
Bortezomib in Combination with Physachenolide C Reduces the Tumorigenic Properties of KRASmut/P53mut Lung Cancer Cells by Inhibiting c-FLIP

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. In Vitro Cell Viability
2.3. MTS Cytotoxicity
2.4. In Vitro Cell Migration
2.5. Cell Invasion
2.6. Western Blotting Analysis and Protein Stability
2.7. Metabolic Stress
2.8. CD8+ T Cell Purification and Activation
2.9. Xenograft Lung Tumor Model
2.10. Chronic Toxicity Assessment In Vivo
2.11. Statistical Analysis
3. Results
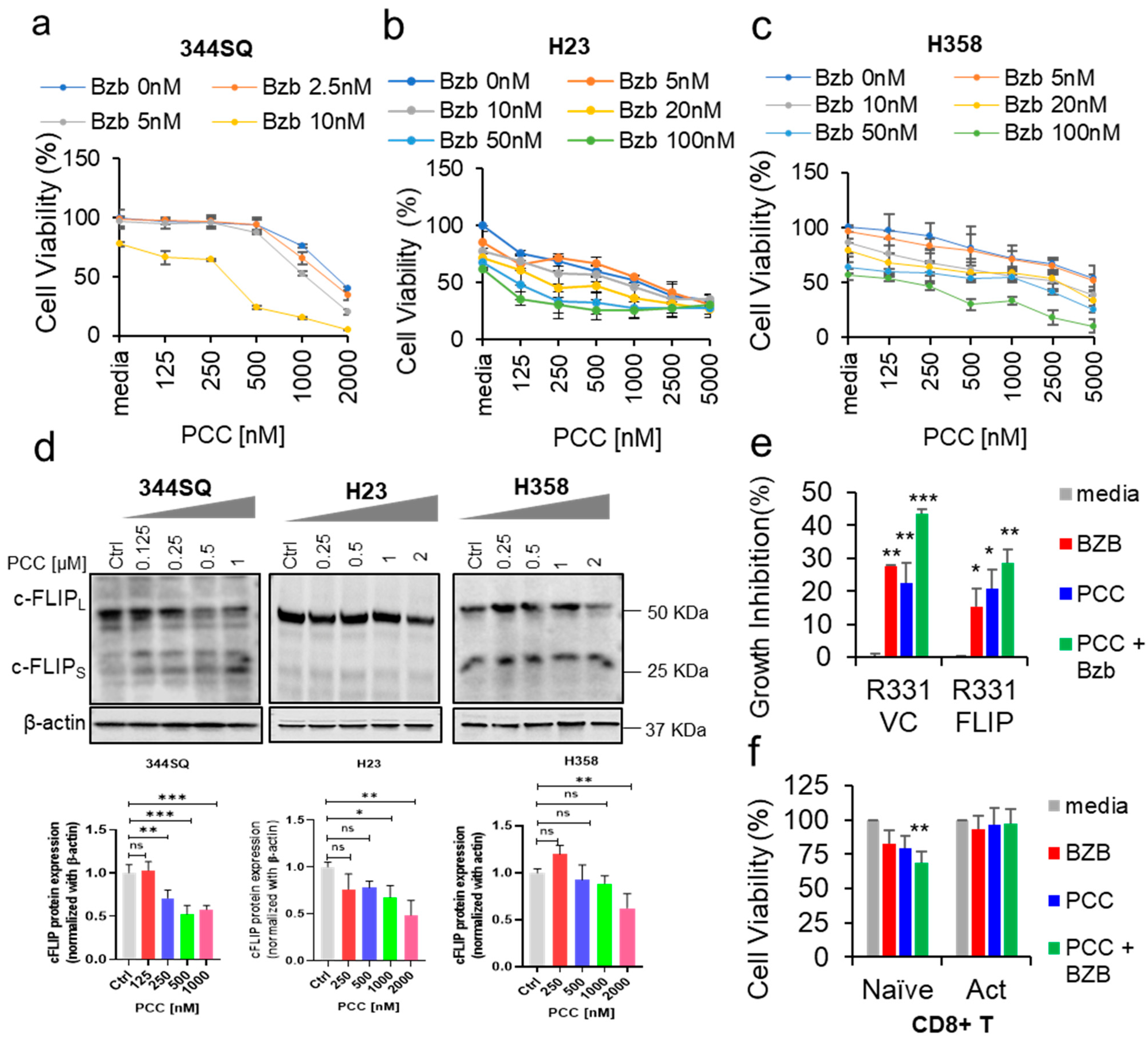
3.1. Effects of Bortezomib and PCC Combination on Lung Cancer Cell Viability
3.2. Role of c-FLIP Overexpression on Cancer Cell Viability
3.3. Bortezomib and PCC Combination on Resting and Activated CD8+ T Cells
3.4. Effect of Bortezomib and PCC Combination on Cell Migration
3.5. Impact of Bortezomib and PCC Combination on Invasive Ability of Lung Cancer Cells
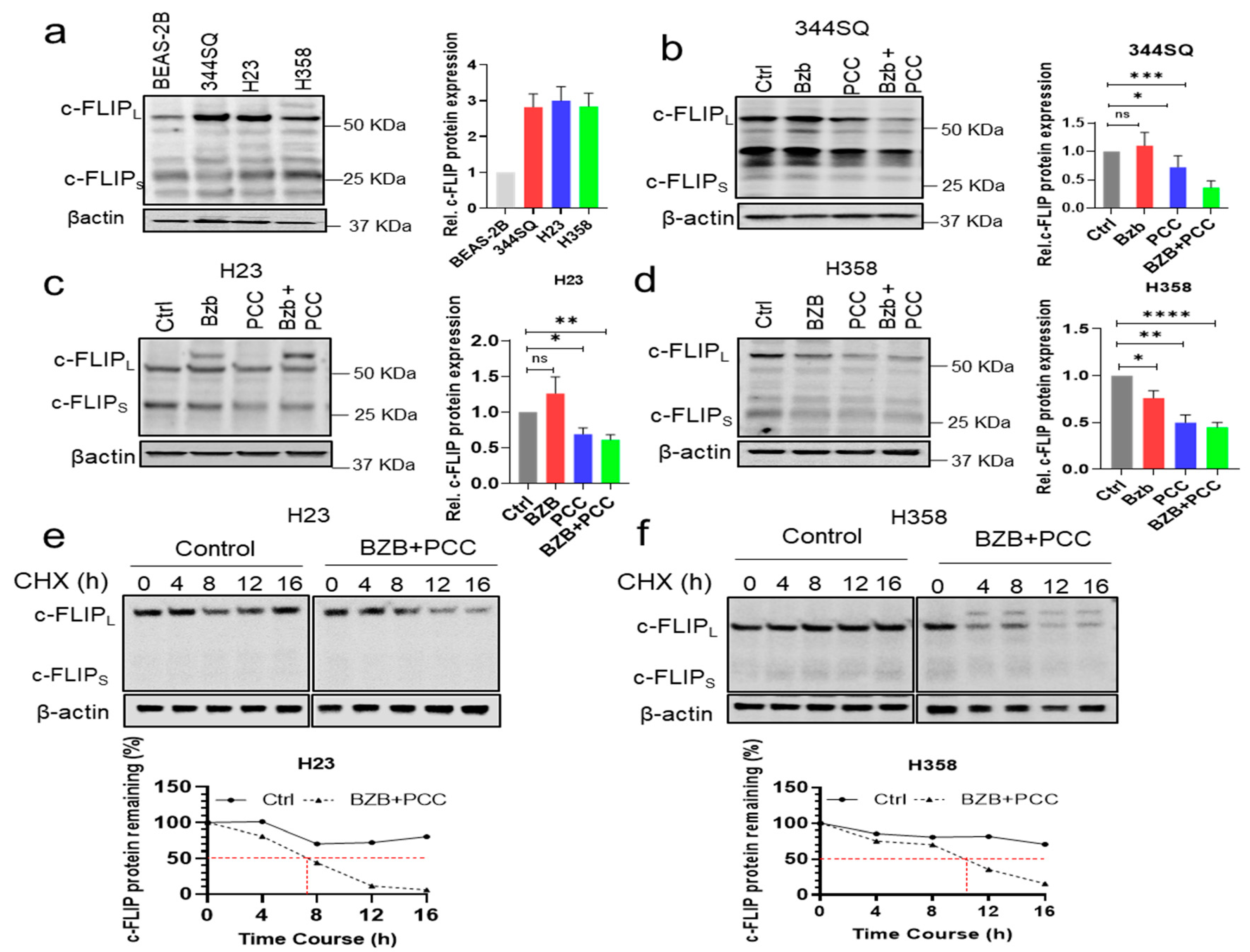
3.6. Effects of Bortezomib and PCC Combination on c-FLIP Protein Expression
3.7. Bortezomib and PCC Combination Reduces c-FLIP Protein Stabilization
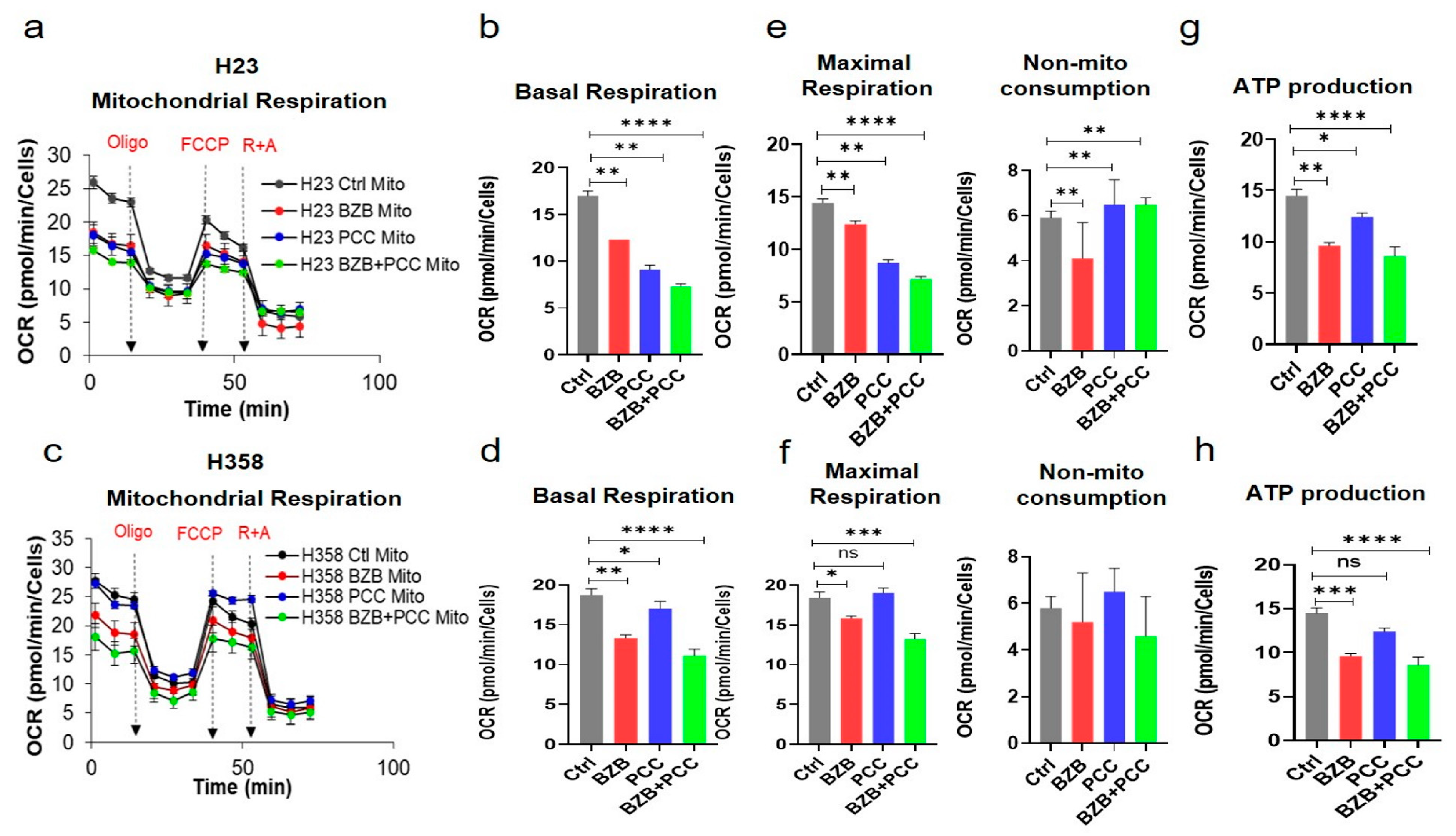
3.8. Effects of Bortezomib and PCC Combination on Mitochondrial Respiration of Cancer Cells
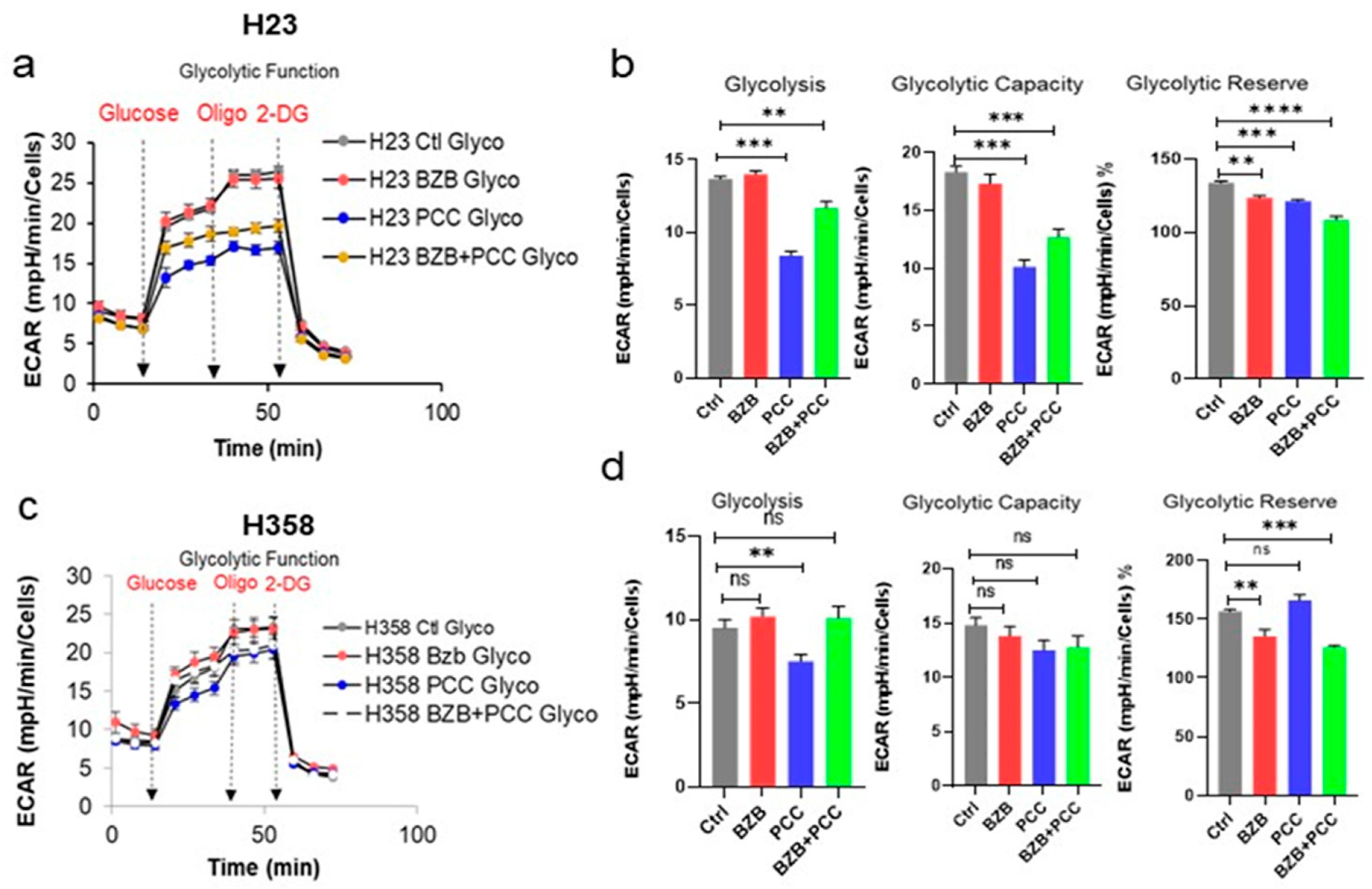
3.9. Effects of Bortezomib and PCC Combination on Glycolytic Function on Cancer Cells
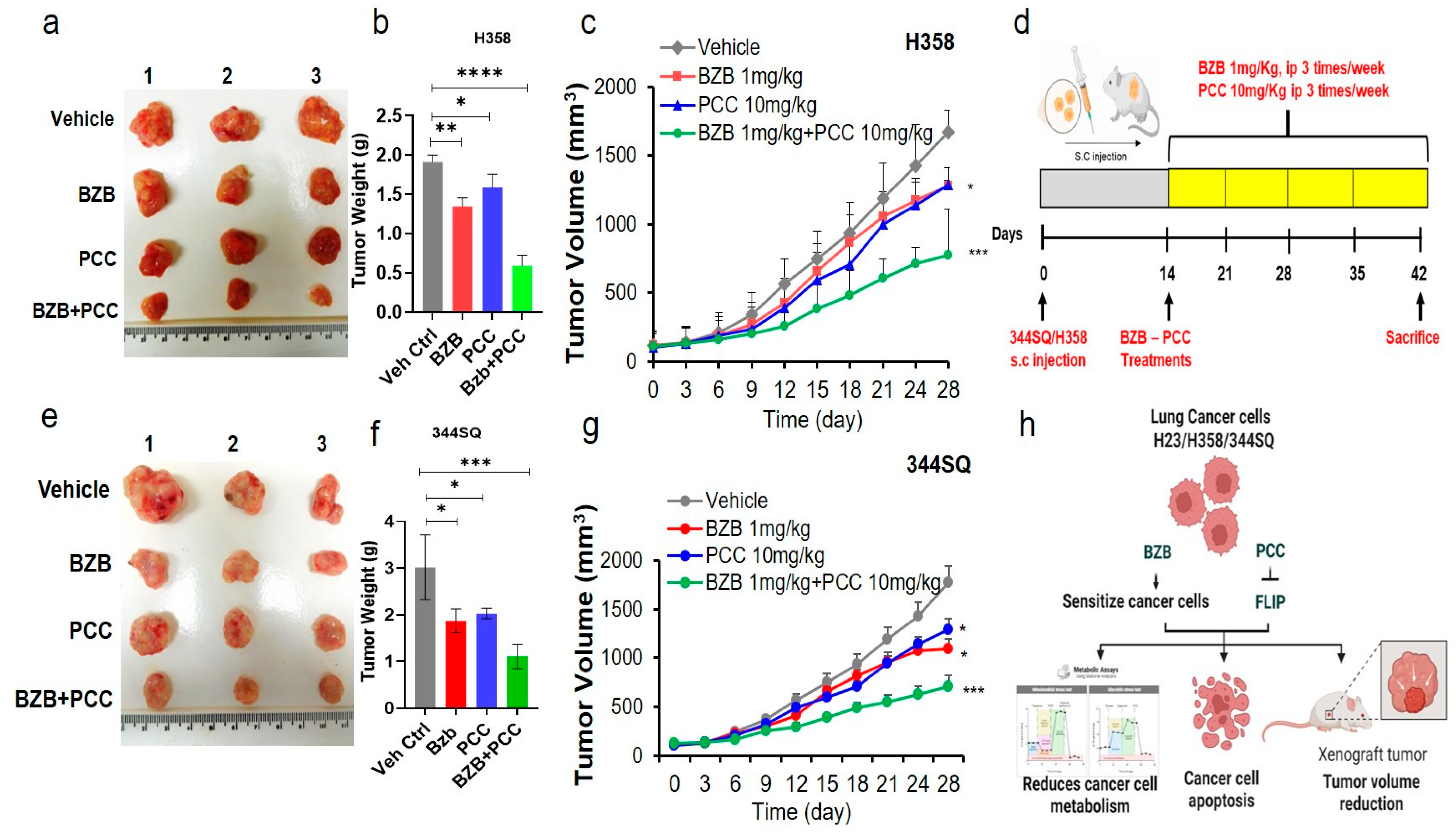
3.10. Antitumor Effects of Bortezomib and PCC Combination in Lung Xenograft Mice
3.11. In Vivo Toxicity Analysis of Bortezomib and PCC Combination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zacksenhaus, E.; Egan, S.E. Progression to Metastasis of Solid Cancer. Cancers 2021, 13, 717. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.; Roth, S.; Kong, J.; Guerra, G.; Narasimhan, V.; Pereira, L.; Desai, J.; Heriot, A.; Ramsay, R. An Update on Immunotherapy for Solid Tumors: A Review. Ann. Surg. Oncol. 2018, 25, 3404–3412. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Mogi, A.; Kuwano, H. TP53 mutations in nonsmall cell lung cancer. J. Biomed. Biotechnol. 2011, 2011, 583929. [Google Scholar] [CrossRef]
- Adderley, H.; Blackhall, F.H.; Lindsay, C.R. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMedicine 2019, 41, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Asma, S.T.; Acaroz, U.; Imre, K.; Morar, A.; Shah, S.R.A.; Hussain, S.Z.; Arslan-Acaroz, D.; Demirbas, H.; Hajrulai-Musliu, Z.; Istanbullugil, F.R.; et al. Natural Products/Bioactive Compounds as a Source of Anticancer Drugs. Cancers 2022, 14, 6203. [Google Scholar] [CrossRef]
- Chen, L.-X.; He, H.; Qiu, F. Natural withanolides: An overview. Nat. Prod. Rep. 2011, 28, 705–740. [Google Scholar] [CrossRef]
- Chang, H.C.; Chang, F.R.; Wang, Y.C.; Pan, M.R.; Hung, W.C.; Wu, Y.C. A bioactive withanolide Tubocapsanolide A inhibits proliferation of human lung cancer cells via repressing Skp2 expression. Mol. Cancer Ther. 2007, 6, 1572–1578. [Google Scholar] [CrossRef]
- Kakar, S.S.; Parte, S.; Carter, K.; Joshua, I.G.; Worth, C.; Rameshwar, P.; Ratajczak, M.Z. Correction: Withaferin A (WFA) inhibits tumor growth and metastasis by targeting ovarian cancer stem cells. Oncotarget 2020, 11, 3103–3104. [Google Scholar] [CrossRef]
- Mayola, E.; Gallerne, C.; Esposti, D.D.; Martel, C.; Pervaiz, S.; Larue, L.; Debuire, B.; Lemoine, A.; Brenner, C.; Lemaire, C. Withaferin A induces apoptosis in human melanoma cells through generation of reactive oxygen species and down-regulation of Bcl-2. Apoptosis 2011, 16, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Tewary, P.; Brooks, A.D.; Xu, Y.M.; Wijeratne, E.M.K.; Babyak, A.L.; Back, T.C.; Chari, R.; Evans, C.N.; Henrich, C.J.; Meyer, T.J.; et al. Small-Molecule Natural Product Physachenolide C Potentiates Immunotherapy Efficacy by Targeting BET Proteins. Cancer Res. 2021, 81, 3374–3386. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Tsai, Y.L.; Wu, Y.C.; Chang, F.R.; Liu, M.H.; Chen, W.Y.; Wu, C.C. Withanolides-induced breast cancer cell death is correlated with their ability to inhibit heat protein 90. PLoS ONE 2012, 7, e37764. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.M.; Brooks, A.D.; Wijeratne, E.M.K.; Henrich, C.J.; Tewary, P.; Sayers, T.J.; Gunatilaka, A.A.L. 17beta-Hydroxywithanolides as Sensitizers of Renal Carcinoma Cells to Tumor Necrosis Factor-alpha Related Apoptosis Inducing Ligand (TRAIL) Mediated Apoptosis: Structure-Activity Relationships. J. Med. Chem. 2017, 60, 3039–3051. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.M.; Liu, M.X.; Grunow, N.; Wijeratne, E.M.K.; Paine-Murrieta, G.; Felder, S.; Kris, R.M.; Gunatilaka, A.A.L. Discovery of Potent 17beta-Hydroxywithanolides for Castration-Resistant Prostate Cancer by High-Throughput Screening of a Natural Products Library for Androgen-Induced Gene Expression Inhibitors. J. Med. Chem. 2015, 58, 6984–6993. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hamza, A.; Zhang, T.; Gu, M.; Zou, P.; Newman, B.; Li, Y.; Gunatilaka, A.A.L.; Zhan, C.G.; Sun, D. Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochem. Pharmaco.l 2010, 79, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Kane, R.; Farrell, A.T.; Abraham, S.; Benson, K.; Brower, M.E.; Bradley, S.; Gobburu, J.V.; Goheer, A.; Lee, S.L.; et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin. Cancer Res. 2004, 10 Pt 1, 3954–3964. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef]
- Hambley, B.; Caimi, P.F.; William, B.M. Bortezomib for the treatment of mantle cell lymphoma: An update. Ther. Adv. Hematol. 2016, 7, 196–208. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Wright, J.; Moskowitz, C.; Muzzy, J.; MacGregor-Cortelli, B.; Stubblefield, M.; Straus, D.; Portlock, C.; Hamlin, P.; Choi, E.; et al. Phase II clinical experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J. Clin. Oncol. 2005, 23, 676–684. [Google Scholar] [CrossRef]
- Shanker, A.; Brooks, A.D.; Tristan, C.A.; Wine, J.W.; Elliott, P.J.; Yagita, H.; Takeda, K.; Smyth, M.J.; Murphy, W.J.; Sayers, T.J. Treating metastatic solid tumors with bortezomib and a tumor necrosis factor-related apoptosis-inducing ligand receptor agonist antibody. J. Natl. Cancer Instig. 2008, 100, 649–662. [Google Scholar] [CrossRef]
- Brooks, A.D.; Jacobsen, K.M.; Li, W.; Shanker, A.; Sayers, T.J. Bortezomib sensitizes human renal cell carcinomas to TRAIL apoptosis through increased activation of caspase-8 in the death-inducing signaling complex. Mol. Cancer Res. 2010, 8, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R. c-FLIP, a master anti-apoptotic regulator. Exp. Oncol. 2012, 34, 176–184. [Google Scholar] [PubMed]
- Melki, M.T.; Saidi, H.; Dufour, A.; Olivo-Marin, J.C.; Gougeon, M.L. Escape of HIV-1-infected dendritic cells from TRAIL-mediated NK cell cytotoxicity during NK-DC cross-talk—A pivotal role of HMGB1. PLoS Pathog. 2010, 6, e1000862. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R.; Day, T.W.; Wu, C.H. Cellular FLICE-like inhibitory protein (C-FLIP): A novel target for cancer therapy. Curr. Cancer Drug Targets 2008, 8, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, M.; Canevari, S.; Mezzanzanica, D. Cellular FLICE-inhibitory protein (c-FLIP) signalling: A key regulator of receptor-mediated apoptosis in physiologic context and in cancer. Int. J. Biochem. Cell Biol. 2010, 42, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Korkolopoulou, P.; Goudopoulou, A.; Voutsinas, G.; Thomas-Tsagli, E.; Kapralos, P.; Patsouris, E.; Saetta, A.A. c-FLIP expression in bladder urothelial carcinomas: Its role in resistance to Fas-mediated apoptosis and clinicopathologic correlations. Urology 2004, 63, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, S.; Song, X.; Sima, N.; Xu, X.; Luo, A.; Chen, G.; Deng, D.; Xu, Q.; Meng, L.; et al. The relationship between c-FLIP expression and human papillomavirus E2 gene disruption in cervical carcinogenesis. Gynecol. Oncol. 2007, 105, 571–577. [Google Scholar] [CrossRef]
- Valente, G.; Manfroi, F.; Peracchio, C.; Nicotra, G.; Castino, R.; Nicosia, G.; Kerim, S.; Isidoro, C. cFLIP expression correlates with tumour progression and patient outcome in non-Hodgkin lymphomas of low grade of malignancy. Br. J. Haematol. 2006, 132, 560–570. [Google Scholar] [CrossRef]
- Li, X.; Pan, X.; Zhang, H.; Lei, D.; Liu, D.; Xu, F.; Luan, X. Overexpression of cFLIP in head and neck squamous cell carcinoma and its clinicopathologic correlations. J. Cancer Res. Clin. Oncol. 2008, 134, 609–615. [Google Scholar] [CrossRef]
- Du, X.; Bao, G.; He, X.; Zhao, H.; Yu, F.; Qiao, Q.; Lu, J.; Ma, Q. Expression and biological significance of c-FLIP in human hepatocellular carcinomas. J. Exp. Clin. Cancer Res. 2009, 28, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.D.; Yu, J.P.; Liu, J.; Luo, H.S.; Chen, H.X.; Yu, H.G. Overexpression of cellular FLICE-inhibitory protein (FLIP) in gastric adenocarcinoma. Clin. Sci. 2004, 106, 397–405. [Google Scholar] [CrossRef] [PubMed]
- McCourt, C.; Maxwell, P.; Mazzucchelli, R.; Montironi, R.; Scarpelli, M.; Salto-Tellez, M.; O’Sullivan, J.M.; Longley, D.B.; Waugh, D.J. Elevation of c-FLIP in castrate-resistant prostate cancer antagonizes therapeutic response to androgen receptor-targeted therapy. Clin. Cancer Res. 2012, 18, 3822–3833. [Google Scholar] [CrossRef]
- Yu, J.W.; Jeffrey, P.D.; Shi, Y. Mechanism of procaspase-8 activation by c-FLIPL. Proc. Natl. Acad. Sci. USA 2009, 106, 8169–8174. [Google Scholar] [CrossRef]
- Seki, N.; Hayakawa, Y.; Brooks, A.D.; Wine, J.; Wiltrout, R.H.; Yagita, H.; Tanner, J.E.; Smyth, M.J.; Sayers, T.J. Tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis is an important endogenous mechanism for resistance to liver metastases in murine renal cancer. Cancer Res. 2003, 63, 207–213. [Google Scholar] [PubMed]
- Engelman, J.A.; Janne, P.A. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res. 2008, 14, 2895–2899. [Google Scholar] [CrossRef]
- Bui, H.T.T.; Le, N.H.; Le, Q.A.; Kim, S.E.; Lee, S.; Kang, D. Synergistic apoptosis of human gastric cancer cells by bortezomib and TRAIL. Int. J. Med. Sci. 2019, 16, 1412. [Google Scholar] [CrossRef]
- Caldiran, F.; Berkel, C.; Yilmaz, E.; Kucuk, B.; Cacan, A.H.; Citli, S.; Canpolat, E.; Cacan, E. Combination treatment of bortezomib and epirubicin increases the expression of TNFRSF10 A/B, and induces TRAIL-mediated cell death in colorectal cancer cells. Biochem. Biophys. Res. Commun. 2023, 675, 33–40. [Google Scholar] [CrossRef]
- Wijeratne, E.M.K.; Xu, Y.-M.; Liu, M.X.; Inacio, M.C.; Brooks, A.D.; Tewary, P.; Sayers, T.J.; Gunatilaka, A.A.L. Ring A/B-modified 17β-hydroxywithanolide analogues as antiproliferative agents for prostate cancer and potentiators of immunotherapy for renal carcinoma and melanoma. J. Nat. Prod. 2021, 84, 3029–3038. [Google Scholar] [CrossRef]
- Mehdizadeh, K.; Ataei, F.; Hosseinkhani, S. Treating MCF7 breast cancer cell with proteasome inhibitor Bortezomib restores apoptotic factors and sensitizes cell to Docetaxel. Med. Oncol. 2021, 38, 64. [Google Scholar] [CrossRef] [PubMed]
- Biltekin, S.N.; Uçar, E.Ö. Separate and Mutual Effects of BIRB796 and Bortezomib on pHsp27 and Viability of U87MG Glioma Cells. Biol. Bull. 2023, 50, 761–772. [Google Scholar] [CrossRef]
- Chougoni, K.K.; Neely, V.L.; Ding, B.; Oduah, E.; Nguyen, K.; Hu, B.; Koblinski, J.; Windle, B.; Harada, H.; Grossman, S. Sensitizing oncogenic mutant p53-expressing non-small cell lung cancer to proteasome inhibitors. Cancer Res. 2023, 83 (Suppl. S7), 3950. [Google Scholar] [CrossRef]
- Roth, W.; Reed, J.C. FLIP protein and TRAIL-induced apoptosis. Vitam. Horm. 2004, 67, 189–206. [Google Scholar] [PubMed]
- Shirley, S.; Micheau, O. Targeting c-FLIP in cancer. Cancer Lett. 2013, 332, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Liu, X.; Fu, H.; Zhou, W.; Zhong, D. 2-Deoxyglucose Suppresses ERK Phosphorylation in LKB1 and Ras Wild-Type Non-Small Cell Lung Cancer Cells. PLoS ONE 2016, 11, e0168793. [Google Scholar] [CrossRef]
- Tung, M.C.; Lin, P.L.; Wang, Y.C.; He, T.Y.; Lee, M.C.; Yeh, S.D.; Chen, C.Y.; Lee, H. Mutant p53 confers chemoresistance in non-small cell lung cancer by upregulating Nrf2. Oncotarget 2015, 6, 41692–41705. [Google Scholar] [CrossRef]
- Park, D.; Shim, E.; Kim, Y.; Kim, Y.M.; Lee, H.; Choe, J.; Kang, D.; Lee, Y.S.; Jeoung, D. C-FLIP promotes the motility of cancer cells by activating FAK and ERK, and increasing MMP-9 expression. Mol. Cells 2008, 25, 184–195. [Google Scholar] [CrossRef]
- Shim, E.; Lee, Y.S.; Kim, H.Y.; Jeoung, D. Down-regulation of c-FLIP increases reactive oxygen species, induces phosphorylation of serine/threonine kinase Akt, and impairs motility of cancer cells. Biotechnol. Lett. 2007, 29, 141–147. [Google Scholar] [CrossRef]
- El-Gazzar, A.; Wittinger, M.; Perco, P.; Anees, M.; Horvat, R.; Mikulits, W.; Grunt, T.W.; Mayer, B.; Krainer, M. The role of c-FLIP(L) in ovarian cancer: Chaperoning tumor cells from immunosurveillance and increasing their invasive potential. Gynecol. Oncol. 2010, 117, 451–459. [Google Scholar] [CrossRef]
- Adams, A.C.; Macy, A.M.; Kang, P.; Castro-Ochoa, K.F.; Wijeratne, E.M.K.; Xu, Y.-M.; Liu, M.X.; Charos, A.; Bosenberg, M.W.; Gunatilaka, A.A.L. Physachenolide C induces complete regression of established murine melanoma tumors via apoptosis and cell cycle arrest. Transl. Oncol. 2022, 15, 101259. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Asanuma, K.; Okamoto, T.; Iino, T.; Hagi, T.; Nakamura, T.; Sudo, A. Combination of Everolimus and Bortezomib Inhibits the Growth and Metastasis of Bone and Soft Tissue Sarcomas via JNK/p38/ERK MAPK and AKT Pathways. Cancers 2023, 15, 2468. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhou, Y.; Zhang, Y.; Hu, H.; Mao, H.-Q.; Selaru, F.M. A combination therapy of bortezomib, CXCR4 inhibitor, and checkpoint inhibitor is effective in cholangiocarcinoma in vivo. Iscience 2023, 26, 106095. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Arrighi, S.; Darwiche, Y.; Deb, S. Comparison of Anticancer Drug Toxicities: Paradigm Shift in Adverse Effect Profile. Life 2021, 12, 48. [Google Scholar] [CrossRef] [PubMed]
- Calixto, J.B. The role of natural products in modern drug discovery. An. Da Acad. Bras. De Ciências 2019, 91 (Suppl. S3), e20190105. [Google Scholar] [CrossRef]
- Satoki, A.; Uchida, M.; Fujiwara, M.; Uesawa, Y.; Shimizu, T. Comprehensive analysis of bortezomib-induced adverse events using the Japanese real-world database. Oncology 2022, 100, 188–194. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanagasabai, T.; Dunbar, Z.; Ochoa, S.G.; Farris, T.; Dhandayuthapani, S.; Wijeratne, E.M.K.; Gunatilaka, A.A.L.; Shanker, A. Bortezomib in Combination with Physachenolide C Reduces the Tumorigenic Properties of KRASmut/P53mut Lung Cancer Cells by Inhibiting c-FLIP. Cancers 2024, 16, 670. https://doi.org/10.3390/cancers16030670
Kanagasabai T, Dunbar Z, Ochoa SG, Farris T, Dhandayuthapani S, Wijeratne EMK, Gunatilaka AAL, Shanker A. Bortezomib in Combination with Physachenolide C Reduces the Tumorigenic Properties of KRASmut/P53mut Lung Cancer Cells by Inhibiting c-FLIP. Cancers. 2024; 16(3):670. https://doi.org/10.3390/cancers16030670
Chicago/Turabian StyleKanagasabai, Thanigaivelan, Zerick Dunbar, Salvador González Ochoa, Tonie Farris, Sivanesan Dhandayuthapani, E. M. Kithsiri Wijeratne, A. A. Leslie Gunatilaka, and Anil Shanker. 2024. "Bortezomib in Combination with Physachenolide C Reduces the Tumorigenic Properties of KRASmut/P53mut Lung Cancer Cells by Inhibiting c-FLIP" Cancers 16, no. 3: 670. https://doi.org/10.3390/cancers16030670
APA StyleKanagasabai, T., Dunbar, Z., Ochoa, S. G., Farris, T., Dhandayuthapani, S., Wijeratne, E. M. K., Gunatilaka, A. A. L., & Shanker, A. (2024). Bortezomib in Combination with Physachenolide C Reduces the Tumorigenic Properties of KRASmut/P53mut Lung Cancer Cells by Inhibiting c-FLIP. Cancers, 16(3), 670. https://doi.org/10.3390/cancers16030670