
Molecular Basis of XRN2-Deficient Cancer Cell Sensitivity to Poly(ADP-ribose) Polymerase Inhibition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Antibodies
2.3. Tissue Culture
2.4. RNAi and Transfection
2.5. DNA Assay
2.6. Colony-Forming Assay
2.7. Western Blot
2.8. Immunofluorescence
2.9. Dot Blot Assay
2.10. Comet Assay
2.11. Statistical Analyses
3. Results
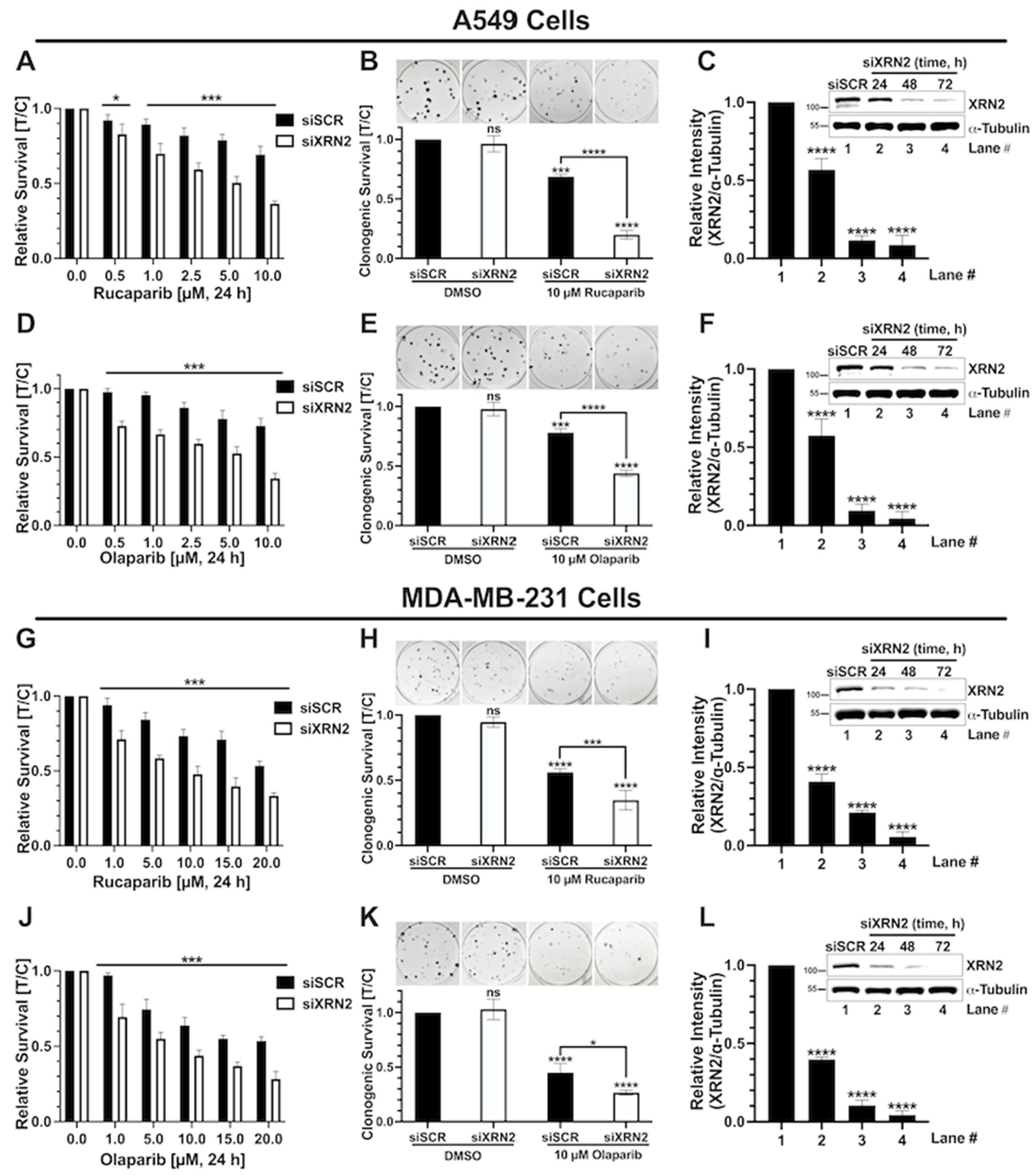
3.1. XRN2 Deficiency Sensitizes Cancer Cells to PARP Inhibition
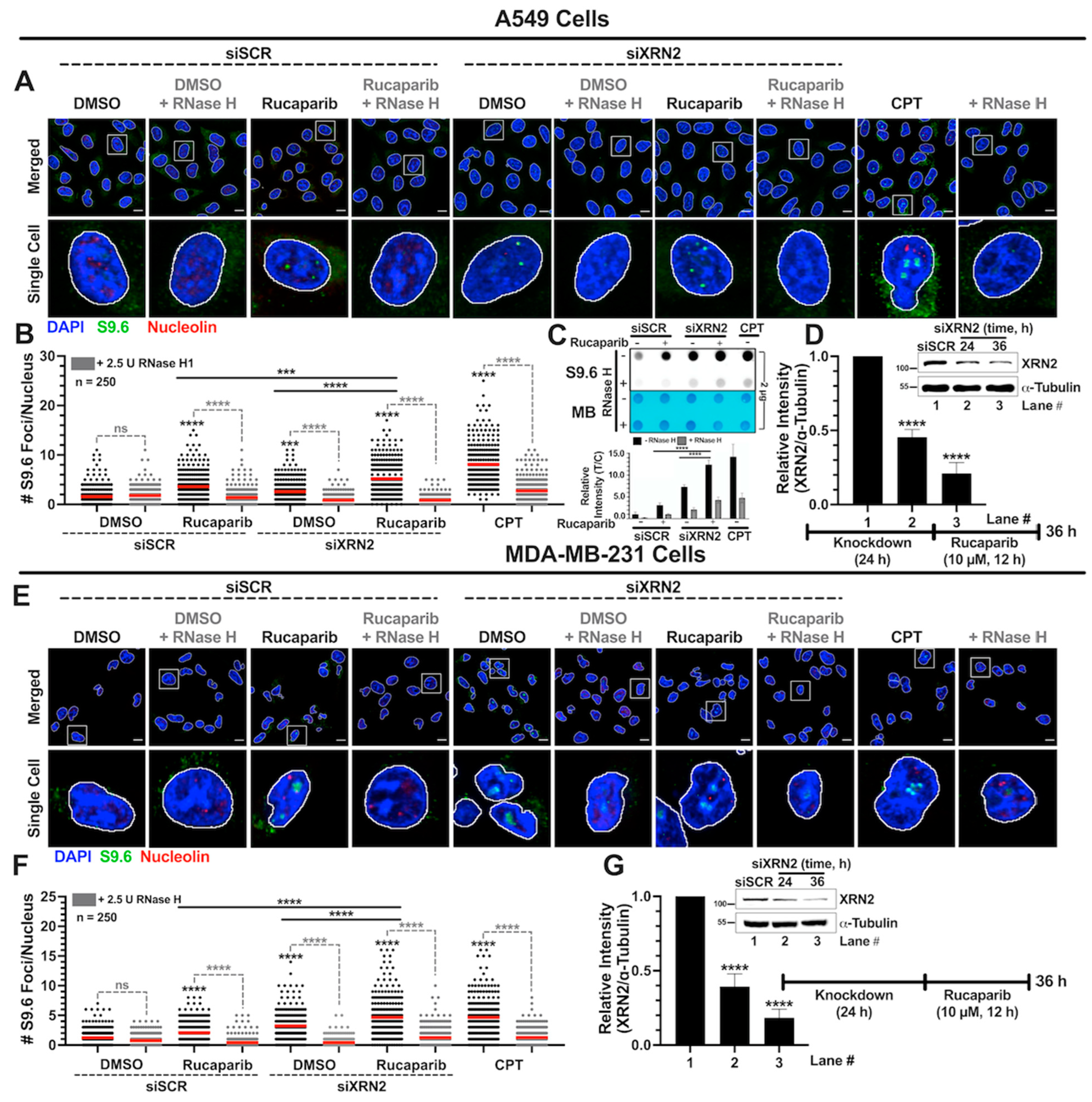
3.2. Simultaneous XRN2 Depletion and PARP Inhibition Enhance R-loop Formation
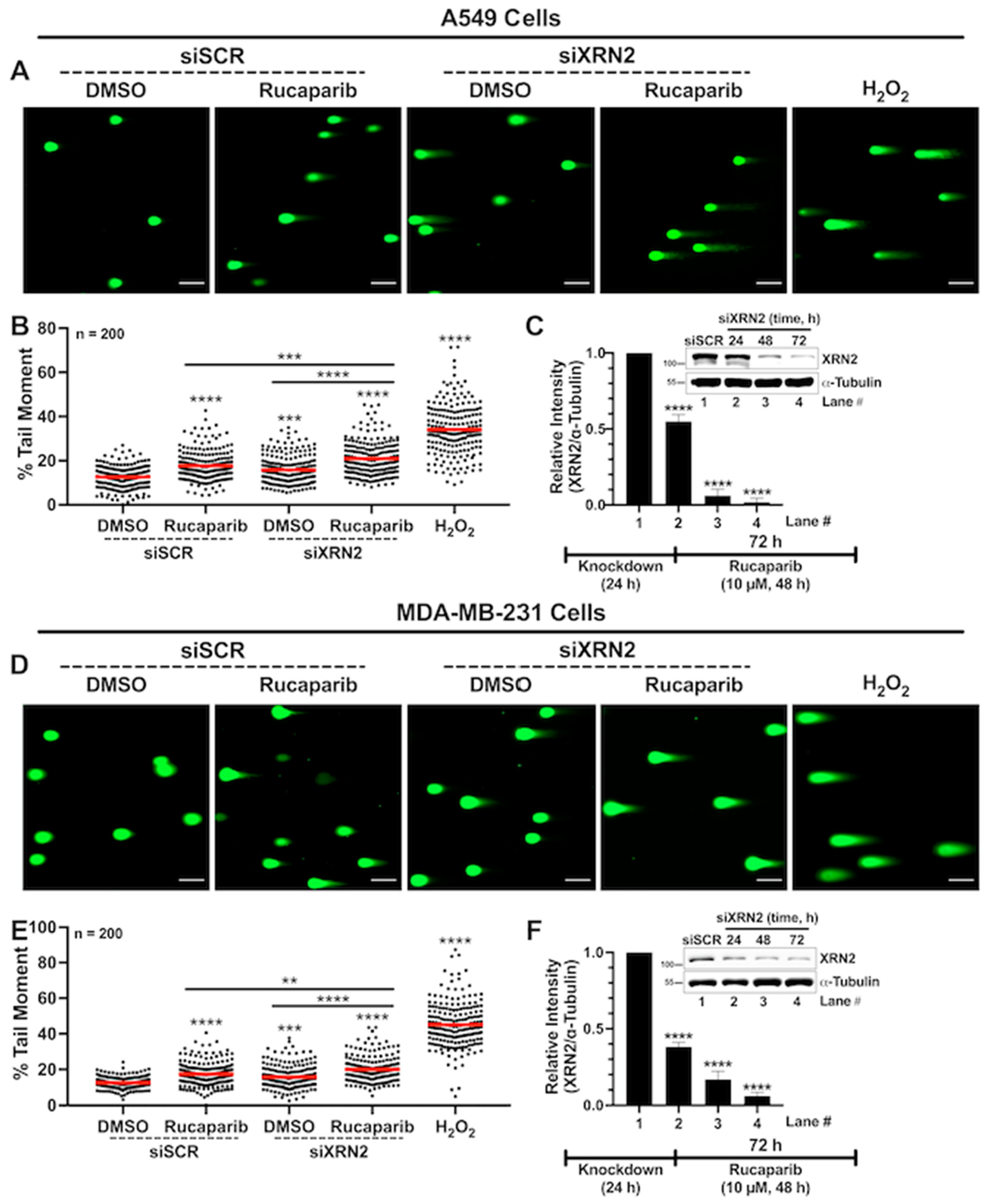
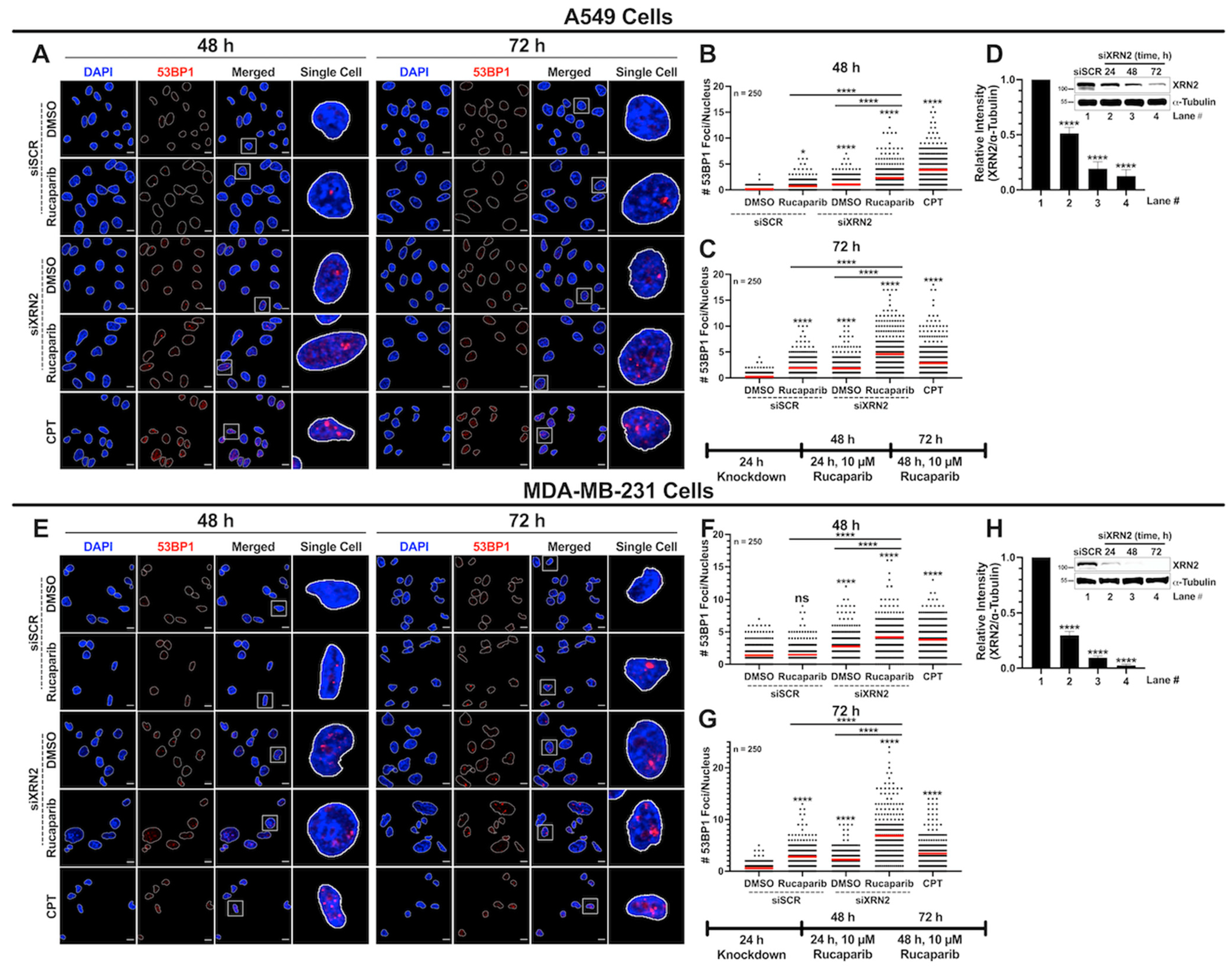
3.3. Concurrent XRN2 Depletion and PARP Inhibition Exacerbate DSB Formation and Downstream Signaling
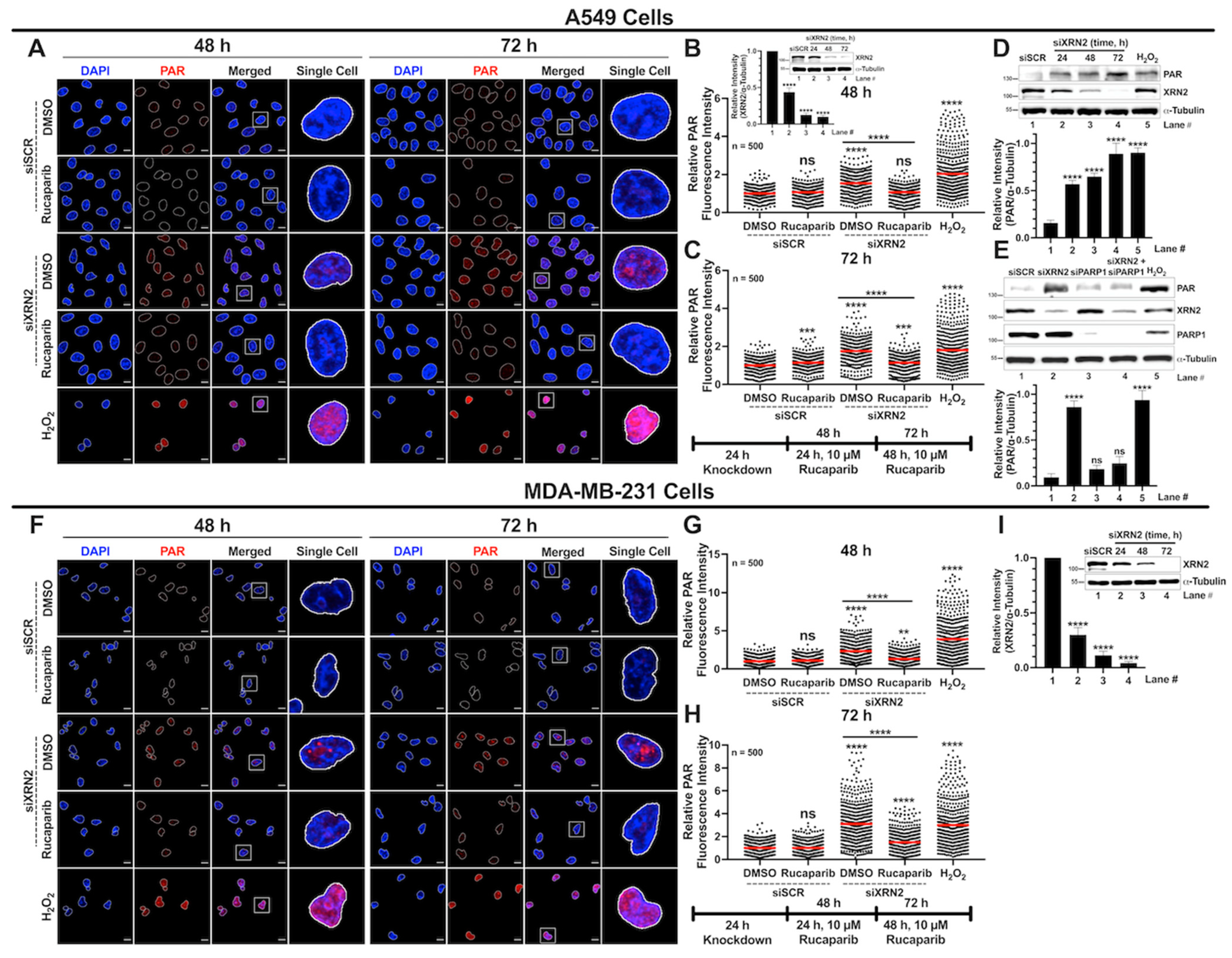
3.4. XRN2 Deficiency Enhances PARP1 Activity in Cancer Cells
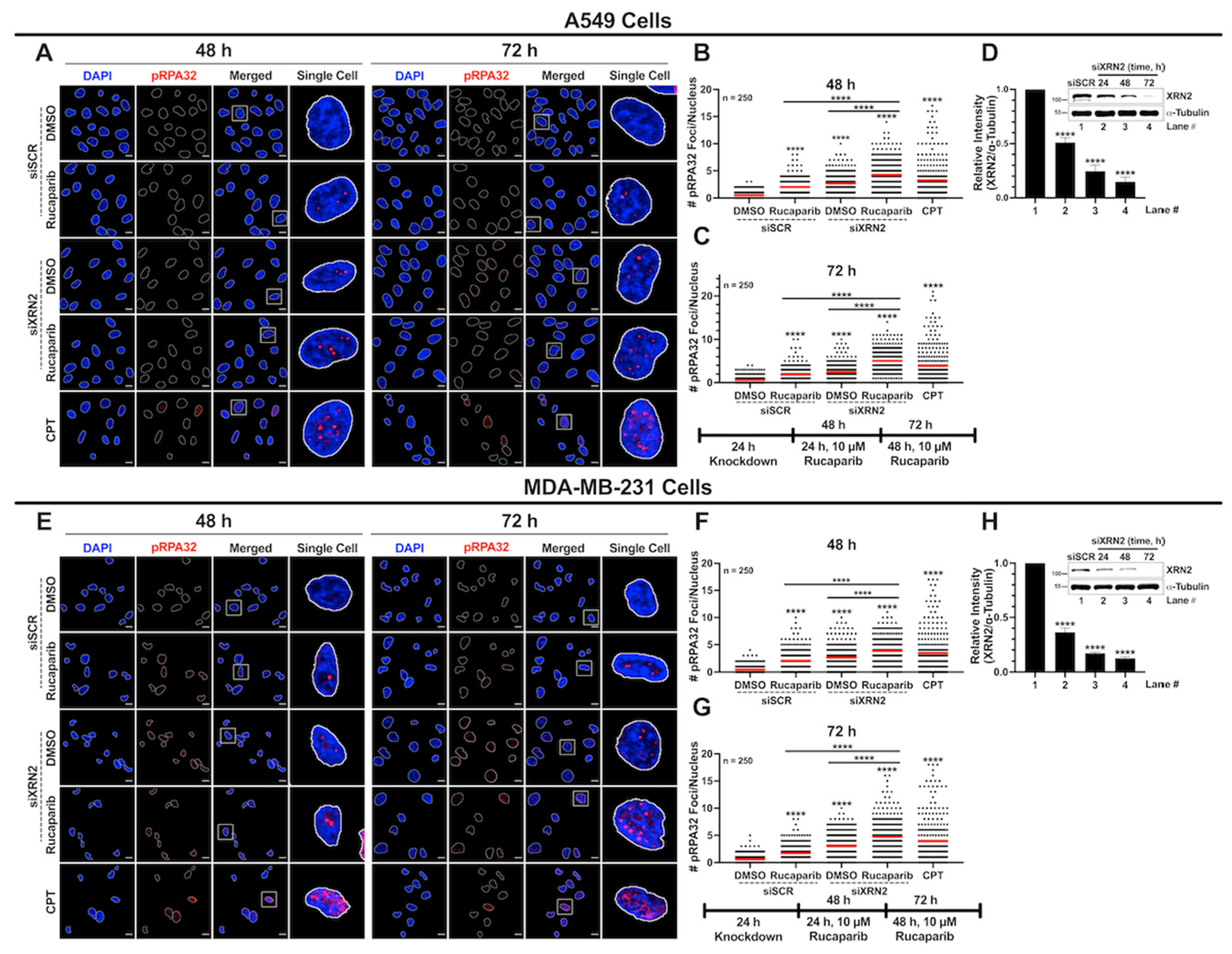
3.5. XRN2 Deficiency Combined with PARP Inhibition Results in Enhanced Replication Stress
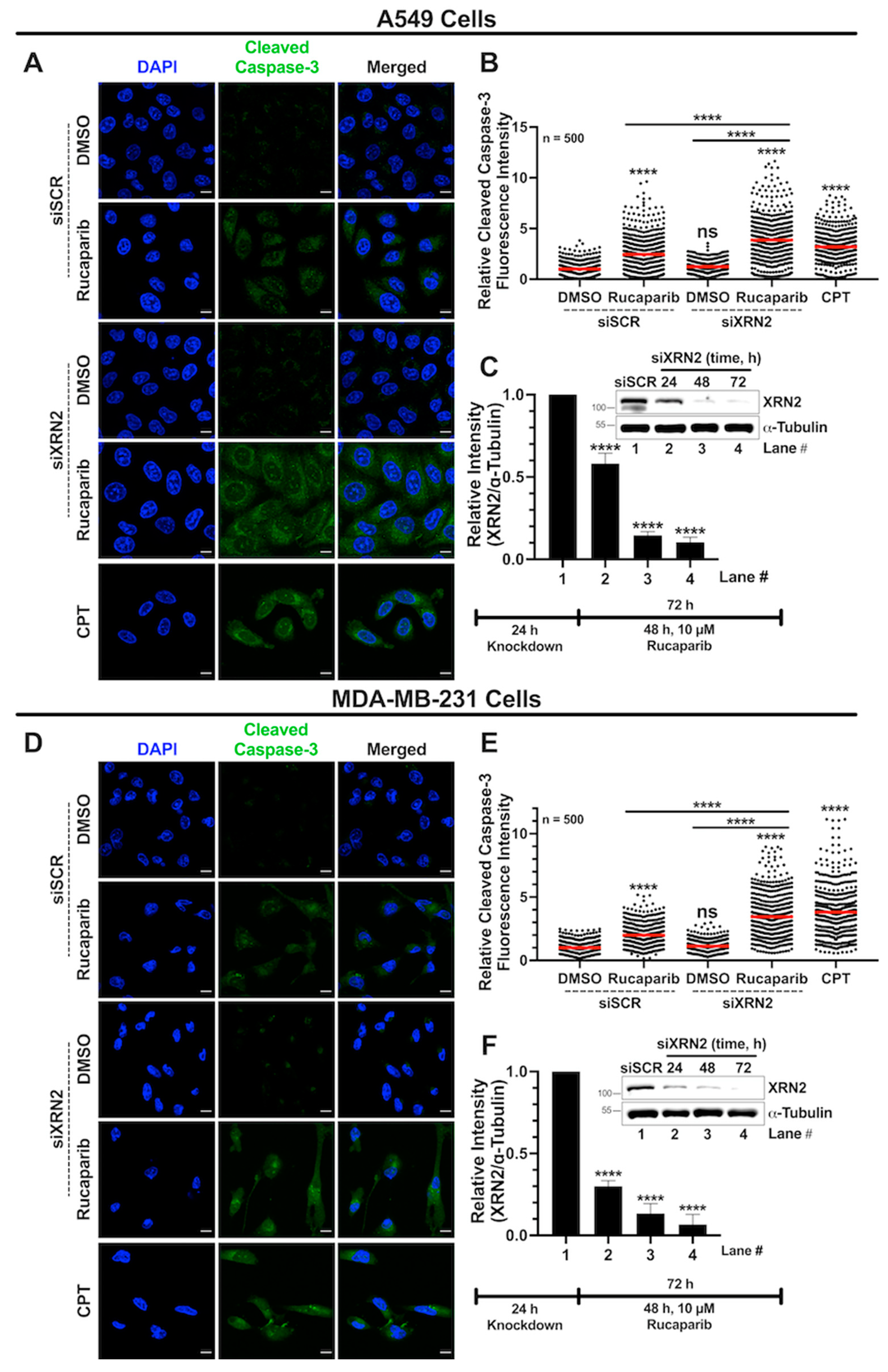
3.6. Combined XRN2 Depletion and PARP Inhibition Activate Caspase-3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| R-loops | RNA–DNA hybrids with a displaced single-stranded DNA |
| DSBs | DNA double-strand breaks |
| HR | Homologous recombination |
| NHEJ | Non-homologous end joining |
| PAR | Poly(ADP-ribose) |
| PARPi | PARP inhibitors |
References
- Madabhushi, R.; Pan, L.; Tsai, L.-H. DNA Damage and Its Links to Neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. R Loops: From Transcription Byproducts to Threats to Genome Stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Hegazy, Y.A.; Fernando, C.M.; Tran, E.J. The balancing act of R-loop biology: The good, the bad, and the ugly. J. Biol. Chem. 2020, 295, 905–913. [Google Scholar] [CrossRef] [PubMed]
- West, S.; Gromak, N.; Proudfoot, N.J. Human 5′→3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 2004, 432, 522–525. [Google Scholar] [CrossRef]
- Fong, N.; Brannan, K.; Erickson, B.; Kim, H.; Cortazar, M.A.; Sheridan, R.M.; Nguyen, T.; Karp, S.; Bentley, D.L. Effects of Transcription Elongation Rate and Xrn2 Exonuclease Activity on RNA Polymerase II Termination Suggest Widespread Kinetic Competition. Mol. Cell 2015, 60, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Manley, J.L. Transcription termination by nuclear RNA polymerases. Genes Dev. 2009, 23, 1247–1269. [Google Scholar] [CrossRef]
- Cortazar, M.A.; Erickson, B.; Fong, N.; Pradhan, S.J.; Ntini, E.; Bentley, D.L. Xrn2 substrate mapping identifies torpedo loading sites and extensive premature termination of RNA pol II transcription. Genes Dev. 2022, 36, 1062–1078. [Google Scholar] [CrossRef]
- Eaton, J.D.; Davidson, L.; Bauer, D.L.; Natsume, T.; Kanemaki, M.T.; West, S. Xrn2 accelerates termination by RNA polymerase II, which is underpinned by CPSF73 activity. Genes Dev. 2018, 32, 127–139. [Google Scholar] [CrossRef]
- Eaton, J.D.; Francis, L.; Davidson, L.; West, S. A unified allosteric/torpedo mechanism for transcriptional termination on human protein-coding genes. Genes Dev. 2020, 34, 132–145. [Google Scholar] [CrossRef]
- Brannan, K.; Kim, H.; Erickson, B.; Glover-Cutter, K.; Kim, S.; Fong, N.; Kiemele, L.; Hansen, K.; Davis, R.; Lykke-Andersen, J.; et al. mRNA Decapping Factors and the Exonuclease Xrn2 Function in Widespread Premature Termination of RNA Polymerase II Transcription. Mol. Cell 2012, 46, 311–324. [Google Scholar] [CrossRef]
- Wagschal, A.; Rousset, E.; Basavarajaiah, P.; Contreras, X.; Harwig, A.; Laurent-Chabalier, S.; Nakamura, M.; Chen, X.; Zhang, K.; Meziane, O.; et al. Microprocessor, Setx, Xrn2, and Rrp6 Co-operate to Induce Premature Termination of Transcription by RNAPII. Cell 2012, 150, 1147–1157. [Google Scholar] [CrossRef]
- Nojima, T.; Gomes, T.; Grosso, A.R.F.; Kimura, H.; Dye, M.J.; Dhir, S.; Carmo-Fonseca, M.; Proudfoot, N.J. Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell 2015, 161, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Davidson, L.; Kerr, A.; West, S. Co-transcriptional degradation of aberrant pre-mRNA by Xrn2. EMBO J. 2012, 31, 2566–2578. [Google Scholar] [CrossRef]
- Kaneko, S.; Rozenblatt-Rosen, O.; Meyerson, M.; Manley, J.L. The multifunctional protein p54nrb/PSF recruits the exonuclease XRN2 to facilitate pre-mRNA 3′ processing and transcription termination. Genes Dev. 2007, 21, 1779–1789. [Google Scholar] [CrossRef]
- Chalamcharla, V.R.; Folco, H.D.; Dhakshnamoorthy, J.; Grewal, S.I.S. Conserved factor Dhp1/Rat1/Xrn2 triggers premature transcription termination and nucleates heterochromatin to promote gene silencing. Proc. Natl. Acad. Sci. USA 2015, 112, 15548–15555. [Google Scholar] [CrossRef]
- Tucker, J.F.; Ohle, C.; Schermann, G.; Bendrin, K.; Zhang, W.; Fischer, T.; Zhang, K. A Novel Epigenetic Silencing Pathway Involving the Highly Conserved 5′-3′ Exoribonuclease Dhp1/Rat1/Xrn2 in Schizosaccharomyces pombe. PLoS Genet. 2016, 12, e1005873. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Pestov, D.G. 5′-end surveillance by Xrn2 acts as a shared mechanism for mammalian pre-rRNA maturation and decay. Nucleic Acids Res. 2011, 39, 1811–1822. [Google Scholar] [CrossRef]
- Patidar, P.L.; Viera, T.; Morales, J.C.; Singh, N.; Motea, E.A.; Khandelwal, M.; Fattah, F.J. XRN2 interactome reveals its synthetic lethal relationship with PARP1 inhibition. Sci. Rep. 2020, 10, 14253. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, P.; James, M.; Vikis, H.G.; Liu, H.; Wen, W.; Franklin, A.; You, M. Genetic variants cis-regulating Xrn2 expression contribute to the risk of spontaneous lung tumor. Oncogene 2010, 29, 1041–1049. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pander, A.; Chinnaiyan, A.M. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Dang, T.T.; Lerner, M.; Saunders, D.; Smith, N.; Gulej, R.; Zalles, M.; Towner, R.A.; Morales, J.C. XRN2 Is Required for Cell Motility and Invasion in Glioblastomas. Cells 2022, 11, 1481. [Google Scholar] [CrossRef]
- Morales, J.C.; Richard, P.; Patidar, P.L.; Motea, E.A.; Dang, T.T.; Manley, J.L.; Boothman, D.A. XRN2 Links Transcription Termination to DNA Damage and Replication Stress. PLoS Genet. 2016, 12, e1006107. [Google Scholar] [CrossRef]
- Dang, T.T.; Morales, J.C. XRN2 Links RNA:DNA Hybrid Resolution to Double Strand Break Repair Pathway Choice. Cancers 2020, 12, 1821. [Google Scholar] [CrossRef]
- Krishnan, R.; Lapierre, M.; Gautreau, B.; Nixon, K.C.J.; El Ghamrasni, S.; Patel, P.S.; Hao, J.; Yerlici, V.T.; Guturi, K.K.N.; St-Germain, J.; et al. RNF8 ubiquitylation of XRN2 facilitates R-loop resolution and restrains genomic instability in BRCA1 mutant cells. Nucleic Acids Res. 2023, 51, 10484–10505. [Google Scholar] [CrossRef]
- Labarca, C.; Paigen, K. A simple, rapid, and sensitive DNA assay procedure. Anal. Biochem. 1980, 102, 344–352. [Google Scholar] [CrossRef]
- Viera, T.; Patidar, P.L. DNA damage induced by KP372-1 hyperactivates PARP1 and enhances lethality of pancreatic cancer cells with PARP inhibition. Sci. Rep. 2020, 10, 20210. [Google Scholar] [CrossRef]
- Smolka, J.A.; Sanz, L.A.; Hartono, S.R.; Chédin, F. Recognition of RNA by the S9.6 antibody creates pervasive artifacts when imaging RNA:DNA hybrids. J. Cell Biol. 2021, 220, e202004079. [Google Scholar] [CrossRef]
- Jia, Z.; Liang, Y.; Ma, B.; Xu, X.; Xiong, J.; Duan, L.; Wang, D. A 5-mC Dot Blot Assay Quantifying the DNA Methylation Level of Chondrocyte Dedifferentiation In Vitro. J. Vis. Exp. 2017, 123, e55565. [Google Scholar]
- Villarreal, O.D.; Mersaoui, S.Y.; Yu, Z.; Masson, J.-Y.; Richard, S. Genome-wide R-loop analysis defines unique roles for DDX5, XRN2, and PRMT5 in DNA/RNA hybrid resolution. Life Sci. Alliance 2020, 3, e202000762. [Google Scholar] [CrossRef]
- Motea, E.A.; Fattah, F.J.; Xiao, L.; Girard, L.; Rommel, A.; Morales, J.C.; Patidar, P.; Zhou, Y.; Porter, A.; Xie, Y.; et al. Kub5-Hera (RPRD1B) Deficiency Promotes “BRCAness” and Vulnerability to PARP Inhibition in BRCA-proficient Breast Cancers. Clin Cancer Res. 2018, 24, 6459–6470. [Google Scholar] [CrossRef]
- Keung, M.Y.; Wu, Y.; Badar, F.; Vadgama, J.V. Response of Breast Cancer Cells to PARP Inhibitors Is Independent of BRCA Status. J. Clin. Med. 2020, 9, 940. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef]
- Hamperl, S.; Cimprich, K.A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair 2014, 19, 84–94. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between Replication and Transcription Complexes Cause Common Fragile Site Instability at the Longest Human Genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef]
- Nagarajan, V.K.; Jones, C.I.; Newbury, S.F.; Green, P.J. XRN 5′-->3′ exoribonucleases: Structure, mechanisms and functions. Biochim. Biophys. Acta 2013, 1829, 590–603. [Google Scholar] [CrossRef]
- Amberg, D.C.; Goldstein, A.L.; Cole, C.N. Isolation and characterization of RAT1: An essential gene of Saccharomyces cerevisiae required for the efficient nucleocytoplasmic trafficking of mRNA. Genes Dev. 1992, 6, 1173–1189. [Google Scholar] [CrossRef]
- Sugano, S.; Shobuike, T.; Takeda, T.; Sugino, A.; Ikeda, H. Molecular analysis of the dhp1 + gene of Schizosaccharomyces pombe: An essential gene that has homology to the DST 2 and RAT 1 genes of Saccharomyces cerevisiae. Mol. Gen. Genet. 1994, 243, 1–8. [Google Scholar] [CrossRef]
- Shobuike, T.; Sugano, S.; Yamashita, T.; Ikeda, H. Characterization of cDNA encoding mouse homolog of fission yeast dhp1+ gene: Structural and functional conservation. Nucleic Acids Res. 1995, 23, 357–361. [Google Scholar] [CrossRef]
- Morales, J.C.; Richard, P.; Rommel, A.; Fattah, F.J.; Motea, E.A.; Patidar, P.L.; Xiao, L.; Leskov, K.; Wu, S.-Y.; Hittelman, W.N.; et al. Kub5-Hera, the human Rtt103 homolog, plays dual functional roles in transcription termination and DNA repair. Nucleic Acids Res. 2014, 42, 4996–5006. [Google Scholar] [CrossRef]
- Reiss, M.; Keegan, J.; Aldrich, A.; Lyons, S.M.; Flynn, R.L. The exoribonuclease XRN2 mediates degradation of the long non-coding telomeric RNA TERRA. FEBS Lett. 2023, 597, 1818–1836. [Google Scholar] [CrossRef]
- Vohhodina, J.; Goehring, L.J.; Liu, B.; Kong, Q.; Botchkarev, V.V.; Huynh, M.; Liu, Z.; Abderazzaq, F.O.; Clark, A.P.; Ficarro, S.B.; et al. BRCA1 binds TERRA RNA and suppresses R-Loop-based telomeric DNA damage. Nat. Commun. 2021, 12, 3542. [Google Scholar] [CrossRef]
- Patidar, P.L.; Motea, E.A.; Fattah, F.J.; Zhou, Y.; Morales, J.C.; Xie, Y.; Garner, H.R.; Boothman, D.A. The Kub5-Hera/RPRD1B interactome: A novel role in preserving genetic stability by regulating DNA mismatch repair. Nucleic Acids Res. 2016, 44, 1718–1731. [Google Scholar] [CrossRef]
- Chédin, F.; Hartono, S.R.; A Sanz, L.; Vanoosthuyse, V. Best practices for the visualization, mapping, and manipulation of R-loops. EMBO J. 2021, 40, e106394. [Google Scholar] [CrossRef]
- Crossley, M.P.; Brickner, J.R.; Song, C.; Zar, S.M.T.; Maw, S.S.; Chedin, F.; Tsai, M.-S.; Cimprich, K. A Catalytically inactive, purified RNase H1: A specific and sensitive probe for RNA-DNA hybrid imaging. J. Cell Biol. 2021, 220, e202101092. [Google Scholar] [CrossRef]
- Hartono, S.R.; Malapert, A.; Legros, P.; Bernard, P.; Chédin, F.; Vanoosthuyse, V. The Affinity of the S9.6 Antibody for Double-Stranded RNAs Impacts the Accurate Mapping of R-Loops in Fission Yeast. J. Mol. Biol. 2018, 430, 272–284. [Google Scholar] [CrossRef]
- Laspata, N.; Kaur, P.; Mersaoui, S.Y.; Muoio, D.; Liu, Z.S.; Bannister, M.H.; Nguyen, H.D.; Curry, C.; Pascal, J.M.; Poirier, G.G.; et al. PARP1 associates with R-loops to promote their resolution and genome stability. Nucleic Acids Res. 2023, 51, 2215–2237. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [PubMed]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Huang, J.T.J.; Hiom, K. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018, 9, 4346. [Google Scholar] [CrossRef] [PubMed]
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284. [Google Scholar] [CrossRef]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viera, T.; Abfalterer, Q.; Neal, A.; Trujillo, R.; Patidar, P.L. Molecular Basis of XRN2-Deficient Cancer Cell Sensitivity to Poly(ADP-ribose) Polymerase Inhibition. Cancers 2024, 16, 595. https://doi.org/10.3390/cancers16030595
Viera T, Abfalterer Q, Neal A, Trujillo R, Patidar PL. Molecular Basis of XRN2-Deficient Cancer Cell Sensitivity to Poly(ADP-ribose) Polymerase Inhibition. Cancers. 2024; 16(3):595. https://doi.org/10.3390/cancers16030595
Chicago/Turabian StyleViera, Talysa, Quinn Abfalterer, Alyssa Neal, Richard Trujillo, and Praveen L. Patidar. 2024. "Molecular Basis of XRN2-Deficient Cancer Cell Sensitivity to Poly(ADP-ribose) Polymerase Inhibition" Cancers 16, no. 3: 595. https://doi.org/10.3390/cancers16030595
APA StyleViera, T., Abfalterer, Q., Neal, A., Trujillo, R., & Patidar, P. L. (2024). Molecular Basis of XRN2-Deficient Cancer Cell Sensitivity to Poly(ADP-ribose) Polymerase Inhibition. Cancers, 16(3), 595. https://doi.org/10.3390/cancers16030595