

Cancer and Cardiovascular Disease: The Conjoined Twins

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Burden and Epidemiology of Cardiovascular Disease and Cancer
1.2. Significance of Co-Occurrence of Cardiovascular Disease and Cancer
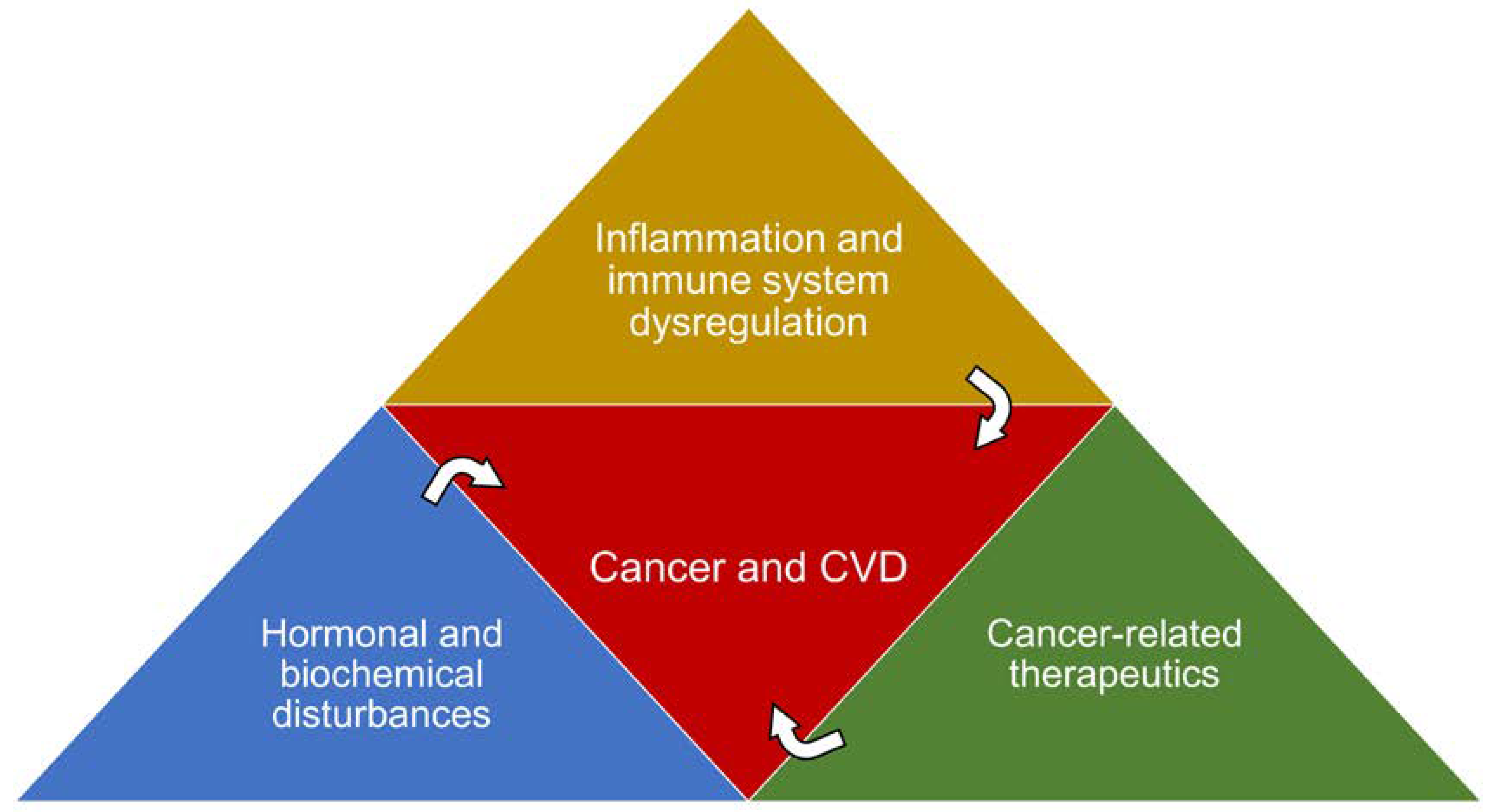
1.3. Shared Risk Factors and Pathophysiological Processes
2. Cardiovascular Health in Cancer Patients
2.1. Role of Cardiovascular Disease Screening
2.2. Cardiovascular Disease Screening in Cancer Patients. Where We Stand, and What Are the Implications?
3. Cardiovascular Interventions in the Armamentarium of Cancer Therapy
3.1. Aspirin
3.2. Angiotensin Converting Enzyme Inhibitors (ACEi) and Angiotensin Receptor Blockers (ARB)
3.3. Beta Blockers (βB)
3.4. Calcium Channel Blockers (CCB)
3.5. Statins (β-Hydroxy β-Methylglutaryl-CoA (HMG-CoA) Reductase Inhibitors)
3.6. Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors (PCSK9-i)
3.7. Sodium-Glucose Co-Transporter-2 Inhibitors (SGLT-2 Inhibitors)
3.8. Exercise
4. Future Perspectives… Will We Be Ready to Change Our Protocols in the Near Future?
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Centers for Disease Control and Prevention. WHO Guidelines. Underlying Cause Death, 1999–2018; CDC WONDER Online Database: Atlanta, GA, USA, 2018. [Google Scholar]
- Weir, H.K. Heart Disease and Cancer Deaths—Trends and Projections in the United States, 1969–2020. Prev. Chronic Dis. 2019, 13, E157. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Murphy, S.L.; Kochanek, K.D.; Arias, E. Deaths: Final data for 2019. Natl. Vital Stat. Rep. 2021, 70, 8. [Google Scholar] [CrossRef]
- The World Health Organization (WHO). Cardiovascular Diseases (CVDs); WHO: Geneva, Switzerland, 2021. [Google Scholar]
- The World Health Organization (WHO). Cancer; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- de Boer, R.A.; Meijers, W.C.; van der Meer, P.; van Veldhuisen, D.J. Cancer and heart disease: Associations and relations. Eur. J. Heart Fail. 2019, 21, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Al-Kindi, S.G.; Oliveira, G.H. Prevalence of Preexisting Cardiovascular Disease in Patients with Different Types of Cancer the Unmet Need for Onco-Cardiology. Mayo Clin. Proc. 2016, 91, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.; Lerman, A.; Sandhu, N.P.; Villarraga, H.R.; Mulvagh, S.L.; Kohli, M. Evaluation and Management of Patients With Heart Disease and Cancer: Cardio-Oncology. Mayo Clin. Proc. 2014, 89, 1287. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, D.; Colombo, A.; Lamantia, G.; Colombo, N.; Civelli, M.; De Giacomi, G.; Pandini, C.; Sandri, M.; Cipolla, C. Cardio-oncology: A new medical issue. Ecancermedicalscience 2008, 2, 126. [Google Scholar] [CrossRef] [PubMed]
- Koutsoukis, A.; Ntalianis, A.; Repasos, E.; Kastritis, E.; Dimopoulos, M.A.; Paraskevaidis, I. Cardio-oncology: A Focus on Cardiotoxicity. Eur. Cardiol. Rev. 2018, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Hershman, D.L.; McBride, R.B.; Eisenberger, A.; Wei, Y.T.; Grann, V.R.; Jacobson, J.S. Doxorubicin, cardiac risk factors, and cardiac toxicity in elderly patients with diffuse B-cell non-Hodgkin’s lymphoma. J. Clin. Oncol. 2008, 26, 3159–3165. [Google Scholar] [CrossRef]
- Wang, S.Y.; Long, J.B.; Hurria, A.; Owusu, C.; Steingart, R.M.; Gross, C.P.; Chen, J. Cardiovascular events, early discontinuation of trastuzumab, and their impact on survival. Breast Cancer Res. Treat. 2014, 146, 411–419. [Google Scholar] [CrossRef]
- Copeland-Halperin, R.S.; Al-Sadawi, M.; Patil, S.; Liu, J.E.; Steingart, R.M.; Dang, C.T.; Yu, A.F. Early Trastuzumab Interruption and Recurrence-Free Survival in ERBB2-Positive Breast Cancer. JAMA Oncol. 2020, 6, 1971. [Google Scholar] [CrossRef] [PubMed]
- Lotrionte, M.; Biondi-zoccai, G.; Abbate, A. Review and Meta-Analysis of Incidence and Clinical Predictors of Anthracycline Cardiotoxicity. Am. J. Cardiol. 2013, 112, 1980–1984. [Google Scholar] [CrossRef] [PubMed]
- Gunaldi, M.; Duman, B.B.; Afsar, C.U.; Paydas, S.; Erkisi, M.; Kara, I.O.; Sahin, B. Risk factors for developing cardiotoxicity of trastuzumab in breast cancer patients: An observational single-centre study. J. Oncol. Pharm. Pract. 2016, 22, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Hamnvik, O.R.; Choueiri, T.K.; Turchin, A.; Mckay, R.R. Clinical Risk Factors for the Development of Hypertension in Patients Treated with Inhibitors of the VEGF Signaling Pathway. Cancer 2015, 121, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K.; et al. Association between Immune Checkpoint Inhibitors with Cardiovascular Events and Atherosclerotic Plaque. Circulation 2020, 142, 2299–2311. [Google Scholar] [CrossRef]
- Wassif, H.; Hussain, M.; Collier, P.H.; Moudgil, R. Immunotherapy-mediated valvulitis: A new cardiovascular immunotherapy-related adverse event. Eur. Hear. J.—Cardiovasc. Imaging 2020, 21, 1102. [Google Scholar] [CrossRef]
- Moslehi, J.; Lichtman, A.H.; Sharpe, A.H.; Galluzzi, L.; Kitsis, R.N. Immune checkpoint inhibitor-associated myocarditis: Manifestations and mechanisms. J. Clin. Investig. 2021, 131, e145186. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.F.; Lei, X.; Haas, A.; Baylis, R.A.; Gao, H.; Luo, L.; Giordano, S.H.; Wehner, M.R.; Nead, K.T.; Leeper, N.J. Risk of Cancer After Diagnosis of Cardiovascular Disease. JACC. CardioOncology 2023, 5, 431–440. [Google Scholar] [CrossRef]
- Koelwyn, G.J.; Aboumsallem, J.P.; Moore, K.J.; de Boer, R.A. Reverse cardio-oncology: Exploring the effects of cardiovascular disease on cancer pathogenesis. J. Mol. Cell. Cardiol. 2022, 163, 1–8. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Libby, P. Inflammation and cardiovascular disease mechanisms. Am. J. Clin. Nutr. 2006, 83, 456S–460S. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet. Oncol. 2014, 15, e493–e503. [Google Scholar] [CrossRef] [PubMed]
- Koene, R.J.; Prizment, A.E.; Blaes, A.; Konety, S.H. Shared Risk Factors in Cardiovascular Disease and Cancer. Circulation 2016, 133, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflamm. 2013, 2013, 139239. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef] [PubMed]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef]
- Katsiki, N.; Mikhailidis, D.P.; Banach, M. Leptin, cardiovascular diseases and type 2 diabetes mellitus. Acta Pharmacol. Sin. 2018, 39, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, M.; Purhonen, A.K.; Jarvelin, M.R.; Rodilla, E.; Pascual, J.; Herzig, K.H. Role of adipokines in obesity-associated hypertension. Acta Physiol. 2010, 200, 107–127. [Google Scholar] [CrossRef] [PubMed]
- de Wit, S.; Glen, C.; de Boer, R.A.; Lang, N.N. Mechanisms shared between cancer, heart failure, and targeted anti-cancer therapies. Cardiovasc. Res. 2023, 118, 3451–3466. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Zwahlen, M.; Minder, C.; Dwyer, S.T.O.; Shalet, S.M.; Egger, M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: Systematic review and meta-regression analysis. Lancet 2004, 363, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Hadi, H.A.R.; Al Suwaidi, J.A. Endothelial dysfunction in diabetes mellitus. Vasc. Health Risk Manag. 2007, 3, 853. [Google Scholar] [PubMed]
- Tabit, C.E.; Chung, W.B.; Hamburg, N.M.; Vita, J.A. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 2010, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.K.; Maurer, H.; Reed, K.; Selagamsetty, R. Diabetes and cancer: Two diseases with obesity as a common risk factor. Diabetes, Obes. Metab. 2014, 16, 97–110. [Google Scholar] [CrossRef]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef]
- Byrne, F.L.; Martin, A.R.; Kosasih, M.; Caruana, B.T.; Farrell, R. The Role of Hyperglycemia in Endometrial Cancer Pathogenesis. Cancers 2020, 12, 1191. [Google Scholar] [CrossRef] [PubMed]
- Kaaks, R.; Lukanova, A.; Kurzer, M.S. Obesity, endogenous hormones, and endometrial cancer risk: A synthetic review. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1531–1543. [Google Scholar]
- Wairagu, P.M.; Phan, A.N.H.; Kim, M.K.; Han, J.; Kim, H.W.; Choi, J.W.; Kim, K.W.; Cha, S.K.; Park, K.H.; Jeong, Y. Insulin priming effect on estradiol-induced breast cancer metabolism and growth. Cancer Biol. Ther. 2015, 16, 484. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Conover, C.A.; Schwartz, R.S. The Insulin-Like Growth Factor Axis. Circ. Res. 2000, 86, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Giovannucci, E. Insulin, Insulin-Like Growth Factors and Colon Cancer: A Review of the Evidence. J. Nutr. 2001, 131, 3109S–3120S. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.H.; LeRoith, D. Obesity, type 2 diabetes, and cancer: The insulin and IGF connection. Endocr. Relat. Cancer 2012, 19, F27–F45. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; LeRoith, D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol. Metab. 2010, 21, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Bao, X.; Hu, M.; Chang, H.; Jiao, M.; Cheng, J.; Xie, L.; Huang, Q.; Li, F.; Li, C.-Y. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 2020, 588, 693–698. [Google Scholar] [CrossRef] [PubMed]
- de Boer, R.A.; Hulot, J.S.; Tocchetti, C.G.; Aboumsallem, J.P.; Ameri, P.; Anker, S.D.; Bauersachs, J.; Bertero, E.; Coats, A.J.S.; Čelutkienė, J.; et al. Common mechanistic pathways in cancer and heart failure. A scientific roadmap on behalf of the Translational Research Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2020, 22, 2272–2289. [Google Scholar] [CrossRef]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol.—Hear. Circ. Physiol. 1994, 267, H742–H750. [Google Scholar] [CrossRef]
- Goodwin, G.W.; Taylor, C.S.; Taegtmeyer, H. Regulation of energy metabolism of the heart during acute increase in heart work. J. Biol. Chem. 1998, 273, 29530–29539. [Google Scholar] [CrossRef]
- De Jong, K.A.; Lopaschuk, G.D. Complex Energy Metabolic Changes in Heart Failure With Preserved Ejection Fraction and Heart Failure With Reduced Ejection Fraction. Can. J. Cardiol. 2017, 33, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Ritterhoff, J.; Young, S.; Villet, O.; Shao, D.; Carnevale Neto, F.; Bettcher, L.F.; Hsu, Y.W.A.; Kolwicz, S.C.; Raftery, D.; Tian, R. Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ. Res. 2020, 126, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Karlstaedt, A.; Moslehi, J.; de Boer, R.A. Cardio-onco-metabolism: Metabolic remodelling in cardiovascular disease and cancer. Nat. Rev. Cardiol. 2022, 19, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Moslehi, J.; Christensen, C.L.; Saha, S.; Tchaicha, J.H.; Ramkissoon, S.H.; Stewart, K.M.; Carretero, J.; Kikuchi, E.; Zhang, H.; et al. D-2-hydroxyglutarate produced by mutant IDH2 causes cardiomyopathy and neurodegeneration in mice. Genes Dev. 2014, 28, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Karlstaedt, A.; Zhang, X.; Vitrac, H.; Harmancey, R.; Vasquez, H.; Wang, J.H.; Goodell, M.A.; Taegtmeyer, H. Oncometabolite d-2-hydroxyglutarate impairs α-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc. Natl. Acad. Sci. USA 2016, 113, 10436–10441. [Google Scholar] [CrossRef] [PubMed]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.M.; Bennett, G.; Coady, S.; D’Agostino, R.B.; Gibbons, R.; Greenland, P.; Lackland, D.T.; Levy, D.; O’Donnell, C.J.; Robinson, J.G.; et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014, 129, S49–S73. [Google Scholar] [CrossRef]
- Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; McBride, P.; Sanford Schwartz, J.; Shero, S.T.; Smith, S.C.; et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults. Am. Hear. Assoc. Task Force Pract. Guidel. Circ. 2014, 129, 1–45. [Google Scholar] [CrossRef]
- Chia, Y.C.; Lim, H.M.; Ching, S.M. Validation of the pooled cohort risk score in an Asian population—A retrospective cohort study. BMC Cardiovasc. Disord. 2014, 14, 163. [Google Scholar] [CrossRef]
- Hanee Henderson, K.; Kaufman, B.G.; Stearns, S.; Couper, D.; Sueta, C.; Kucharska-Newton, A.M.; Foraker, R.E.; Asafu-Adjei, J.; Chang, P. Prevention Validation of the Atherosclerotic Cardiovascular Disease (Ascvd) Pooled Cohort Risk Equations by Education Level: The Atherosclerosis Risk in Communities (Aric) Study. J. Am. Coll. Cardiol. 2016, 5, 1842. [Google Scholar] [CrossRef]
- Rana, J.S.; Tabada, G.H.; Solomon, M.D.; Lo, J.C.; Jaffe, M.G.; Sung, S.H.; Ballantyne, C.M.; Go, A.S. Accuracy of the Atherosclerotic Cardiovascular Risk Equation in a Large Contemporary, Multiethnic Population. J. Am. Coll. Cardiol. 2016, 67, 2118–2130. [Google Scholar] [CrossRef]
- Conroy, R.M.; Pyörälä, K.; Fitzgerald, A.P.; Sans, S.; Menotti, A.; De Backer, G.; De Bacquer, D.; Ducimetière, P.; Jousilahti, P.; Keil, U.; et al. Estimation of ten-year risk of fatal cardiovascular disease in Europe: The SCORE project on behalf of the SCORE project group. Eur. Heart J. 2003, 24, 987–1003. [Google Scholar] [CrossRef] [PubMed]
- Meijers, W.C.; de Boer, R.A. Common risk factors for heart failure and cancer. Cardiovasc. Res. 2019, 115, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Hooning, M.J.; Botma, A.; Aleman, B.M.P.; Baaijens, M.H.A.; Bartelink, H.; Klijn, J.G.M.; Taylor, C.W.; van Leeuwen, F.E. Long-term risk of cardiovascular disease in 10-year survivors of breast cancer. J. Natl. Cancer Inst. 2007, 99, 365–375. [Google Scholar] [CrossRef] [PubMed]
- van Nimwegen, F.A.; Schaapveld, M.; Janus, C.P.M.; Krol, A.D.G.; Petersen, E.J.; Raemaekers, J.M.M.; Kok, W.E.M.; Aleman, B.M.P.; Leeuwen, F.E. van Cardiovascular Disease After Hodgkin Lymphoma Treatment: 40-Year Disease Risk. JAMA Intern. Med. 2015, 175, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Boyne, D.J.; Mickle, A.T.; Brenner, D.R.; Friedenreich, C.M.; Cheung, W.Y.; Tang, K.L.; Wilson, T.A.; Lorenzetti, D.L.; James, M.T.; Ronksley, P.E.; et al. Long-term risk of cardiovascular mortality in lymphoma survivors: A systematic review and meta-analysis. Cancer Med. 2018, 7, 4801. [Google Scholar] [CrossRef]
- Vaughn, D.J.; Palmer, S.C.; Carver, J.R.; Jacobs, L.A.; Mohler, E.R. Cardiovascular risk in long-term survivors of testicular cancer. Cancer 2008, 112, 1949–1953. [Google Scholar] [CrossRef]
- Dixon, J.M.; Farrell, C.; Jones, A.; Leonard, R.; Murray, N.; Palmieri, C.; Plummer, C.J.; Stanley, A.; Verrill, M.W. Expert opinion on the use of anthracyclines in patients with advanced breast cancer at cardiac risk. Ann. Oncol. 2009, 20, 816–827. [Google Scholar] [CrossRef]
- Rubio-Infante, N.; Ramírez-Flores, Y.A.; Castillo, E.C.; Lozano, O.; García-Rivas, G.; Torre-Amione, G. Cardiotoxicity associated with immune checkpoint inhibitor therapy: A meta-analysis. Eur. J. Heart Fail. 2021, 23, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Vater, L.B.; Lefebvre, B.; Turk, A.; Clasen, S.C. Fluoropyrimidine Cardiotoxicity: Incidence, Outcomes, and Safety of Rechallenge. Curr. Oncol. Rep. 2022, 24, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Armenian, S.H.; Lacchetti, C.; Barac, A.; Carver, J.; Constine, L.S.; Denduluri, N.; Dent, S.; Douglas, P.S.; Durand, J.-B.; Ewer, M.; et al. Prevention and Monitoring of Cardiac Dysfunction in Survivors of Adult Cancers: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2016, 35, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Mehta, L.S.; Watson, K.E.; Barac, A.; Beckie, T.M.; Bittner, V.; Cruz-Flores, S.; Dent, S.; Kondapalli, L.; Ky, B.; Okwuosa, T.; et al. Cardiovascular Disease and Breast Cancer: Where These Entities Intersect: A Scientific Statement From the American Heart Association. Circulation 2018, 137, e30–e66. [Google Scholar] [CrossRef] [PubMed]
- Handy, C.E.; Quispe, R.; Pinto, X.; Blaha, M.J.; Blumenthal, R.S.; Michos, E.D.; Lima, J.A.C.; Guallar, E.; Ryu, S.; Cho, J.; et al. Synergistic Opportunities in the Interplay Between Cancer Screening and Cardiovascular Disease Risk Assessment. Circulation 2018, 138, 727–734. [Google Scholar] [CrossRef]
- Richards, M.A.; Stockton, D.; Babb, P.; Coleman, M.P. How many deaths have been avoided through improvements in cancer survival? BMJ 2000, 320, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Enright, K.A.; Krzyzanowska, M.K. Control of cardiovascular risk factors among adult cancer survivors: A population-based survey. Cancer Causes Control 2010, 21, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Gernaat, S.A.M.; Boer, J.M.A.; van den Bongard, D.H.J.; Maas, A.H.E.M.; van der Pol, C.C.; Bijlsma, R.M.; Grobbee, D.E.; Verkooijen, H.M.; Peeters, P.H. The risk of cardiovascular disease following breast cancer by Framingham risk score. Breast Cancer Res. Treat. 2018, 170, 119–127. [Google Scholar] [CrossRef]
- Gernaat, S.A.M.; Išgum, I.; de Vos, B.D.; Takx, R.A.P.; Young-Afat, D.A.; Rijnberg, N.; Grobbee, D.E.; van der Graaf, Y.; de Jong, P.A.; Leiner, T.; et al. Automatic Coronary Artery Calcium Scoring on Radiotherapy Planning CT Scans of Breast Cancer Patients: Reproducibility and Association with Traditional Cardiovascular Risk Factors. PLoS ONE 2016, 11, e0167925. [Google Scholar] [CrossRef]
- Sturgeon, K.M.; Deng, L.; Bluethmann, S.M.; Zhou, S.; Trifiletti, D.M.; Jiang, C.; Kelly, S.P.; Zaorsky, N.G. A population-based study of cardiovascular disease mortality risk in US cancer patients. Eur. Heart J. 2019, 40, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Rauzi, F.; Kirkby, N.S.; Edin, M.L.; Whiteford, J.; Zeldin, D.C.; Mitchell, J.A.; Warner, T.D. Aspirin inhibits the production of proangiogenic 15(S)-HETE by platelet cyclooxygenase-1. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 4256–4266. [Google Scholar] [CrossRef]
- Davì, G.; Patrono, C. Platelet activation and atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494. [Google Scholar] [CrossRef] [PubMed]
- Levine, G.N.; Bates, E.R.; Bittl, J.A.; Brindis, R.G.; Fihn, S.D.; Fleisher, L.A.; Granger, C.B.; Lange, R.A.; Mack, M.J.; Mauri, L.; et al. 2016 ACC/AHA Guideline Focused Update on Duration of Dual Antiplatelet Therapy in Patients With Coronary Artery Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines: An Update of th. Circulation 2016, 134, e123–e155. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Peto, R.; Hennekens, C.; Doll, R.; Bubes, V.; Buring, J.; Dushkesas, R.; Gaziano, M.; Brennan, P.; Meade, T.; et al. Aspirin in the primary and secondary prevention of vascular disease: Collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009, 373, 1849–1860. [Google Scholar]
- Patrono, C. Low-dose aspirin in primary prevention: Cardioprotection, chemoprevention, both, or neither? Eur. Heart J. 2013, 34, 3403–3411. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Patrono, C. Aspirin and Cancer. J. Am. Coll. Cardiol. 2016, 68, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Yang, V.W.; Hylind, L.M.; Krush, A.J.; Petersen, G.M.; Trimbath, J.D.; Piantadosi, S.; Garrett, E.; Geiman, D.E.; Hubbard, W.; et al. Primary chemoprevention of familial adenomatous polyposis with sulindac. N. Engl. J. Med. 2002, 346, 1054–1059. [Google Scholar] [CrossRef]
- Nugent, K.P.; Farmer, K.C.; Spigelman, A.D.; Williams, C.B.; Phillips, R.K. Randomized controlled trial of the effect of sulindac on duodenal and rectal polyposis and cell proliferation in patients with familial adenomatous polyposis. Br. J. Surg. 1993, 80, 1618–1619. [Google Scholar] [CrossRef]
- Bresalier, R.S.; Sandler, R.S.; Quan, H.; Bolognese, J.A.; Oxenius, B.; Horgan, K.; Lines, C.; Riddell, R.; Morton, D.; Lanas, A.; et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N. Engl. J. Med. 2005, 352, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; McMurray, J.J.V.; Pfeffer, M.A.; Wittes, J.; Fowler, R.; Finn, P.; Anderson, W.F.; Zauber, A.; Hawk, E.; Bertagnolli, M. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N. Engl. J. Med. 2005, 352, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-L.; Lin, J.-X.; Zheng, C.-H.; Li, P.; Xie, J.-W.; Wang, J.; Lu, J.; Chen, Q.-Y.; Cao, L.; Lin, M.; et al. Relationship between aspirin use of esophageal, gastric and colorectal cancer patient survival: A meta-analysis. BMC Cancer 2020, 20, 638. [Google Scholar] [CrossRef]
- Miller, E.J.; Patell, R.; Uhlmann, E.J.; Ren, S.; Southard, H.; Elavalakanar, P.; Weber, G.M.; Neuberg, D.; Zwicker, J.I. Antiplatelet medications and risk of intracranial hemorrhage in patients with metastatic brain tumors. Blood Adv. 2022, 6, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Streicher, S.A.; Yu, H.; Lu, L.; Kidd, M.S.; Risch, H.A. Case-control study of aspirin use and risk of pancreatic cancer. CancerEpidemiol. Biomark. Prev. 2014, 23, 1254–1263. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, R.; Yu, L.; Xiao, J.; Zhou, X.; Li, X.; Song, P.; Li, X. Aspirin Use and Common Cancer Risk: A Meta-Analysis of Cohort Studies and Randomized Controlled Trials. Front. Oncol. 2021, 11, 690219. [Google Scholar] [CrossRef] [PubMed]
- Algra, A.M.; Rothwell, P.M. Effects of regular aspirin on long-term cancer incidence and metastasis: A systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012, 13, 518–527. [Google Scholar] [CrossRef]
- Burn, J.; Gerdes, A.; Macrae, F.; Mecklin, J.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Morrison, P.J.; Murday, V.; et al. Long-term eff ect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Fowkes, F.G.R.; Belch, J.F.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Eff ect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Flossmann, E.; Rothwell, P.M. Effect of aspirin on long-term risk of colorectal cancer: Consistent evidence from randomised and observational studies. Lancet 2007, 369, 1603–1613. [Google Scholar] [CrossRef]
- Jacobs, E.J.; Thun, M.J.; Bain, E.B.; Rodriguez, C.; Henley, S.J.; Calle, E.E. A Large Cohort Study of Long-Term Daily Use of Adult-Strength Aspirin and Cancer Incidence. JNCI J. Natl. Cancer Inst. 2007, 99, 608–615. [Google Scholar] [CrossRef]
- Thun, M.J.; Namboodiri, M.M.; Heath, C.W.J. Aspirin use and reduced risk of fatal colon cancer. N. Engl. J. Med. 1991, 325, 1593–1596. [Google Scholar] [CrossRef]
- Gallo, G.; Volpe, M.; Rubattu, S. Angiotensin Receptor Blockers in the Management of Hypertension: A Real-World Perspective and Current Recommendations. Vasc. Health Risk Manag. 2022, 18, 507–515. [Google Scholar] [CrossRef]
- Wong, J.; Patel, R.A.; Kowey, P.R. The clinical use of angiotensin-converting enzyme inhibitors. Prog. Cardiovasc. Dis. 2004, 47, 116–130. [Google Scholar] [CrossRef]
- Messerli, F.H.; Bangalore, S.; Bavishi, C.; Rimoldi, S.F. Angiotensin-Converting Enzyme Inhibitors in Hypertension: To Use or Not to Use? J. Am. Coll. Cardiol. 2018, 71, 1474–1482. [Google Scholar] [CrossRef]
- Laghlam, D.; Jozwiak, M.; Nguyen, L.S. Renin-Angiotensin-Aldosterone System and Immunomodulation: A State-of-the-Art Review. Cells 2021, 10, 1767. [Google Scholar] [CrossRef]
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef]
- Campbell, D.J. Circulating and tissue angiotensin systems. J. Clin. Investig. 1987, 79, 1–6. [Google Scholar] [CrossRef]
- Vargas Vargas, R.A.; Varela Millán, J.M.; Fajardo Bonilla, E. Renin-angiotensin system: Basic and clinical aspects-A general perspective. Endocrinol. Diabetes Nutr. 2022, 69, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.S.; Chappell, M.C. A local pancreatic renin-angiotensin system: Endocrine and exocrine roles. Int. J. Biochem. Cell Biol. 2003, 35, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Capettini, L.S.A.; Montecucco, F.; Mach, F.; Stergiopulos, N.; Santos, R.A.S.; da Silva, R.F. Role of renin-angiotensin system in inflammation, immunity and aging. Curr. Pharm. Des. 2012, 18, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.D.; Binger, K.J. Macrophage heterogeneity and renin-angiotensin system disorders. Pflugers Arch. 2017, 469, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Rasha, F.; Ramalingam, L.; Gollahon, L.; Rahman, R.L.; Rahman, S.M.; Menikdiwela, K.; Moustaid-Moussa, N. Mechanisms linking the renin-angiotensin system, obesity, and breast cancer. Endocr. Relat. Cancer 2019, 26, R653–R672. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Thomas, W.G.; Hannan, R.D. The renin-angiotensin system and cancer: Old dog, new tricks. Nat. Rev. Cancer 2010, 10, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Hayashi, I.; Yamashina, S.; Itoman, M.; Majima, M. Blockade of angiotensin AT1a receptor signaling reduces tumor growth, angiogenesis, and metastasis. Biochem. Biophys. Res. Commun. 2002, 294, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, A.; Kosaka, T.; Asano, T.; Asano, T.; Seta, K.; Kawai, T.; Hayakawa, M. Angiotensin II type I antagonist prevents pulmonary metastasis of murine renal cancer by inhibiting tumor angiogenesis. Cancer Res. 2002, 62, 4176–4179. [Google Scholar] [PubMed]
- Attoub, S.; Gaben, A.M.; Al-Salam, S.; Al Sultan, M.A.H.; John, A.; Nicholls, M.G.; Mester, J.; Petroianu, G. Captopril as a potential inhibitor of lung tumor growth and metastasis. Ann. N. Y. Acad. Sci. 2008, 1138, 65–72. [Google Scholar] [CrossRef]
- Christian, J.B.; Lapane, K.L.; Hume, A.L.; Eaton, C.B.; Weinstock, M.A. Association of ACE inhibitors and angiotensin receptor blockers with keratinocyte cancer prevention in the randomized VATTC trial. J. Natl. Cancer Inst. 2008, 100, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, G.; García Rodríguez, L.A.; Ruigómez, A.; Johansson, S.; Wallander, M.A.; Frithz, G.; Svärdsudd, K. Association between Captopril, Other Antihypertensive Drugs and Risk of Prostate Cancer. Prostate 2004, 58, 50–56. [Google Scholar] [CrossRef]
- Wilop, S.; Von Hobe, S.; Crysandt, M.; Esser, A.; Osieka, R.; Jost, E. Impact of angiotensin I converting enzyme inhibitors and angiotensin II type 1 receptor blockers on survival in patients with advanced non-small-cell lung cancer undergoing first-line platinum-based chemotherapy. J. Cancer Res. Clin. Oncol. 2009, 135, 1429–1435. [Google Scholar] [CrossRef]
- Lever, A.F.; Hole, D.J.; Gillis, C.R.; McCallum, I.R.; McInnes, G.T.; MacKinnon, P.L.; Meredith, P.A.; Murray, L.S.; Reid, J.L.; Robertson, J.W.K. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet 1998, 352, 179–184. [Google Scholar] [CrossRef]
- Lanas, A.; Alcedo González, J. [Chemoprevention in adenocarcinoma of the esophagus]. Acta Gastroenterol. Latinoam. 2007, 37, 37–48. [Google Scholar] [PubMed]
- Cheung, K.S.; Chan, E.W.; Seto, W.K.; Wong, I.C.K.; Leung, W.K. ACE (Angiotensin-Converting Enzyme) Inhibitors/Angiotensin Receptor Blockers Are Associated with Lower Colorectal Cancer Risk: A Territory-Wide Study with Propensity Score Analysis. Hypertension 2020, 76, 968–975. [Google Scholar] [CrossRef]
- Liu, H.; Naxerova, K.; Pinter, M.; Incio, J.; Lee, H.; Shigeta, K.; Ho, W.W.; Crain, J.A.; Jacobson, A.; Michelakos, T.; et al. Use of Angiotensin System Inhibitors Is Associated with Immune Activation and Longer Survival in Nonmetastatic Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 5959–5969. [Google Scholar] [CrossRef] [PubMed]
- Koay, E.J.; Truty, M.J.; Cristini, V.; Thomas, R.M.; Chen, R.; Chatterjee, D.; Kang, Y.; Bhosale, P.R.; Tamm, E.P.; Crane, C.H.; et al. Transport properties of pancreatic cancer describe gemcitabine delivery and response. J. Clin. Investig. 2014, 124, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cao, J.; Melamed, A.; Worley, M.; Gockley, A.; Jones, D.; Nia, H.T.; Zhang, Y.; Stylianopoulos, T.; Kumar, A.S.; et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2019, 116, 2210–2219. [Google Scholar] [CrossRef] [PubMed]
- Lewinter, C.; Nielsen, T.H.; Edfors, L.R.; Linde, C.; Bland, J.M.; LeWinter, M.; Cleland, J.G.F.; Køber, L.; Braunschweig, F.; Mansson-Broberg, A. A systematic review and meta-analysis of beta-blockers and renin–angiotensin system inhibitors for preventing left ventricular dysfunction due to anthracyclines or trastuzumab in patients with breast cancer. Eur. Heart J. 2021, 43, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, K.B.; Hicks, B.; Azoulay, L.; Pottegård, A. Use of ACE (Angiotensin-Converting Enzyme) Inhibitors and Risk of Lung Cancer: A Nationwide Nested Case-Control Study. Circ. Cardiovasc. Qual. Outcomes 2021, 14, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Yoshimasa, T.; Yagura, T.; Yamamoto, I. Non-homogeneous distribution of beta 1- and beta 2-adrenoceptors in various human tissues. Life Sci. 1993, 52, 1063–1070. [Google Scholar] [CrossRef]
- Bangalore, S.; Messerli, F.H.; Kostis, J.B.; Pepine, C.J. Cardiovascular protection using beta-blockers: A critical review of the evidence. J. Am. Coll. Cardiol. 2007, 50, 563–572. [Google Scholar] [CrossRef]
- do Vale, G.T.; Ceron, C.S.; Gonzaga, N.A.; Simplicio, J.A.; Padovan, J.C. Three Generations of β-blockers: History, Class Differences and Clinical Applicability. Curr. Hypertens. Rev. 2019, 15, 22–31. [Google Scholar] [CrossRef]
- Thaker, H.; Li, Y.; Gershenson, D.M.; Lutgendorf, S.; Cole, S.W. Stress Hormone—Mediated Invasion of Ovarian Cancer Cells. Clin. Cancer Res. 2011, 12, 369–375. [Google Scholar] [CrossRef]
- Chakroborty, D.; Sarkar, C.; Basu, B.; Dasgupta, P.S.; Basu, S. Catecholamines regulate tumor angiogenesis. Cancer Res. 2009, 69, 3727–3730. [Google Scholar] [CrossRef] [PubMed]
- Krizanova, O.; Babula, P.; Pacak, K. Stress, catecholaminergic system and cancer. Stress 2016, 19, 419–428. [Google Scholar] [CrossRef]
- Lutgendorf, S.K.; Cole, S.; Costanzo, E.; Bradley, S.; Coffin, J.; Jabbari, S.; Rainwater, K.; Ritchie, J.M.; Yang, M.; Sood, A.K. Stress-related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines. Clin. Cancer Res. 2003, 9, 4514–4521. [Google Scholar]
- Perron, L.; Bairati, I.; Harel, F.; Meyer, F. Antihypertensive drug use and the risk of prostate cancer (Canada). Cancer Causes Control 2004, 15, 535–541. [Google Scholar] [CrossRef]
- Algazi, M.; Plu-Bureau, G.; Flahault, A.; Dondon, M.G.; Lê, M.G. [Could treatments with beta-blockers be associated with a reduction in cancer risk?]. Rev. Epidemiol. Sante Publique 2004, 52, 53–65. [Google Scholar] [CrossRef]
- Bangalore, S.; Kumar, S.; Kjeldsen, S.E.; Makani, H.; Grossman, E.; Wetterslev, J.; Gupta, A.K.; Sever, P.S.; Gluud, C.; Messerli, F.H. Antihypertensive drugs and risk of cancer: Network meta-analyses and trial sequential analyses of 324,168 participants from randomised trials. Lancet. Oncol. 2011, 12, 65–82. [Google Scholar] [CrossRef]
- Rodriguez, C.; Jacobs, E.J.; Deka, A.; Patel, A.V.; Bain, E.B.; Thun, M.J.; Calle, E.E. Use of blood-pressure-lowering medication and risk of prostate cancer in the Cancer Prevention Study II Nutrition Cohort. Cancer Causes Control 2009, 20, 671–679. [Google Scholar] [CrossRef]
- Elliott, W.J.; Ram, C.V.S. Calcium channel blockers. J. Clin. Hypertens. 2011, 13, 687–689. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P. Calcium channel blockers, apoptosis and cancer: Is there a biologic relationship? J. Am. Coll. Cardiol. 1999, 34, 1857–1866. [Google Scholar] [CrossRef]
- Taylor, J.T.; Zeng, X.-B.; Pottle, J.E.; Lee, K.; Wang, A.R.; Yi, S.G.; Scruggs, J.A.S.; Sikka, S.S.; Li, M. Calcium signaling and T-type calcium channels in cancer cell cycling. World J. Gastroenterol. 2008, 14, 4984–4991. [Google Scholar] [CrossRef] [PubMed]
- Panner, A.; Wurster, R.D. T-type calcium channels and tumor proliferation. Cell Calcium 2006, 40, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, S.-L.; Wang, N.; Zhang, B.-B.; Li, M. Blockade of T-type Ca(2+) channels inhibits human ovarian cancer cell proliferation. Cancer Investig. 2011, 29, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, P.J.; McCloskey, K.D. Ca(V) channels and cancer: Canonical functions indicate benefits of repurposed drugs as cancer therapeutics. Eur. Biophys. J. 2016, 45, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zeng, X.; Zhang, R.; Huang, J.; Kuang, X.; Yang, J.; Liu, J.; Tawfik, O.; Thrasher, J.B.; Li, B. Cav1.3 channel α1D protein is overexpressed and modulates androgen receptor transactivation in prostate cancers. Urol. Oncol. 2014, 32, 524–536. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Lai, M.-D.; Phan, N.N.; Sun, Z.; Lin, Y.-C. Meta-Analysis of Public Microarray Datasets Reveals Voltage-Gated Calcium Gene Signatures in Clinical Cancer Patients. PLoS ONE 2015, 10, e0125766. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [CrossRef] [PubMed]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef]
- Chimento, A.; Casaburi, I.; Avena, P.; Trotta, F.; De Luca, A.; Rago, V.; Pezzi, V.; Sirianni, R. Cholesterol and Its Metabolites in Tumor Growth: Therapeutic Potential of Statins in Cancer Treatment. Front. Endocrinol. 2018, 9, 807. [Google Scholar] [CrossRef]
- Boudreau, D.M.; Yu, O.; Johnson, J. Statin Use and Cancer Risk: A Comprehensive Review. Expert Opin. Drug Saf. 2010, 9, 603. [Google Scholar] [CrossRef] [PubMed]
- Vanova, K.; Boukalova, S.; Gbelcova, H.; Muchova, L.; Neuzil, J.; Gurlich, R.; Ruml, T.; Vitek, L. Heme oxygenase is not involved in the anti-proliferative effects of statins on pancreatic cancer cells. BMC Cancer 2016, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, L.B.; Zhang, J.; Tang, D.P.; Wei, J.J.; Zhuang, Z.H. Simvastatin, but not pravastatin, inhibits the proliferation of esophageal adenocarcinoma and squamous cell carcinoma cells: A cell-molecular study. Lipids Health Dis. 2018, 17, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Fang, Z.; Sun, W.; Clark, L.H.; Stine, J.E.; Tran, A.Q.; Sullivan, S.A.; Gilliam, T.P.; Zhou, C.; Bae-Jump, V.L. Atorvastatin exhibits anti-tumorigenic and anti-metastatic effects in ovarian cancer in vitro. Am. J. Cancer Res. 2017, 7, 2478. [Google Scholar] [PubMed]
- Wang, S.T.; Ho, H.J.; Lin, J.T.; Shieh, J.J.; Wu, C.Y. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017, 8, e2626. [Google Scholar] [CrossRef] [PubMed]
- Schointuch, M.N.; Gilliam, T.P.; Stine, J.E.; Han, X.; Zhou, C.; Gehrig, P.A.; Kim, K.; Bae-Jump, V.L. Simvastatin, an HMG-CoA reductase inhibitor, exhibits anti-metastatic and anti-tumorigenic effects in endometrial cancer. Gynecol. Oncol. 2014, 134, 346. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.; Eberg, M.; Benayoun, S.; Aprikian, A.; Batist, G.; Suissa, S.; Azoulay, L. Use of statins and the risk of death in patients with prostate cancer. J. Clin. Oncol. 2014, 32, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.; Klein, E.A.; Li, J.; Moussa, A.S.; Jones, J.S. Statin use and risk of prostate cancer in a population of men who underwent biopsy. J. Urol. 2011, 186, 86–90. [Google Scholar] [CrossRef]
- Murtola, T.J.; Tammela, T.L.J.; Lahtela, J.; Auvinen, A. Cholesterol-Lowering Drugs and Prostate Cancer Risk: A Population-based Case-Control Study. Cancer Epidemiol. Prev. Biomark. 2007, 16, 2226–2232. [Google Scholar] [CrossRef]
- Bansal, D.; Undela, K.; D’Cruz, S.; Schifano, F. Statin use and risk of prostate cancer: A meta-analysis of observational studies. PLoS ONE 2012, 7, e46691. [Google Scholar] [CrossRef]
- Prabhu, N.; Kapur, N.; Catalona, W.; Leikin, R.; Helenowski, I.; Jovanovich, B.; Gurley, M.; Okwuosa, T.M.; Kuzel, T.M. Statin use and risk of prostate cancer biochemical recurrence after radical prostatectomy. Urol. Oncol. Semin. Orig. Investig. 2021, 39, 130.e9–130.e15. [Google Scholar] [CrossRef] [PubMed]
- Scosyrev, E.; Tobis, S.; Donsky, H.; Wu, G.; Joseph, J.; Rashid, H.; Messing, E. Statin use and the risk of biochemical recurrence of prostate cancer after definitive local therapy: A meta-analysis of eight cohort studies. BJU Int. 2013, 111, E71–E77. [Google Scholar] [CrossRef]
- Allott, E.H.; Howard, L.E.; Cooperberg, M.R.; Kane, C.J.; Aronson, W.J.; Terris, M.K.; Amling, C.L.; Freedland, S.J. Postoperative statin use and risk of biochemical recurrence following radical prostatectomy: Results from the Shared Equal Access Regional Cancer Hospital (SEARCH) database. BJU Int. 2014, 114, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Singh, S. Statins are associated with reduced risk of gastric cancer: A systematic review and meta-analysis. Ann. Oncol. 2013, 24, 1721–1730. [Google Scholar] [CrossRef] [PubMed]
- Lakha, F.; Theodoratou, E.; Farrington, S.M.; Tenesa, A.; Cetnarskyj, R.; Din, F.V.N.; Porteous, M.E.; Dunlop, M.G.; Campbell, H. Statin use and association with colorectal cancer survival and risk: Case control study with prescription data linkage. BMC Cancer 2012, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Baba, Y.; Ng, K.; Giovannucci, E.; Fuchs, C.S.; Ogino, S.; Chan, A.T. Statin Use and Colorectal Cancer Risk According to Molecular Subtypes in Two Large Prospective Cohort Studies. Cancer Prev. Res. 2011, 4, 1808–1815. [Google Scholar] [CrossRef]
- Ahern, T.P.; Pedersen, L.; Tarp, M.; Cronin-Fenton, D.P.; Garne, J.P.; Silliman, R.A.; Sørensen, H.T.; Lash, T.L. Statin Prescriptions and Breast Cancer Recurrence Risk: A Danish Nationwide Prospective Cohort Study. JNCI J. Natl. Cancer Inst. 2011, 103, 1461–1468. [Google Scholar] [CrossRef]
- Brewer, T.M.; Masuda, H.; Liu, D.D.; Shen, Y.; Liu, P.; Iwamoto, T.; Kai, K.; Barnett, C.M.; Woodward, W.A.; Reuben, J.M.; et al. Statin use in primary inflammatory breast cancer: A cohort study. Br. J. Cancer 2013, 109, 318–324. [Google Scholar] [CrossRef]
- Nickels, S.; Vrieling, A.; Seibold, P.; Heinz, J.; Obi, N.; Flesch-Janys, D.; Chang-Claude, J. Mortality and Recurrence Risk in Relation to the Use of Lipid-Lowering Drugs in a Prospective Breast Cancer Patient Cohort. PLoS ONE 2013, 8, 75088. [Google Scholar] [CrossRef]
- Nowakowska, M.K.; Lei, X.; Thompson, M.T.; Shaitelman, S.F.; Wehner, M.R.; Woodward, W.A.; Giordano, S.H.; Nead, K.T. Association of statin use with clinical outcomes in patients with triple-negative breast cancer. Cancer 2021, 127, 4142–4150. [Google Scholar] [CrossRef]
- Van Wyhe, R.D.; Rahal, O.M.; Woodward, W.A. Effect of statins on breast cancer recurrence and mortality: A review. Breast Cancer 2017, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.H.; Lee, S.H.; Kim, J.Y.; Ahn, J.S.; Park, Y.H.; Im, Y.H. Statins affect ETS1-overexpressing triple-negative breast cancer cells by restoring DUSP4 deficiency. Sci. Rep. 2016, 6, 33035. [Google Scholar] [CrossRef] [PubMed]
- Za, Y.C.C.E.; Wu, S.; Huang, L.; Buquet, C.; Shen, R.; Nzalez, B.S.G.; Latorre, E.A.G.; Boyer, O.; Varin, R.; Jiménez-Zamudio, L.A.; et al. Synergistic promoting effects of pentoxifylline and simvastatin on the apoptosis of triple-negative MDA-MB-231 breast cancer cells. Int. J. Oncol. 2018, 52, 1246–1254. [Google Scholar]
- Lin, Z.; Zhang, Z.; Jiang, X.; Kou, X.; Bao, Y.; Liu, H.; Sun, F.; Ling, S.; Qin, N.; Jiang, L.; et al. Mevastatin blockade of autolysosome maturation stimulates LBH589-induced cell death in triple-negative breast cancer cells. Oncotarget 2017, 8, 17833. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Kim, H.S.; Kim, J.H.; Lee, J. The Effect of Statin Added to Systemic Anticancer Therapy: A Meta-Analysis of Randomized, Controlled Trials. J. Clin. Med. 2018, 7, 325. [Google Scholar] [CrossRef] [PubMed]
- Neilan, T.G.; Quinaglia, T.; Onoue, T.; Mahmood, S.S.; Drobni, Z.D.; Gilman, H.K.; Smith, A.; Heemelaar, J.C.; Brahmbhatt, P.; Ho, J.S.; et al. Atorvastatin for Anthracycline-Associated Cardiac Dysfunction: The STOP-CA Randomized Clinical Trial. JAMA 2023, 330, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Soccio, R.E.; Duncan, E.M.; Sehayek, E.; Breslow, J.L. Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J. Lipid Res. 2003, 44, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Jasmin, S.B.; Stifani, S.; Basak, A.; Prat, A.; Chrétien, M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Ferri, N.; Tibolla, G.; Pirillo, A.; Cipollone, F.; Mezzetti, A.; Pacia, S.; Corsini, A.; Catapano, A.L. Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis 2012, 220, 381–386. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Breslow, J.L. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. USA 2004, 101, 7100–7105. [Google Scholar] [CrossRef] [PubMed]
- McNutt, M.C.; Kwon, H.J.; Chen, C.; Chen, J.R.; Horton, J.D.; Lagace, T.A. Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J. Biol. Chem. 2009, 284, 10561–10570. [Google Scholar] [CrossRef] [PubMed]
- Surdo, P.L.; Bottomley, M.J.; Calzetta, A.; Settembre, E.C.; Cirillo, A.; Pandit, S.; Ni, Y.G.; Hubbard, B.; Sitlani, A.; Carfí, A. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 2011, 12, 1300. [Google Scholar] [CrossRef]
- Horton, J.D.; Cohen, J.C.; Hobbs, H.H. PCSK9: A convertase that coordinates LDL catabolism. J. Lipid Res. 2009, 50, S172–S177. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S. PCSK9 inhibitors: Clinical evidence and implementation. Nat. Rev. Cardiol. 2018, 16, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Mcgovern, T.J. Tertiary Pharmacology/Toxicology Review; US Food and Drug Administration: Silver Spring, MD, USA, 2015. [Google Scholar]
- Rosenson, R.S.; Hegele, R.A.; Fazio, S.; Cannon, C.P. The Evolving Future of PCSK9 Inhibitors. J. Am. Coll. Cardiol. 2018, 72, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.A.; Mellis, S.; Yancopoulos, G.D.; Stahl, N.; Logan, D.; Smith, W.B.; Lisbon, E.; Gutierrez, M.; Webb, C.; Wu, R.; et al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N. Engl. J. Med. 2012, 366, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Manniello, M.; Pisano, M. Alirocumab (Praluent): First in the New Class of PCSK9 Inhibitors. Pharm. Ther. 2016, 41, 28. [Google Scholar]
- Vega, A. Center for Drug Evaluation and Research Application Number: 125559orig1s000 Summary Review; US Food and Drug Administration: Silver Spring, MD, USA, 2014. [Google Scholar]
- Roth, E.M.; McKenney, J.M.; Hanotin, C.; Asset, G.; Stein, E.A. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N. Engl. J. Med. 2012, 367, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.; Olsson, A.G.; Scott, R.; Kim, J.B.; Xue, A.; Gebski, V.; Wasserman, S.M.; Stein, E.A. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: The GAUSS randomized trial. JAMA 2012, 308, 2497–2506. [Google Scholar] [CrossRef]
- Giugliano, R.P.; Desai, N.R.; Kohli, P.; Rogers, W.J.; Somaratne, R.; Huang, F.; Liu, T.; Mohanavelu, S.; Hoffman, E.B.; McDonald, S.T.; et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): A randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet 2012, 380, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- Koren, M.J.; Scott, R.; Kim, J.B.; Knusel, B.; Liu, T.; Lei, L.; Bolognese, M.; Wasserman, S.M. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): A randomised, double-blind, placebo-controlled, phase 2 study. Lancet 2012, 380, 1995–2006. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Stein, E.; Dufour, R.; Turner, T.; Civeira, F.; Burgess, L.; Langslet, G.; Scott, R.; Olsson, A.G.; Sullivan, D.; et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Koren, M.J.; Lundqvist, P.; Bolognese, M.; Neutel, J.M.; Monsalvo, M.L.; Yang, J.; Kim, J.B.; Scott, R.; Wasserman, S.M.; Bays, H. Anti-PCSK9 monotherapy for hypercholesterolemia: The MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J. Am. Coll. Cardiol. 2014, 63, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.; Colquhoun, D.; Sullivan, D.; Civeira, F.; Rosenson, R.S.; Watts, G.F.; Bruckert, E.; Cho, L.; Dent, R.; Knusel, B.; et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: The GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J. Am. Coll. Cardiol. 2014, 63, 2541–2548. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Smith, R.J.; Hiatt, W.R. Reducing LDL with PCSK9 Inhibitors—The Clinical Benefit of Lipid Drugs. N. Engl. J. Med. 2015, 373, 1588–1591. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Blom, D.J.; Hala, T.; Bolognese, M.; Lillestol, M.J.; Toth, P.D.; Burgess, L.; Ceska, R.; Roth, E.; Koren, M.J.; Ballantyne, C.M.; et al. A 52-Week Placebo-Controlled Trial of Evolocumab in Hyperlipidemia. N. Engl. J. Med. 2014, 370, 1809–1819. [Google Scholar] [CrossRef]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Huang, C.; Lu, Y.; Xue, G.; Guo, X.; Wang, A.; Yang, M.; Qian, J.; Dong, C.; et al. Cholesterol negatively regulates IL-9-producing CD8+ T cell differentiation and antitumor activity. J. Exp. Med. 2018, 215, 1555–1569. [Google Scholar] [CrossRef]
- Naslavsky, N.; Weigert, R.; Donaldson, J.G. Characterization of a Nonclathrin Endocytic Pathway: Membrane Cargo and Lipid Requirements. Mol. Biol. Cell 2004, 15, 3542. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Cui, Y.; Cao, L.; Zhang, Y.; Yin, Y.; Hu, X. PCSK9 regulates apoptosis in human lung adenocarcinoma A549 cells via endoplasmic reticulum stress and mitochondrial signaling pathways. Exp. Ther. Med. 2017, 13, 1993–1999. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Hiwasa, T.; Oshima, Y.; Yajima, S.; Suzuki, T.; Nanami, T.; Sumazaki, M.; Shiratori, F.; Funahashi, K.; Li, S.-Y.; et al. Association of Serum Anti-PCSK9 Antibody Levels with Favorable Postoperative Prognosis in Esophageal Cancer. Front. Oncol. 2021, 11, 708039. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Grossi, F.; Carbone, F.; Vecchié, A.; Minetti, S.; Bardi, N.; Elia, E.; Ansaldo, A.M.; Ferrara, D.; Rijavec, E.; et al. Serum PCSK9 levels at the second nivolumab cycle predict overall survival in elderly patients with NSCLC: A pilot study. Cancer Immunol. Immunother. 2019, 68, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Nik, M.E.; Jaafari, M.R.; Banach, M.; Sahebkar, A. Effects of immunization against PCSK9 in an experimental model of breast cancer. Arch. Med. Sci. 2019, 15, 570. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Nik, M.E.; Jaafari, M.R.; Banach, M.; Sahebkar, A. Potential anti-tumor effect of a nanoliposomal antiPCSK9 vaccine in mice bearing colorectal cancer. Arch. Med. Sci. 2019, 15, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Nowak, C.; Ärnlöv, J. A Mendelian randomization study of the effects of blood lipids on breast cancer risk. Nat. Commun. 2018, 9, 3957. [Google Scholar] [CrossRef] [PubMed]
- Tamargo, J. Heart Failure and Arrhythmias Sodium—Glucose Cotransporter 2 Inhibitors in Heart Failure: Potential Mechanisms of Action, Adverse Effects and Future Developments Heart Failure and Arrhythmias. Eur Cardiol. 2019, 14, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Fathi, A.; Vickneson, K.; Singh, J.S. SGLT2-inhibitors; more than just glycosuria and diuresis. Heart Fail. Rev. 2021, 26, 623–642. [Google Scholar] [CrossRef]
- Cowie, M.R. SGLT2 inhibitors: Mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 2020, 17, 761–772. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner-La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Kondo, H.; Takahashi, N. Reduced hospitalization for heart failure using anti-diabetic drug dapagliflozin: Implications of DECLARE-TIMI 58 for the basic science community. Cardiovasc. Res. 2019, 115, e54–e57. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Lachin, J.M.; Inzucchi, S.E.; Fitchett, D.; Mattheus, M.; George, J.; Woerle, H.J.; Broedl, U.C.; von Eynatten, M.; Zinman, B. Empagliflozin and Clinical Outcomes in Patients With Type 2 Diabetes Mellitus, Established Cardiovascular Disease, and Chronic Kidney Disease. Circulation 2018, 137, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Naznin, F.; Sakoda, H.; Okada, T.; Tsubouchi, H.; Waise, T.M.Z. Canagli fl ozin, a sodium glucose cotransporter 2 inhibitor, attenuates obesity-induced in fl ammation in the nodose ganglion, hypothalamus, and skeletal muscle of mice. Eur. J. Pharmacol. 2017, 794, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Yagi, S.; Hirata, Y.; Ise, T.; Yamada, H.; Fukuda, D.; Matsuura, T.; Yamaguchi, K.; Tobiume, T.; Soeki, T.; Wakatsuki, T.; et al. Canagliflozin reduces epicardial fat in patients with type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2017, 9, 78. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef]
- Rajeev, S.P.; Cuthbertson, D.J.; Wilding, J.P.H. Energy balance and metabolic changes with sodium-glucose co-transporter 2 inhibition. Diabetes. Obes. Metab. 2016, 18, 125–134. [Google Scholar] [CrossRef]
- Solini, A.; Giannini, L.; Seghieri, M.; Vitolo, E.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. Dapagliflozin acutely improves endothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: A pilot study. Cardiovasc. Diabetol. 2017, 16, 138. [Google Scholar] [CrossRef]
- Lambers Heerspink, H.J.; de Zeeuw, D.; Wie, L.; Leslie, B.; List, J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes. Obes. Metab. 2013, 15, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Dutka, M.; Bobiński, R.; Francuz, T.; Garczorz, W.; Zimmer, K.; Ilczak, T.; Ćwiertnia, M.; Hajduga, M.B. SGLT-2 Inhibitors in Cancer Treatment-Mechanisms of Action and Emerging New Perspectives. Cancers 2022, 14, 5811. [Google Scholar] [CrossRef] [PubMed]
- Dicembrini, I.; Nreu, B.; Mannucci, E.; Monami, M. Sodium-glucose co-transporter-2 (SGLT-2) inhibitors and cancer: A meta-analysis of randomized controlled trials. Diabetes. Obes. Metab. 2019, 21, 1871–1877. [Google Scholar] [CrossRef] [PubMed]
- Ptaszynska, A.; Cohen, S.M.; Messing, E.M.; Reilly, T.P.; Johnsson, E.; Johnsson, K. Assessing Bladder Cancer Risk in Type 2 Diabetes Clinical Trials: The Dapagliflozin Drug Development Program as a “Case Study”. Diabetes Ther. Res. Treat. Educ. Diabetes Relat. Disord. 2015, 6, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Gamez, D.; Deb, S. SGLT2 Inhibitors as Potential Anticancer Agents. Biomedicines 2023, 11, 1867. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, Z.; Jing, D.; Huang, X.; Ren, D.; Shao, Z.; Zhang, Z. SGLT2 inhibitor activates the STING/IRF3/IFN-β pathway and induces immune infiltration in osteosarcoma. Cell Death Dis. 2022, 13, 523. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhu, J.; Yu, S.-J.; Ma, H.-L.; Chen, J.; Ding, X.-F.; Chen, G.; Liang, Y.; Zhang, Q. Sodium-glucose co-transporter-2 (SGLT-2) inhibition reduces glucose uptake to induce breast cancer cell growth arrest through AMPK/mTOR pathway. Biomed. Pharmacother. 2020, 132, 110821. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.H.; Chen, Y.L.; Chen, L.J.; Chu, P.Y.; Hsieh, F.S.; Tsai, M.H.; Shih, C.T.; Chao, T.I.; Huang, C.Y.; Chen, K.F. Canagliflozin inhibits growth of hepatocellular carcinoma via blocking glucose-influx-induced β-catenin activation. Cell Death Dis. 2019, 10, 420. [Google Scholar] [CrossRef]
- Okada, J.; Yamada, E.; Saito, T.; Yokoo, H.; Osaki, A.; Shimoda, Y.; Ozawa, A.; Nakajima, Y.; Pessin, J.E.; Okada, S.; et al. Dapagliflozin Inhibits Cell Adhesion to Collagen I and IV and Increases Ectodomain Proteolytic Cleavage of DDR1 by Increasing ADAM10 Activity. Molecules 2020, 25, 495. [Google Scholar] [CrossRef]
- Osataphan, S.; Macchi, C.; Singhal, G.; Chimene-Weiss, J.; Sales, V.; Kozuka, C.; Dreyfuss, J.M.; Pan, H.; Tangcharoenpaisan, Y.; Morningstar, J.; et al. SGLT2 inhibition reprograms systemic metabolism via FGF21-dependent and -independent mechanisms. JCI Insight 2019, 4, e123130. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.J.; Smart, N.A. Exercise therapy and autonomic function in heart failure patients: A systematic review and meta-analysis. Hear. Fail. Rev. 2018, 23, 91–108. [Google Scholar] [CrossRef] [PubMed]
- Ashor, A.W.; Lara, J.; Siervo, M.; Celis-Morales, C.; Oggioni, C.; Jakovljevic, D.G.; Mathers, J.C. Exercise Modalities and Endothelial Function: A Systematic Review and Dose–Response Meta-Analysis of Randomized Controlled Trials. Sport. Med. 2014, 45, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Asimakis, G.K.; Inners-McBride, K.; Medellin, G.; Conti, V.R. Ischemic preconditioning attenuates acidosis and postischemic dysfunction in isolated rat heart. Am. J. Physiol. 1992, 263, H887–H894. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.V.; Yang, X.M.; Downey, J.M. Smaller infarct after preconditioning does not predict extent of early functional improvement of reperfused heart. Am. J. Physiol. 1999, 277, H1754–H1761. [Google Scholar] [CrossRef] [PubMed]
- Parratt, J.; Vegh, A. Pronounced antiarrhythmic effects of ischemic preconditioning. Cardioscience 1994, 5, 9–18. [Google Scholar] [PubMed]
- Shiki, K.; Hearse, D.J. Preconditioning of ischemic myocardium: Reperfusion-induced arrhythmias. Am. J. Physiol. 1987, 253, H1470–H1476. [Google Scholar] [CrossRef]
- Fiuza-Luces, C.; Santos-Lozano, A.; Joyner, M.; Carrera-Bastos, P.; Picazo, O.; Zugaza, J.L.; Izquierdo, M.; Ruilope, L.M.; Lucia, A. Exercise benefits in cardiovascular disease: Beyond attenuation of traditional risk factors. Nat. Rev. Cardiol. 2018, 15, 731–743. [Google Scholar] [CrossRef]
- Cormie, P.; Zopf, E.M.; Zhang, X.; Schmitz, K.H. The Impact of Exercise on Cancer Mortality, Recurrence, and Treatment-Related Adverse Effects. Epidemiol. Rev. 2017, 39, 71–92. [Google Scholar] [CrossRef]
- Hojman, P.; Gehl, J.; Christensen, J.F.; Pedersen, B.K. Molecular Mechanisms Linking Exercise to Cancer Prevention and Treatment. Cell Metab. 2018, 27, 10–21. [Google Scholar] [CrossRef]
- Moore, S.C.; Lee, I.M.; Weiderpass, E.; Campbell, P.T.; Sampson, J.N.; Kitahara, C.M.; Keadle, S.K.; Arem, H.; De Gonzalez, A.B.; Hartge, P.; et al. Association of Leisure-Time Physical Activity with Risk of 26 Types of Cancer in 1.44 Million Adults. JAMA Intern. Med. 2016, 176, 816–825. [Google Scholar] [CrossRef]
- Ashcraft, K.A.; Peace, R.M.; Betof, A.S.; Dewhirst, M.W.; Jones, L.W. Efficacy and Mechanisms of Aerobic Exercise on Cancer Initiation, Progression, and Metastasis: A Critical Systematic Review of In Vivo Preclinical Data. Cancer Res. 2016, 76, 4032–4050. [Google Scholar] [CrossRef]
- Aveseh, M.; Nikooie, R.; Aminaie, M. Exercise-induced changes in tumour LDH-B and MCT1 expression are modulated by oestrogen-related receptor alpha in breast cancer-bearing BALB/c mice. J. Physiol. 2015, 593, 2635–2648. [Google Scholar] [CrossRef]
- Loughney, L.; West, M.A.; Kemp, G.J.; Grocott, M.P.W.; Jack, S. Exercise intervention in people with cancer undergoing neoadjuvant cancer treatment and surgery: A systematic review. Eur. J. Surg. Oncol. J. Eur. Soc. Surg. Oncol. Br. Assoc. Surg. Oncol. 2016, 42, 28–38. [Google Scholar] [CrossRef]
- Schmitz, K.H.; Courneya, K.S.; Matthews, C.; Demark-Wahnefried, W.; Galvão, D.A.; Pinto, B.M.; Irwin, M.L.; Wolin, K.Y.; Segal, R.J.; Lucia, A.; et al. American College of Sports Medicine roundtable on exercise guidelines for cancer survivors. Med. Sci. Sports Exerc. 2010, 42, 1409–1426. [Google Scholar] [CrossRef]
- Iannaccone, M.; D’Ascenzo, F.; Vadalà, P.; Wilton, S.B.; Noussan, P.; Colombo, F.; Raposeiras Roubín, S.; Abu Assi, E.; González-Juanatey, J.R.; Simao Henriques, J.P.; et al. Prevalence and outcome of patients with cancer and acute coronary syndrome undergoing percutaneous coronary intervention: A BleeMACS substudy. Eur. Heart J. Acute Cardiovasc. Care 2018, 7, 631–638. [Google Scholar] [CrossRef]
- Bharadwaj, A.; Potts, J.; Mohamed, M.O.; Parwani, P.; Swamy, P.; Lopez-Mattei, J.C.; Rashid, M.; Kwok, C.S.; Fischman, D.L.; Vassiliou, V.S.; et al. Acute myocardial infarction treatments and outcomes in 6.5 million patients with a current or historical diagnosis of cancer in the USA. Eur. Heart J. 2020, 41, 2183–2193. [Google Scholar] [CrossRef] [PubMed]
- Leiva, O.; AbdelHameid, D.; Connors, J.M.; Cannon, C.P.; Bhatt, D.L. Common Pathophysiology in Cancer, Atrial Fibrillation, Atherosclerosis, and Thrombosis: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncology 2021, 3, 619–634. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Medication | Potential Cancer Targets | Cancer Outcomes |
|---|---|---|
| Aspirin | CRC, esophageal, pancreatic, brain, lung, stomach, and prostate cancers |
|
| Angiotensin converting enzyme inhibitors (ACEi) Angiotensin receptor blockers (ARB) | Non-small-cell lung cancer Esophageal, pancreatic, and colon, prostate cancers and melanoma |
|
| Beta blockers (βB) | Breast, prostate, and ovarian cancers |
|
| Calcium channel blockers (CCB) | Breast, brain, colorectal, gastric, ovarian, and prostate tumors as well as leukemia |
|
| Statin | CRC, gastric, prostate and breast cancers |
|
| Proprotein convertase subtilisin kexin type 9 inhibitors (PCSK9-i) | Non-small-cell lung cancer, breast and colon cancers |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zmaili, M.; Alzubi, J.; Alkhayyat, M.; Albakri, A.; Alkhalaileh, F.; Longinow, J.; Moudgil, R. Cancer and Cardiovascular Disease: The Conjoined Twins. Cancers 2024, 16, 1450. https://doi.org/10.3390/cancers16081450
Zmaili M, Alzubi J, Alkhayyat M, Albakri A, Alkhalaileh F, Longinow J, Moudgil R. Cancer and Cardiovascular Disease: The Conjoined Twins. Cancers. 2024; 16(8):1450. https://doi.org/10.3390/cancers16081450
Chicago/Turabian StyleZmaili, Mohammad, Jafar Alzubi, Motasem Alkhayyat, Almaza Albakri, Feras Alkhalaileh, Joshua Longinow, and Rohit Moudgil. 2024. "Cancer and Cardiovascular Disease: The Conjoined Twins" Cancers 16, no. 8: 1450. https://doi.org/10.3390/cancers16081450
APA StyleZmaili, M., Alzubi, J., Alkhayyat, M., Albakri, A., Alkhalaileh, F., Longinow, J., & Moudgil, R. (2024). Cancer and Cardiovascular Disease: The Conjoined Twins. Cancers, 16(8), 1450. https://doi.org/10.3390/cancers16081450