (MGMT). Cancer Biol. Ther. 2010, 9, 49–55. [Google Scholar] [CrossRef]
- Spratt, T.E.; Campbell, C.R. Synthesis of oligodeoxynucleotides containing analogs of O6-methylguanine and reaction with O6-alkylguanine-DNA alkyltransferase. Biochemistry 1994, 33, 11364–11371. [Google Scholar] [CrossRef] [PubMed]
- Spratt, T.E.; de los Santos, H. Reaction of O6-alkylguanine-DNA alkyltransferase with O6-methylguanine analogues: Evidence that the oxygen of O6-methylguanine is protonated by the protein to effect methyl transfer. Biochemistry 1992, 31, 3688–3694. [Google Scholar] [CrossRef] [PubMed]
- Swann, P.F.; Waters, T.R.; Moulton, D.C.; Xu, Y.Z.; Zheng, Q.; Edwards, M.; Mace, R. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 1996, 273, 1109–1111. [Google Scholar] [CrossRef]
- Waters, T.R.; Swann, P.F. Cytotoxic mechanism of 6-thioguanine: HMutSalpha, the human mismatch binding heterodimer, binds to DNA containing S6-methylthioguanine. Biochemistry 1997, 36, 2501–2506. [Google Scholar] [CrossRef] [PubMed]
- Dolan, M.E.; Pegg, A.E. O6-benzylguanine and its role in chemotherapy. Clin. Cancer Res. 1997, 3, 837–847. [Google Scholar] [PubMed]
- Dolan, M.E.; Moschel, R.C.; Pegg, A.E. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc. Natl. Acad. Sci. USA 1990, 87, 5368–5372. [Google Scholar] [CrossRef]
- Shibata, T.; Glynn, N.; McMurry, T.B.; McElhinney, R.S.; Margison, G.P.; Williams, D.M. Novel synthesis of O6-alkylguanine containing oligodeoxyribonucleotides as substrates for the human DNA repair protein, O6-methylguanine DNA methyltransferase (MGMT). Nucleic Acids Res. 2006, 34, 1884–1891. [Google Scholar] [CrossRef]
- Kaina, B.; Muhlhausen, U.; Piee-Staffa, A.; Christmann, M.; Garcia Boy, R.; Rosch, F.; Schirrmacher, R. Inhibition of O6-methylguanine-DNA methyltransferase by glucose-conjugated inhibitors: Comparison with nonconjugated inhibitors and effect on fotemustine and temozolomide-induced cell death. J. Pharmacol. Exp. Ther. 2004, 311, 585–593. [Google Scholar] [CrossRef]
- Middleton, M.R.; Margison, G.P. Improvement of chemotherapy efficacy by inactivation of a DNA-repair pathway. Lancet Oncol. 2003, 4, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Bobola, M.S.; Silber, J.R.; Ellenbogen, R.G.; Geyer, J.R.; Blank, A.; Goff, R.D. O6-methylguanine-DNA methyltransferase, O6-benzylguanine, and resistance to clinical alkylators in pediatric primary brain tumor cell lines. Clin. Cancer Res. 2005, 11, 2747–2755. [Google Scholar] [CrossRef] [PubMed]
- Koch, D.; Hundsberger, T.; Boor, S.; Kaina, B. Local intracerebral administration of O(6)-benzylguanine combined with systemic chemotherapy with temozolomide of a patient suffering from a recurrent glioblastoma. J. Neurooncol. 2007, 82, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Broniscer, A.; Gururangan, S.; MacDonald, T.J.; Goldman, S.; Packer, R.J.; Stewart, C.F.; Wallace, D.; Danks, M.K.; Friedman, H.S.; Poussaint, T.Y.; et al. Phase I trial of single-dose temozolomide and continuous administration of O6-benzylguanine in children with brain tumors: A pediatric brain tumor consortium report. Clin. Cancer Res. 2007, 13, 6712–6718. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.A.; Desjardins, A.; Weingart, J.; Brem, H.; Dolan, M.E.; Delaney, S.M.; Vredenburgh, J.; Rich, J.; Friedman, A.H.; Reardon, D.A.; et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J. Clin. Oncol. 2005, 23, 7178–7187. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase I trial of temozolomide plus O6-benzylguanine 5-day regimen with recurrent malignant glioma. Neuro-Oncology 2009, 11, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase II trial of temozolomide plus O6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J. Clin. Oncol. 2009, 27, 1262–1267. [Google Scholar] [CrossRef]
- Verbeek, B.; Southgate, T.D.; Gilham, D.E.; Margison, G.P. O6-Methylguanine-DNA methyltransferase inactivation and chemotherapy. Br. Med. Bull. 2008, 85, 17–33. [Google Scholar] [CrossRef]
- Barvaux, V.A.; Lorigan, P.; Ranson, M.; Gillum, A.M.; McElhinney, R.S.; McMurry, T.B.; Margison, G.P. Sensitization of a human ovarian cancer cell line to temozolomide by simultaneous attenuation of the Bcl-2 antiapoptotic protein and DNA repair by O6-alkylguanine-DNA alkyltransferase. Mol. Cancer Ther. 2004, 3, 1215–1220. [Google Scholar] [CrossRef]
- Clemons, M.; Kelly, J.; Watson, A.J.; Howell, A.; McElhinney, R.S.; McMurry, T.B.; Margison, G.P. O6-(4-bromothenyl)guanine reverses temozolomide resistance in human breast tumour MCF-7 cells and xenografts. Br. J. Cancer 2005, 93, 1152–1156. [Google Scholar] [CrossRef]
- Middleton, M.R.; Kelly, J.; Thatcher, N.; Donnelly, D.J.; McElhinney, R.S.; McMurry, T.B.; McCormick, J.E.; Margison, G.P. O(6)-(4-bromothenyl)guanine improves the therapeutic index of temozolomide against A375M melanoma xenografts. Int. J. Cancer 2000, 85, 248–252. [Google Scholar] [CrossRef]
- Turriziani, M.; Caporaso, P.; Bonmassar, L.; Buccisano, F.; Amadori, S.; Venditti, A.; Cantonetti, M.; D’Atri, S.; Bonmassar, E. O6-(4-bromothenyl)guanine (PaTrin-2), a novel inhibitor of O6-alkylguanine DNA alkyl-transferase, increases the inhibitory activity of temozolomide against human acute leukaemia cells in vitro. Pharmacol. Res. 2006, 53, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.J.; Middleton, M.R.; McGown, G.; Thorncroft, M.; Ranson, M.; Hersey, P.; McArthur, G.; Davis, I.D.; Thomson, D.; Beith, J.; et al. O(6)-methylguanine-DNA methyltransferase depletion and DNA damage in patients with melanoma treated with temozolomide alone or with lomeguatrib. Br. J. Cancer 2009, 100, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.J.; Sabharwal, A.; Thorncroft, M.; McGown, G.; Kerr, R.; Bojanic, S.; Soonawalla, Z.; King, A.; Miller, A.; Waller, S.; et al. Tumor O(6)-methylguanine-DNA methyltransferase inactivation by oral lomeguatrib. Clin. Cancer Res. 2010, 16, 743–749. [Google Scholar] [CrossRef]
- Ranson, M.; Hersey, P.; Thompson, D.; Beith, J.; McArthur, G.A.; Haydon, A.; Davis, I.D.; Kefford, R.F.; Mortimer, P.; Harris, P.A.; et al. Randomized trial of the combination of lomeguatrib and temozolomide compared with temozolomide alone in chemotherapy naive patients with metastatic cutaneous melanoma. J. Clin. Oncol. 2007, 25, 2540–2545. [Google Scholar] [CrossRef] [PubMed]
- Khan, O.A.; Ranson, M.; Michael, M.; Olver, I.; Levitt, N.C.; Mortimer, P.; Watson, A.J.; Margison, G.P.; Midgley, R.; Middleton, M.R. A phase II trial of lomeguatrib and temozolomide in metastatic colorectal cancer. Br. J. Cancer 2008, 98, 1614–1618. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Villaruz, L.; Tarhini, A.; Moschos, S.; Sulecki, M.; Viverette, F.; Shipe-Spotloe, J.; Radkowski, R.; Kirkwood, J.M. Inhibition of DNA repair with MGMT pseudosubstrates: Phase I study of lomeguatrib in combination with dacarbazine in patients with advanced melanoma and other solid tumours. Br. J. Cancer 2011, 105, 773–777. [Google Scholar] [CrossRef]
- Pauly, G.T.; Loktionova, N.A.; Fang, Q.; Vankayala, S.L.; Guida, W.C.; Pegg, A.E. Substitution of aminomethyl at the meta-position enhances the inactivation of O6-alkylguanine-DNA alkyltransferase by O6-benzylguanine. J. Med. Chem. 2008, 51, 7144–7153. [Google Scholar] [CrossRef]
- Sun, G.; Zhao, L.; Zhong, R.; Peng, Y. The specific role of O(6)-methylguanine-DNA methyltransferase inhibitors in cancer chemotherapy. Future Med. Chem. 2018, 10, 1971–1996. [Google Scholar] [CrossRef]
- Wang, C.; Abegg, D.; Hoch, D.G.; Adibekian, A. Chemoproteomics-Enabled Discovery of a Potent and Selective Inhibitor of the DNA Repair Protein MGMT. Angew. Chem. Int. Ed. 2016, 55, 2911–2915. [Google Scholar] [CrossRef]
- Melikishvili, M.; Rodgers, D.W.; Fried, M.G. 6-Carboxyfluorescein and structurally similar molecules inhibit DNA binding and repair by O(6)-alkylguanine DNA alkyltransferase. DNA Repair 2011, 10, 1193–1202. [Google Scholar] [CrossRef]
- Goder, A.; Nagel, G.; Kraus, A.; Dorsam, B.; Seiwert, N.; Kaina, B.; Fahrer, J. Lipoic acid inhibits the DNA repair protein O 6-methylguanine-DNA methyltransferase (MGMT) and triggers its depletion in colorectal cancer cells with concomitant autophagy induction. Carcinogenesis 2015, 36, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xu-Welliver, M.; Kanugula, S.; Pegg, A.E. Inactivation and degradation of O(6)-alkylguanine-DNA alkyltransferase after reaction with nitric oxide. Cancer Res. 2002, 62, 3037–3043. [Google Scholar] [PubMed]
- Paranjpe, A.; Zhang, R.; Ali-Osman, F.; Bobustuc, G.C.; Srivenugopal, K.S. Disulfiram is a direct and potent inhibitor of human O6-methylguanine-DNA methyltransferase (MGMT) in brain tumor cells and mouse brain and markedly increases the alkylating DNA damage. Carcinogenesis 2014, 35, 692–702. [Google Scholar] [CrossRef]
- Geiger, H.; Schleimer, D.; Nattamai, K.J.; Dannenmann, S.R.; Davies, S.M.; Weiss, B.D. Mutagenic potential of temozolomide in bone marrow cells in vivo. Blood 2006, 107, 3010–3011. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, J.; Eichhorn, U.; Wiessler, M.; Kaina, B. Inactivation of O(6)-methylguanine-DNA methyltransferase by glucose-conjugated inhibitors. Int. J. Cancer 2001, 93, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Tomaszowski, K.H.; Schirrmacher, R.; Kaina, B. Multidrug Efflux Pumps Attenuate the Effect of MGMT Inhibitors. Mol. Pharm. 2015, 12, 3924–3934. [Google Scholar] [CrossRef]
- Ladino, C.A.; Chari, R.V.; Bourret, L.A.; Kedersha, N.L.; Goldmacher, V.S. Folate-maytansinoids: Target-selective drugs of low molecular weight. Int. J. Cancer 1997, 73, 859–864. [Google Scholar] [CrossRef]
- Javanmard, S.; Loktionova, N.A.; Fang, Q.; Pauly, G.T.; Pegg, A.E.; Moschel, R.C. Inactivation of O6-alkylguanine-DNA alkyltransferase by folate esters of O6-benzyl-2′-deoxyguanosine and of O6-[4-(hydroxymethyl)benzyl]guanine. J. Med. Chem. 2007, 50, 5193–5201. [Google Scholar] [CrossRef]
- Nelson, M.E.; Loktionova, N.A.; Pegg, A.E.; Moschel, R.C. 2-amino-O4-benzylpteridine derivatives: Potent inactivators of O6-alkylguanine-DNA alkyltransferase. J. Med. Chem. 2004, 47, 3887–3891. [Google Scholar] [CrossRef]
- Zhu, R.; Baumann, R.P.; Penketh, P.G.; Shyam, K.; Sartorelli, A.C. Hypoxia-selective O6-alkylguanine-DNA alkyltransferase inhibitors: Design, synthesis, and evaluation of 6-(benzyloxy)-2-(aryldiazenyl)-9H-purines as prodrugs of O6-benzylguanine. J. Med. Chem. 2013, 56, 1355–1359. [Google Scholar] [CrossRef]
- Zhu, R.; Liu, M.C.; Luo, M.Z.; Penketh, P.G.; Baumann, R.P.; Shyam, K.; Sartorelli, A.C. 4-nitrobenzyloxycarbonyl derivatives of O(6)-benzylguanine as hypoxia-activated prodrug inhibitors of O(6)-alkylguanine-DNA alkyltransferase (AGT), which produces resistance to agents targeting the O-6 position of DNA guanine. J. Med. Chem. 2011, 54, 7720–7728. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Loktionova, N.A.; Pegg, A.E.; Moschel, R.C. Beta-glucuronidase-cleavable prodrugs of O6-benzylguanine and O6-benzyl-2′-deoxyguanosine. J. Med. Chem. 2005, 48, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.Y.; Kau, Y.C.; Liu, S.J. Advanced interstitial chemotherapy for treating malignant glioma. Expert Opin. Drug Deliv. 2016, 13, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Guerin, C.; Olivi, A.; Weingart, J.D.; Lawson, H.C.; Brem, H. Recent advances in brain tumor therapy: Local intracerebral drug delivery by polymers. Investig. New Drugs 2004, 22, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, B.D.; Blanco, E.; Gao, J. Polymer implants for intratumoral drug delivery and cancer therapy. J. Pharm. Sci. 2008, 97, 1681–1702. [Google Scholar] [CrossRef] [PubMed]
- Weingart, J.; Grossman, S.A.; Carson, K.A.; Fisher, J.D.; Delaney, S.M.; Rosenblum, M.L.; Olivi, A.; Judy, K.; Tatter, S.B.; Dolan, M.E. Phase I trial of polifeprosan 20 with carmustine implant plus continuous infusion of intravenous O6-benzylguanine in adults with recurrent malignant glioma: New approaches to brain tumor therapy CNS consortium trial. J. Clin. Oncol. 2007, 25, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, I.; Veevnik, D.; Fedulov, A.; Yurkshtovich, N.; Yurkshtovich, T.; Pejler, G.; Lokot, I. Local delivery of temozolomide via a biologically inert carrier (Temodex) prolongs survival in glioma patients, irrespectively of the methylation status of MGMT. Neoplasma 2019, 66, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shen, J.; Zhang, L.; Wang, L.; Xu, H.; Han, Y.; Jia, J.; Lu, Y.; Yu, R.; Liu, H. Injectable postoperative enzyme-responsive hydrogels for reversing temozolomide resistance and reducing local recurrence after glioma operation. Biomater. Sci. 2020, 8, 5306–5316. [Google Scholar] [CrossRef]
- Kievit, F.M.; Wang, F.Y.; Fang, C.; Mok, H.; Wang, K.; Silber, J.R.; Ellenbogen, R.G.; Zhang, M. Doxorubicin loaded iron oxide nanoparticles overcome multidrug resistance in cancer in vitro. J. Control. Release 2011, 152, 76–83. [Google Scholar] [CrossRef]
- Stephen, Z.R.; Kievit, F.M.; Veiseh, O.; Chiarelli, P.A.; Fang, C.; Wang, K.; Hatzinger, S.J.; Ellenbogen, R.G.; Silber, J.R.; Zhang, M. Redox-responsive magnetic nanoparticle for targeted convection-enhanced delivery of O6-benzylguanine to brain tumors. ACS Nano 2014, 8, 10383–10395. [Google Scholar] [CrossRef] [PubMed]
- Stephen, Z.R.; Gebhart, R.N.; Jeon, M.; Blair, A.A.; Ellenbogen, R.G.; Silber, J.R.; Zhang, M. pH-Sensitive O6-Benzylguanosine Polymer Modified Magnetic Nanoparticles for Treatment of Glioblastomas. Bioconjug. Chem. 2017, 28, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Rait, A.; Kim, E.; Pirollo, K.F.; Nishida, M.; Farkas, N.; Dagata, J.A.; Chang, E.H. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano 2014, 8, 5494–5514. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhou, Y.; Chen, J.; Huang, N.; Wang, Z.; Cheng, Y. Gene Therapy for Drug-Resistant Glioblastoma via Lipid-Polymer Hybrid Nanoparticles Combined with Focused Ultrasound. Int. J. Nanomed. 2021, 16, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Allay, J.A.; Dumenco, L.L.; Koc, O.N.; Liu, L.; Gerson, S.L. Retroviral transduction and expression of the human alkyltransferase cDNA provides nitrosourea resistance to hematopoietic cells. Blood 1995, 85, 3342–3351. [Google Scholar] [CrossRef] [PubMed]
- Maze, R.; Carney, J.P.; Kelley, M.R.; Glassner, B.J.; Williams, D.A.; Samson, L. Increasing DNA repair methyltransferase levels via bone marrow stem cell transduction rescues mice from the toxic effects of 1,3-bis(2-chloroethyl)-1-nitrosourea, a chemotherapeutic alkylating agent. Proc. Natl. Acad. Sci. USA 1996, 93, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Moritz, T.; Mackay, W.; Glassner, B.J.; Williams, D.A.; Samson, L. Retrovirus-mediated expression of a DNA repair protein in bone marrow protects hematopoietic cells from nitrosourea-induced toxicity in vitro and in vivo. Cancer Res. 1995, 55, 2608–2614. [Google Scholar]
- Davis, B.M.; Roth, J.C.; Liu, L.; Xu-Welliver, M.; Pegg, A.E.; Gerson, S.L. Characterization of the P140K, PVP(138-140)MLK, and G156A O6-methylguanine-DNA methyltransferase mutants: Implications for drug resistance gene therapy. Hum. Gene Ther. 1999, 10, 2769–2778. [Google Scholar] [CrossRef]
- Davis, B.M.; Reese, J.S.; Koc, O.N.; Lee, K.; Schupp, J.E.; Gerson, S.L. Selection for G156A O6-methylguanine DNA methyltransferase gene-transduced hematopoietic progenitors and protection from lethality in mice treated with O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 1997, 57, 5093–5099. [Google Scholar]
- Koc, O.N.; Reese, J.S.; Davis, B.M.; Liu, L.; Majczenko, K.J.; Gerson, S.L. DeltaMGMT-transduced bone marrow infusion increases tolerance to O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea and allows intensive therapy of 1,3-bis(2-chloroethyl)-1-nitrosourea-resistant human colon cancer xenografts. Hum. Gene Ther. 1999, 10, 1021–1030. [Google Scholar] [CrossRef]
- Cabrini, G.; Fabbri, E.; Lo Nigro, C.; Dechecchi, M.C.; Gambari, R. Regulation of expression of O6-methylguanine-DNA methyltransferase and the treatment of glioblastoma (Review). Int. J. Oncol. 2015, 47, 417–428. [Google Scholar] [CrossRef]
- Chiou, G.Y.; Chien, C.S.; Wang, M.L.; Chen, M.T.; Yang, Y.P.; Yu, Y.L.; Chien, Y.; Chang, Y.C.; Shen, C.C.; Chio, C.C.; et al. Epigenetic regulation of the miR142-3p/interleukin-6 circuit in glioblastoma. Mol. Cell 2013, 52, 693–706. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Yarmishyn, A.A.; Wang, M.L.; Chen, H.Y.; Chiou, S.H.; Yang, Y.P.; Lin, C.F.; Huang, P.I.; Chen, Y.W.; Ma, H.I.; et al. MicroRNA-142-3p is involved in regulation of MGMT expression in glioblastoma cells. Cancer Manag. Res. 2018, 10, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.T.; Chen, X.B.; Liu, H.L. Up-regulation of miR-370-3p restores glioblastoma multiforme sensitivity to temozolomide by influencing MGMT expression. Sci. Rep. 2016, 6, 32972. [Google Scholar] [CrossRef] [PubMed]
- Nadaradjane, A.; Briand, J.; Bougras-Cartron, G.; Disdero, V.; Vallette, F.M.; Frenel, J.S.; Cartron, P.F. miR-370-3p Is a Therapeutic Tool in Anti-glioblastoma Therapy but Is Not an Intratumoral or Cell-free Circulating Biomarker. Mol. Ther. Nucleic Acids 2018, 13, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, C.; Mangani, D.; Roscigno, G.; Romano, G.; Diaz-Lagares, A.; Iaboni, M.; Donnarumma, E.; Fiore, D.; De Marinis, P.; Soini, Y.; et al. MiR-221/222 target the DNA methyltransferase MGMT in glioma cells. PLoS ONE 2013, 8, e74466. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, J.; Hoadley, K.; Kushwaha, D.; Ramakrishnan, V.; Li, S.; Kang, C.; You, Y.; Jiang, C.; Song, S.W.; et al. miR-181d: A predictive glioblastoma biomarker that downregulates MGMT expression. Neuro-Oncology 2012, 14, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Natsume, A.; Toda, H.; Iwamizu, H.; Sugita, T.; Hachisu, R.; Watanabe, R.; Yuki, K.; Motomura, K.; Bankiewicz, K.; et al. Efficient delivery of liposome-mediated MGMT-siRNA reinforces the cytotoxity of temozolomide in GBM-initiating cells. Gene Ther. 2010, 17, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, S.; Cui, X.; Wang, Q.; Yang, E.; Tong, F.; Hong, B.; Xiao, M.; Xin, L.; Xu, C.; et al. A novel compound EPIC-0412 reverses temozolomide resistance via inhibiting DNA repair/MGMT in glioblastoma. Neuro-Oncology 2023, 25, 857–870. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Mitra, S. Regulation of the human O(6)-methylguanine-DNA methyltransferase gene by transcriptional coactivators cAMP response element-binding protein-binding protein and p300. J. Biol. Chem. 2000, 275, 34197–34204. [Google Scholar] [CrossRef]
- Alonso, M.M.; Gomez-Manzano, C.; Bekele, B.N.; Yung, W.K.; Fueyo, J. Adenovirus-based strategies overcome temozolomide resistance by silencing the O6-methylguanine-DNA methyltransferase promoter. Cancer Res. 2007, 67, 11499–11504. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Deng, Z.; Wang, H.; Li, X.; Sun, T.; Tao, Z.; Yao, L.; Jin, Y.; Wang, X.; Yang, L.; et al. MGMT autoantibodies as a potential prediction of recurrence and treatment response biomarker for glioma patients. Cancer Med. 2019, 8, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, K.; Wang, H.; Chen, G.; Liu, Y.; Li, W.; Zhou, Y. Experimental study of selective MGMT peptides mimicking TMZ drug resistance in glioma. Biochem. Biophys. Rep. 2022, 32, 101386. [Google Scholar] [CrossRef]
- Lin, K.; Gueble, S.E.; Sundaram, R.K.; Huseman, E.D.; Bindra, R.S.; Herzon, S.B. Mechanism-based design of agents that selectively target drug-resistant glioma. Science 2022, 377, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Heras, G.; Castro-Robles, B.; Romero-Sanchez, C.M.; Carrion, B.; Barbella-Aponte, R.; Sandoval, H.; Segura, T. Involvement of N-methylpurine DNA glycosylase in resistance to temozolomide in patient-derived glioma cells. Sci. Rep. 2020, 10, 22185. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Bronstein, S.M.; Skopek, T.R.; Swenberg, J.A. Efficient repair of O6-ethylguanine, but not O4-ethylthymine or O2-ethylthymine, is dependent upon O6-alkylguanine-DNA alkyltransferase and nucleotide excision repair activities in human cells. Cancer Res. 1992, 52, 2008–2011. [Google Scholar]
- Aloisi, C.M.N.; Escher, N.A.; Kim, H.S.; Geisen, S.M.; Fontana, G.A.; Yeo, J.E.; Scharer, O.D.; Sturla, S.J. A combination of direct reversion and nucleotide excision repair counters the mutagenic effects of DNA carboxymethylation. DNA Repair 2022, 110, 103262. [Google Scholar] [CrossRef]
- Tubbs, J.L.; Latypov, V.; Kanugula, S.; Butt, A.; Melikishvili, M.; Kraehenbuehl, R.; Fleck, O.; Marriott, A.; Watson, A.J.; Verbeek, B.; et al. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature 2009, 459, 808–813. [Google Scholar] [CrossRef]
- Pearson, S.J.; Ferguson, J.; Santibanez-Koref, M.; Margison, G.P. Inhibition of O6-methylguanine-DNA methyltransferase by an alkyltransferase-like protein from Escherichia coli. Nucleic Acids Res. 2005, 33, 3837–3844. [Google Scholar] [CrossRef]
- Gupta, D.; Heinen, C.D. The mismatch repair-dependent DNA damage response: Mechanisms and implications. DNA Repair 2019, 78, 60–69. [Google Scholar] [CrossRef]
- Fink, D.; Aebi, S.; Howell, S.B. The role of DNA mismatch repair in drug resistance. Clin. Cancer Res. 1998, 4, 1–6. [Google Scholar] [PubMed]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [PubMed]
- Duckett, D.R.; Drummond, J.T.; Murchie, A.I.; Reardon, J.T.; Sancar, A.; Lilley, D.M.; Modrich, P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc. Natl. Acad. Sci. USA 1996, 93, 6443–6447. [Google Scholar] [CrossRef] [PubMed]
- Mojas, N.; Lopes, M.; Jiricny, J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 2007, 21, 3342–3355. [Google Scholar] [CrossRef]
- Kaina, B.; Ziouta, A.; Ochs, K.; Coquerelle, T. Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex-, Mex+ and methylation-tolerant mismatch repair compromised cells: Facts and models. Mutat. Res. 1997, 381, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Karran, P.; Marinus, M.G. Mismatch correction at O6-methylguanine residues in E. coli DNA. Nature 1982, 296, 868–869. [Google Scholar] [CrossRef]
- Yoshioka, K.; Yoshioka, Y.; Hsieh, P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol. Cell 2006, 22, 501–510. [Google Scholar] [CrossRef]
- Lin, D.P.; Wang, Y.; Scherer, S.J.; Clark, A.B.; Yang, K.; Avdievich, E.; Jin, B.; Werling, U.; Parris, T.; Kurihara, N.; et al. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004, 64, 517–522. [Google Scholar] [CrossRef]
- Yang, G.; Scherer, S.J.; Shell, S.S.; Yang, K.; Kim, M.; Lipkin, M.; Kucherlapati, R.; Kolodner, R.D.; Edelmann, W. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell 2004, 6, 139–150. [Google Scholar] [CrossRef]
- Gupta, D.; Lin, B.; Cowan, A.; Heinen, C.D. ATR-Chk1 activation mitigates replication stress caused by mismatch repair-dependent processing of DNA damage. Proc. Natl. Acad. Sci. USA 2018, 115, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Gupta, D.; Heinen, C.D. Human pluripotent stem cells have a novel mismatch repair-dependent damage response. J. Biol. Chem. 2014, 289, 24314–24324. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, R.P.; Isogawa, A.; Paulo, J.A.; Onizuka, K.; Takahashi, T.; Amunugama, R.; Duxin, J.P.; Fujii, S. Crosstalk between repair pathways elicits double-strand breaks in alkylated DNA and implications for the action of temozolomide. Elife 2021, 10, e69544. [Google Scholar] [CrossRef]
- Fujii, S.; Sobol, R.W.; Fuchs, R.P. Double-strand breaks: When DNA repair events accidentally meet. DNA Repair 2022, 112, 103303. [Google Scholar] [CrossRef] [PubMed]
- Debiak, M.; Nikolova, T.; Kaina, B. Loss of ATM sensitizes against O6-methylguanine triggered apoptosis, SCEs and chromosomal aberrations. DNA Repair 2004, 3, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Philip, S.; Swaminathan, S.; Kuznetsov, S.G.; Kanugula, S.; Biswas, K.; Chang, S.; Loktionova, N.A.; Haines, D.C.; Kaldis, P.; Pegg, A.E.; et al. Degradation of BRCA2 in alkyltransferase-mediated DNA repair and its clinical implications. Cancer Res. 2008, 68, 9973–9981. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MGMT | ED50 (µM) |
|---|---|
| N-hMGMT | 0.15 ± 0.01 [57] |
| N-hMGMT/C-hMGMT | 0.20 ± 0.02 [57] |
| hMGMT | 0.24 ± 0.02 [57] |
| Ogt | 600 [73] |
| Ada | >1 mM (no repair) [73] |
| YMGMT * | >1 mM (no repair) [73] |
| # | Template | 3D Model | Confidence | Template Information |
|---|---|---|---|---|
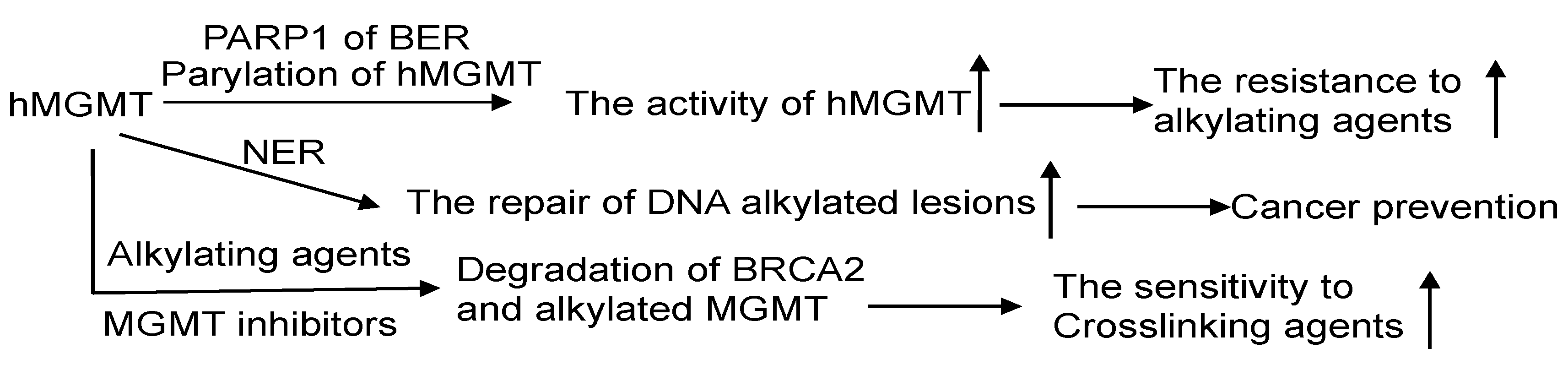
| 1 | d1qnta2 |  | 100 | Fold: Ribonuclease H-like motif Family: Methylated DNA-protein cysteine methyltransferase domain |
| 2 | C1t39A |  | 98.7 | PDB header: transferase/DNA PDB title: human O6-alkylguanine-DNA alkyltransferase covalently crosslinked to DNA |
| 3 | C4bhcA |  | 97.7 | PDB header: transferase PDB title: crystal structure of the m.tuberculosis O6-methylguanine DNA methyltransferase r371 variant |
| The Category of Strategies | Compounds or Molecules | Effects and Advantage |
|---|---|---|
| Non caner-selective inhibitors | O6-BG and O6-4-BTG | Efficiently inhibit the activity of MGMT in mammalian cells, animal models and patients [147,148,149,150]. |
| Meta-substitution of O6-BG | Improve the water solubility and more potent inhibitors of MGMT in mammalian cells [168]. | |
| 6-(benzyloxy)pyrimidine derivatives | Inhibit the activity of MGMT [13,169]. | |
| Acrolein and chloromethyltriazoles | React with cysteine residues to inhibit the activity of MGMT in cells efficiently [170]. | |
| 6-carboxyflfluorescein | Inhibit the activity of MGMT with a non-covalent manner [171]. | |
| Lipoic acid | A natural compound that efficiently inhibits the activity of MGMT and enhances the cytotoxic effects of TMZ in cancer cells that are resistant to TMZ [172]. | |
| Nitric oxide and disulfiram | Inactivate MGMT by interacting with Cys145 [173,174]. | |
| Cancer-selective inhibitors | O6-BG-Glu and O6-BTG-Glu | Conjugate a glucose group with an MGMT inhibitor. They inhibit the activity of MGMT in various cancer cell lines [150,176,177]. |
| O4-benzylfolate | Conjugate a folate group to an MGMT inhibitor. It is a more potent inhibitor than O6-BG and enhances BCNU-induced cell death dependent on the expression of the α-folate receptor [179,180]. | |
| O6-benzyl-2′-deoxyguanosine | Increase MGMT inhibitory activity in tumor cells and improve the water solubility [179]. | |
| Glucuronic acid linked prodrugs of O6-BG and O6-benzyl-2′-deoxyguanosine | These prodrugs are stable and less active. O6-BG and O6-benzyl-2′-deoxyguanosine can be released after the removal of beta-glucuronidase [181,182]. They may be useful in cancer cells that liberate beta-glucuronidase. | |
| Local drug delivery | Gliadel (BCNU wafers) | The first clinical application of polymer drug delivery to treat brain tumors [185,186]. |
| Encapsulated TMZ with inert matrix | Demonstrated superior efficacy compared to standard therapy alone, resulting in a remarkable increase in overall survival for GBM patients [188]. | |
| Injectable hydrogel capable of delivering TMZ and O6-BG | Demonstrated effectiveness in reducing the recurrence of TMZ-resistant glioma after surgery and enhancing the inhibitory efficiency against tumors [189]. | |
| Nanoparticle-based delivery of TMZ and O6-BG | These delivery systems successfully deplete MGMT and significantly enhance the therapeutic effect of TMZ in cancer cells and animal models [191,192,193,194]. | |
| Targeting the expression of MGMT | miRNAs including miR-142-3p, miR-181d, miR-370-3p | Downregulate the expression of MGMT and enhance the sensitivity to TMZ in GBM cell lines [38,201,202,203,204,205,206,207]. |
| EPIC-0412 | Enhances the chemotherapeutic effect of TMZ by epigenetically silencing the expression of MGMT [209]. | |
| Oncolytic viruses | Downregulate the expression of MGMT [210,211]. | |
| Others | Autoantibodies | May overcome the resistance to TMZ in glioma cells [213]. |
| New agent-KL-50 | Induce cell killing selectively in MGMT-silenced tumors, independent of MMR [214]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, Q. The Versatile Attributes of MGMT: Its Repair Mechanism, Crosstalk with Other DNA Repair Pathways, and Its Role in Cancer. Cancers 2024, 16, 331. https://doi.org/10.3390/cancers16020331
Fang Q. The Versatile Attributes of MGMT: Its Repair Mechanism, Crosstalk with Other DNA Repair Pathways, and Its Role in Cancer. Cancers. 2024; 16(2):331. https://doi.org/10.3390/cancers16020331
Chicago/Turabian StyleFang, Qingming. 2024. "The Versatile Attributes of MGMT: Its Repair Mechanism, Crosstalk with Other DNA Repair Pathways, and Its Role in Cancer" Cancers 16, no. 2: 331. https://doi.org/10.3390/cancers16020331
APA StyleFang, Q. (2024). The Versatile Attributes of MGMT: Its Repair Mechanism, Crosstalk with Other DNA Repair Pathways, and Its Role in Cancer. Cancers, 16(2), 331. https://doi.org/10.3390/cancers16020331