

The Role of Hepatitis Viruses as Drivers of Hepatocancerogenesis

Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Epidemiology
1.2. Non-Viral Co-Factors
1.3. Viral Pathogenesis
2. Hepatitis C Virus
2.1. Mechanisms of Hepatocarcinogenesis
2.2. Incidence and Risk Factors of HCC in Patients with HCV
2.3. HCC Risk in Patients with SVR
3. Hepatitis B Virus
3.1. Mechanisms of Hepatocarcinogenesis
3.2. Incidence and Risk Factors of HCC in Patients with HBV
3.3. HCC Risk in Untreated and Treated Patients

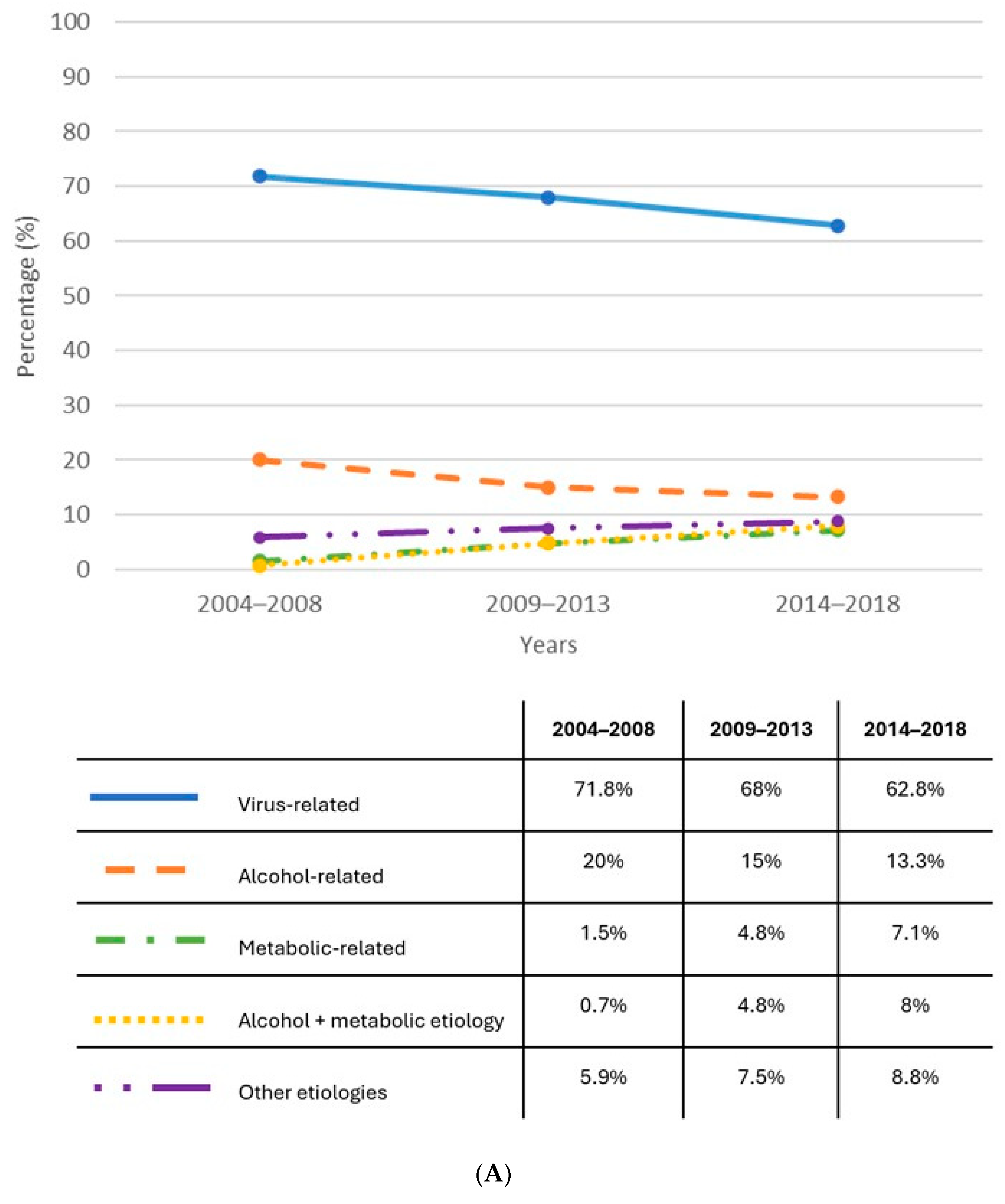{kind=link}
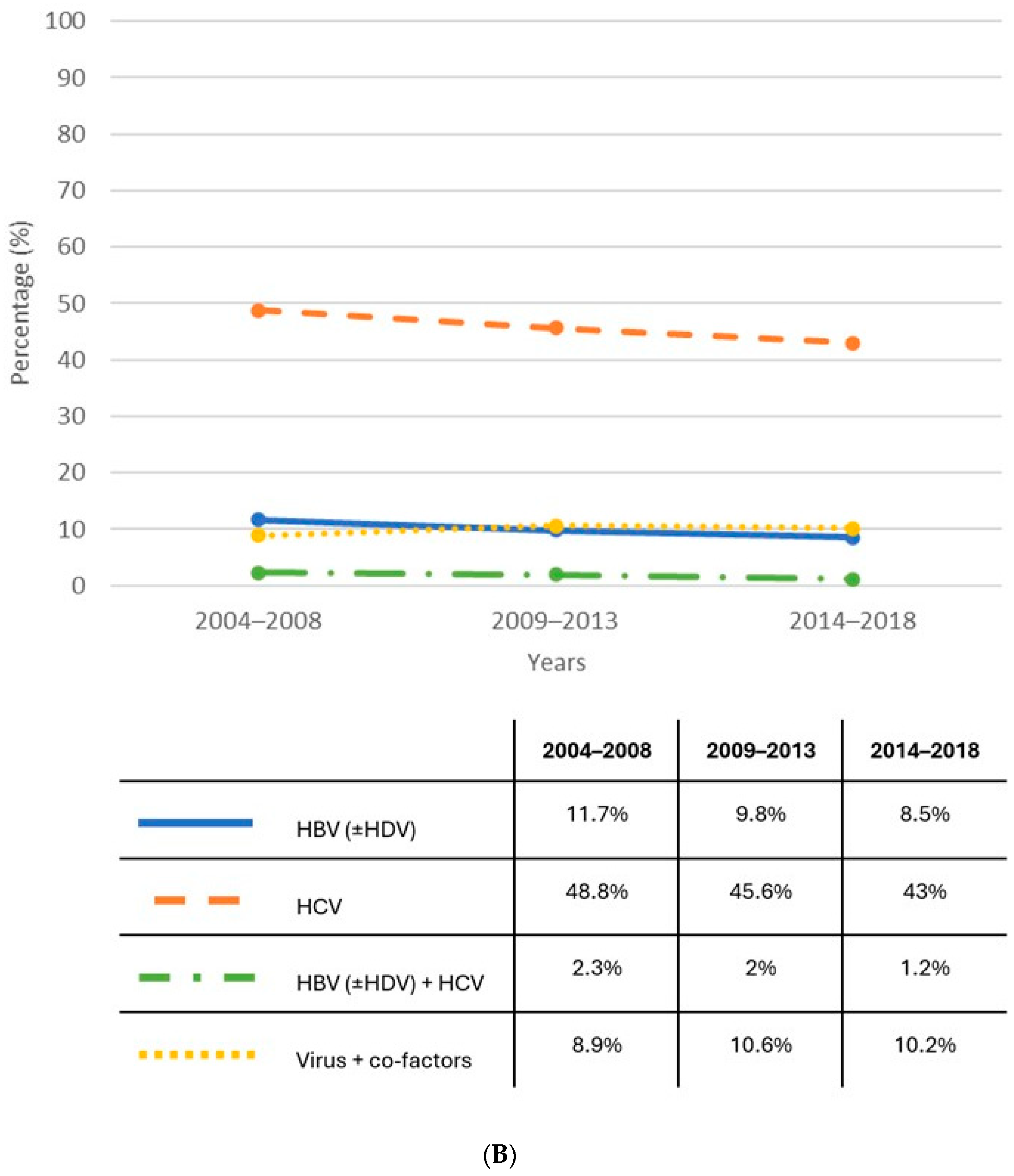{kind=link}
| Author, Journal | Country | Methodology | Patient Characteristic | Findings |
|---|---|---|---|---|
| Papatheodoridis GV, J Hepatol 2020 [66] | Europe | Retrospective | 370 (26.9%) patients with cirrhosis All treated patients | HCC occurrence: 1.2% of patients with chronic hepatitis vs. 5.75% of patients with cirrhosis |
| Liu K, APT 2019 [68] | China | Retrospective | 797 patients treated with TDF vs. 291 untreated patients 53.7% patients with cirrhosis | 5-year cumulative probability of HCC: 14.9% in untreated patients vs. 9.8% in patients treated with TDF |
| Choi J, Jama Oncol 2019 [70] | Korea | Retrospective | 11,464 patients treated with ETV and 12,692 patients treated with TDF Cirrhosis: 26.1% in ETV vs. 27.5% in TDF | Annual incidence rate of HCC: 1.06 per 100 p/y in ETV vs. 0.64 in TDF groups |
| Pol S, APT 2021 [73] | France | Retrospective | 814 patients treated with ETV and 986 patients treated with TDF Cirrhosis: 9% in both groups | HCC incidence rate: 1.6 per 100 p/y in ETV vs. 1.8 per 100 p/y in TDF groups (not a statistically significant difference) |
3.4. Occult Hepatitis B Virus Infection (OBI) and HCC Risk
4. Hepatitis D Virus
4.1. Mechanisms of Hepatocarcinogenesis
4.2. HCC Risk in Patients with HBV/HDV
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younossi, Z.M.; Wong, G.; Anstee, Q.M.; Henry, L. The Global Burden of Liver Disease. Clin. Gastroenterol. Hepatol. 2023, 21, 1978–1991. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Net. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; El-Serag, H.B. Epidemiology of hepatocellular carcinoma: Consider the population. J. Clin. Gastroenterol. 2013, 47, 2–6. [Google Scholar] [CrossRef]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2019, 72, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Garuti, F.; Neri, A.; Avanzato, F.; Gramenzi, A.; Rampoldi, D.; Rucci, P.; Farinati, F.; Giannini, E.G.; Piscaglia, F.; Rapaccini, G.L.; et al. on behalf of the ITA.LI.CA study group. The changing scenario of hepatocellular carcinoma in Italy: An update. Liver Int. 2021, 41, 585–597. [Google Scholar] [CrossRef]
- AIOM. Available online: www.iss.it/documents/20126/8404074/LG97_AISF-AIOM_Epatocarcinoma (accessed on 12 April 2024).
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.-L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Zhang, X.; Guan, L.; Tian, H.; Zeng, Z.; Chen, J.; Huang, D.; Sun, J.; Guo, J.; Cui, H.; Li, Y. Risk Factors and Prevention of Viral Hepatitis-related Hepatocellular carcinoma. Front. Oncol. 2021, 11, 686962. [Google Scholar] [CrossRef] [PubMed]
- Donato, F.; Tagger, A.; Gelatti, U.; Parrinello, G.; Boffetta, P.; Albertini, A.; Decarli, A.; Trevisi, P.; Ribero, M.L.; Martelli, C.; et al. Alcohol and Hepatocellular Carcinoma: The Effect of Lifetime Intake and Hepatitis Virus Infections in Men and Women. Am. J. Epidemiol. 2002, 155, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Stroffolini, T.; Stroffolini, G. A Historical Overview on the Role of Hepatitis B and C Viruses as Aetiological Factors for Hepatocellular Carcinoma. Cancers 2023, 15, 2388. [Google Scholar] [CrossRef] [PubMed]
- Vandenbulcke, H.; Moreno, C.; Colle, I.; Knebel, J.-F.; Francque, S.; Sersté, T.; George, C.; de Galocsy, C.; Laleman, W.; Delwaide, J.; et al. Alcohol intake increases the risk of HCC in hepatitis C virus-related compensated cirrhosis: A prospective study. J. Hepatol. 2016, 65, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.-C.; Lee, Y.-C.A.; Hashibe, M.; Dai, M.; Zheng, T.; Boffetta, P. Interaction between cigarette smoking and hepatitis B and C virus infection on the risk of liver cancer: A meta-analysis. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Baecker, A.; Wu, M.; Zhou, J.-Y.; Yang, J.; Han, R.-Q.; Wang, P.-H.; Jin, Z.-Y.; Liu, A.-M.; Gu, X.; et al. Interaction between tobacco smoking and hepatitis B virus infection on the risk of liver cancer in a Chinese population. Int. J. Cancer 2018, 142, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.W.; Lin, C.L.; Liu, C.J.; Yang, S.H.; Tseng, Y.L.; Wu, C.F. Influence of Metabolic Risk Factors on Risk of Hepatocellular Carcinoma and Liver-Related Death in Men With Chronic Hepatitis B: A Large Cohort Study. Gastroenterology 2017, 153, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Vetrano, E.; Rinaldi, L.; Mormone, A.; Giorgione, C.; Galiero, R.; Caturano, A.; Nevola, R.; Marfella, R.; Sasso, F.C. Non-alcoholic Fatty Liver Disease, Type 2 Diabetes, and Non-viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies. Biomedicines 2023, 11, 468. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.S.; Jun, B.G.; Yi, S.W. Impact of diabetes, obesity, and dyslipidemia on the risk of hepatocellular carcinoma in patients with chronic liver diseases. Clin. Mol. Hepatol. 2022, 28, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Kang, D.; Cao, W.; Wang, Y.; Liu, Z. Diabetes mellitus and risk of hepatocellular carcinoma: A systematic review and meta-analysis. Diabetes Metab. Res. Rev. 2012, 28, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, X.; Gong, G.; Ben, Q.; Qiu, W.; Chen, Y.; Li, G.; Wang, L. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: A systematic review and meta-analysis of cohort studies. Int. J. Cancer 2012, 130, 1639–1648. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Tran, T.; Everhart, J.E. Diabetes Increases the Risk of Chronic Liver Disease and Hepatocellular Carcinoma. Gastroenterology 2004, 126, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Min, W.; Yifei, L.; Xian, Y.; Ligang, Y.; Shaokang, W.; Guiju, S. Associations between diabetes mellitus and the risk of hepatocellular carcinoma in Asian individuals with hepatitis B and C infection: Systematic review and a meta-analysis of cohort studies. Eur. J. Cancer Prev. 2022, 31, 107–116. [Google Scholar]
- Tan, Y.; Wei, S.; Zhang, W.; Yang, J.; Yan, L. Type 2 diabetes mellitus increases the risk of hepatocellular carcinoma in subjects with chronic hepatitis B virus infection: A meta-analysis and systematic review. Cancer Manag. Res. 2019, 11, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Dyal, H.K.; Aguilar, M.; Bartos, G.; Holt, E.W.; Bhuket, T.; Liu, B.; Cheung, R.; Wong, R.J. Diabetes Mellitus Increases the Risk of Hepatocellular Carcinoma in Chronic Hepatitis C Virus Patients: A Systematic Review. Dig. Dis. Sci. 2016, 61, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Tourkochristou, E.; Assimakopoulos, S.F.; Thomopoulos, K.; Marangos, M.; Triantos, C. NAFLD and HBV interplay-related mechanisms underlying liver disease progression. Front. Immunol. 2022, 13, 965548. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Choi, S.; Park, S.M. Association of high body mass index and hepatocellular carcinoma in patients with chronic hepatitis B virus infection: A Korean population-based cohort study. JAMA Oncol. 2018, 4, 737–739. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Splan, M.F.; Weiss, N.S.; McDonald, G.B.; Beretta, L.; Lee, S.P. Incidence and predictors of hepatocellular carcinoma in patients with cirrhosis. Clin. Gastroenterol. Hepatol. 2007, 5, 938–945. [Google Scholar] [CrossRef] [PubMed]
- N’Kontchou, G.; Paries, J.; Htar, M.T.T.; Ganne-Carrie, N.; Costentin, L.; Grando-Lemaire, V.; Trinchet, J.C.; Beaugrand, M. Risk factors for hepatocellular carcinoma in patients with alcoholic or viral C cirrhosis. Clin. Gastroenterol. Hepatol. 2006, 4, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Sandhu, S.; Lai, J.P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular Carcinoma in Cirrhosis: Incidence and Risk Factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cacciapuoti, C. Hepatitis C virus genotypes distribution: An epidemiological up-date in Europe. Infect. Agent. Cancer 2016, 11, 53. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Murata, M.; Yoshida, K.; Sekimoto, G.; Uemura, Y.; Sakaida, N.; Kaibori, M.; Kamiyama, Y.; Nishizawa, M.; Fujisawa, J.; et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007, 46, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Tang, Q.; Chen, M.; Chen, W.; Lu, Y.; Liu, Z.; He, Z. Hepatocellular Senescence: Immunosurveillance and Future Senescence-Induced Therapy in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 589908. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Shi, Y.; Zhang, M.; Goswami, S.; Afridi, S.; Meng, L.; Ma, J.; Chen, Y.; Lin, Y.; Zhang, J.; et al. Global immune characterization of HBV/HCV-related hepatocellular carcinoma identifies macrophage and T-cell subsets associated with disease progression. Cell Discov. 2020, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Shirvani-Dastgerdi, E.; Schwartz, R.E.; Ploss, A. Hepatocarcinogenesis associated with hepatitis B, delta and C viruses. Curr. Opin. Virol. 2016, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.J.; Meng, Z.; Zhou, Y.; Cheng, D.; Ye, H.E.; Zhou, Q.B.; Deng, X.G.; Chen, R.F. Hepatitis C virus core protein regulates OCT4 expression and promotes cell cycle progression in hepatocellular carcinoma. Oncol. Rep. 2016, 36, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.M. Hepatitis C virus-induced activation of β-catenin promotes c-Myc expression and a cascade of pro-carcinogenetic events. Oncogene 2013, 32, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- D’souza, S.; Lau, K.C.K.; Coffin, C.S.; Patel, T.R. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J. Gastroenterol. 2020, 26, 5759–5783. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Tang, Q.B.; Wang, J.; Zhou, L.; Huang, W.L.; Liu, R.Y.; Chen, R.F. Hepatitis C virus core protein induces malignant transformation of biliary epithelial cells by activating nuclear factor-kappa B pathway. J. Gastroenterol. Hepatol. 2010, 25, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Selimovic, D.; El-Khattouti, A.; Ghozlan, H.; Haikel, Y.; Abdelkader, O.; Hassan, M. Hepatitis C virus-related hepatocellular carcinoma: An insight into molecular mechanisms and therapeutic strategies. World J. Hepatol. 2012, 4, 342–355. [Google Scholar] [CrossRef]
- Park, K.J.; Choi, S.H.; Choi, D.; Park, J.M.; Yie, S.W.; Lee, S.Y.; Hwang, S.B. Hepatitis C virus NS5A protein modulates c-Jun N-terminal kinase through interaction with tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 2003, 278, 30711–30718. [Google Scholar] [CrossRef]
- Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; Artman, A.; et al. Global Burden of Disease Liver Cancer Collaboration. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [PubMed]
- Tsukuma, H.; Hiyama, T.; Tanaka, S.; Nakao, M.; Yabuchi, T.; Kitamura, T.; Nakanishi, K.; Fujimoto, I.; Inoue, A.; Yamazaki, H.; et al. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N. Engl. J. Med. 1993, 328, 1797–1801. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Nakata, K.; Omagari, K.; Furukawa, R.; Kusumoto, Y.; Mori, I.; Tajima, H.; Tanioka, H.; Yano, M.; Nagataki, S. Risk of hepatocellular carcinoma in patients with cirrhosis in Japan. Cancer 1994, 74, 2234–2238. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Green, P.; Lowy, E.; Mun, E.J.; Berry, K. Differences in hepatocellular carcinoma risk, predictors and trends over time according to etiology of cirrhosis. PLoS ONE 2018, 13, e0204412. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL recommendations on treatment of hepatitis C: Final update of the series. J. Hepatol. 2020, 73, 1170–1218. [Google Scholar] [CrossRef] [PubMed]
- Calvaruso, V.; Cabibbo, G.; Cacciola, I.; Petta, S.; Madonia, S.; Bellia, A.; Tinè, F.; Distefano, M.; Licata, A.; Giannitrapani, L.; et al. on Behalf of Rete Sicilia Selezione Terapia–HCV (RESIST-HCV) Incidence of Hepatocellular Carcinoma in Patients With HCV-Associated Cirrhosis Treated with Direct-Acting Antiviral Agents. Gastroenterology 2018, 155, 411–421. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated with Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Kanwal, F.; Richardson, P.; Kramer, J. Risk of hepatocellular carcinoma after sustained virological response in Veterans with hepatitis C virus infection. Hepatology 2016, 64, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Morisco, F.; Federico, A.; Marignani, M.; Cannavò, M.; Pontillo, G.; Guarino, M.; Dallio, M.; Pegini, P.; Benigno, R.G.; Lombardo, F.L.; et al. Risk Factors for Liver Decompensation and HCC in HCV-Cirrhotic Patients after DAAs: A Multicenter Prospective Study. Cancers 2021, 13, 3810. [Google Scholar] [CrossRef] [PubMed]
- Kondili, L.A.; Quaranta, M.G.; Cavalletto, L.; Calvaruso, V.; Ferrigno, L.; D’Ambrosio, R.; Simonelli, I.; Brancaccio, G.; Raimondo, G.; Brunetto, M.R.; et al. Profiling the risk of hepatocellular carcinoma after long-term HCV eradication in patients with liver cirrhosis in the PITER cohort. Dig. Liver Dis. 2023, 55, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Asch, S.M.; Cao, Y.; Li, L.; El-Serag, H.B. Long-Term Risk of Hepatocellular Carcinoma in HCV Patients Treated with Direct Acting Antiviral Agents. Hepatology 2020, 71, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Vutien, P.; Kim, N.J.; Moon, A.M.; Johnson, K.M.; Berry, K.; Green, P.K.; Ioannou, G.N. Hepatocellular carcinoma risk decreases as time accrues following hepatitis C virus eradication. Aliment. Pharmacol. Ther. 2024, 59, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Datfar, T.; Doulberis, M.; Papaefthymiou, A.; Hines, I.N.; Manzini, G. Viral Hepatitis and Hepatocellular Carcinoma: State of the Art. Pathogens 2021, 10, 1366. [Google Scholar] [CrossRef] [PubMed]
- Sivasudhan, E.; Blake, N.; Lu, Z.; Meng, J.; Rong, R. Hepatitis B Viral Protein HBx and the Molecular Mechanisms Modulating the Hallmarks of Hepatocellular Carcinoma: A Comprehensive Review. Cells 2022, 11, 741. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, Y.; Feng, X.; Wang, X.; Wu, M.; Gong, L.; Shu, B.; Lu, Q.; Dong, J. HBx acts as an oncogene and promotes the invasion and metastasis of hepatocellular carcinoma both in vivo and vitro. Dig. Liver Dis. 2021, 53, 360–366. [Google Scholar] [CrossRef]
- Kwon, J.A.; Rho, H.M. Transcriptional repression of the human p53 gene by hepatitis B viral core protein (HBc) in human liver cells. Biol. Chem. 2003, 384, 203–212. [Google Scholar] [CrossRef]
- Liu, W.; Guo, T.F.; Jing, Z.T.; Yang, Z.; Liu, L.; Yang, Y.P.; Lin, X.; Tong, Q.Y. Hepatitis B virus core protein promotes hepatocarcinogenesis by enhancing Src expression and activating the Src/PI3K/Akt pathway. FASEB J. 2018, 32, 3033–3046. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.L.K.; Wong, M.L.; Hui, A.Y.; Hung, L.C.T.; Chan, F.K.L.; Sung, J.J.Y. Hepatitis B virus genotype C takes a more aggressive disease course than hepatitis B virus genotype B in hepatitis B e Antigen-positive patients. J. Clin. Microbiol. 2003, 41, 1277–1279. [Google Scholar] [CrossRef]
- Chen, C.J.; Yang, H.I.; Su, J.; Jen, C.L.; You, S.L.; Lu, S.N.; Huang, G.T.; Iloeje, U.H.; REVEAL-HBV Study Group. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006, 295, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Tarocchi, M.; Polvani, S.; Marroncini, G.; Galli, A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J. Gastroenterol. 2014, 20, 11630–11640. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.J.; Yang, Y.W.; You, S.L.; Lai, M.S.; Chen, C.J. Thirty-year outcomes of the national hepatitis B immunization program in Taiwan. JAMA 2013, 310, 974–976. [Google Scholar] [CrossRef] [PubMed]
- Raffetti, E.; Fattovich, G.; Donato, F. Incidence of hepatocellular carcinoma in untreated subjects with chronic hepatitis B: A systematic review and meta-analysis. Liver Int. 2016, 36, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Papatheodoridis, G.V.; Sypsa, V.; Dalekos, G.N.; Yurdaydin, C.; Van Boemmel, F.; Buti, M.; Calleja, J.L.; Chi, H.; Goulis, J.; Manolakopoulos, S.; et al. Hepatocellular carcinoma prediction beyond year 5 of oral therapy in a large cohort of Caucasian patients with chronic hepatitis B. J. Hepatol. 2020, 72, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Kao, J.H. Review article: The prevention of hepatitis B-related hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2018, 48, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Choi, J.; Le, A.; Yip, T.C.; Wong, V.W.; Chan, S.L.; Chan, H.L.; Nguyen, M.H.; Lim, Y.S.; Wong, G.L. Tenofovir disoproxil fumarate reduces hepatocellular carcinoma, decompensation and death in chronic hepatitis B patients with cirrhosis. Aliment. Pharmacol. Ther. 2019, 50, 1037–1048. [Google Scholar] [CrossRef]
- Tseng, C.H.; Tseng, C.M.; Wu, J.L.; Hsu, Y.C.; El-Serag, H.B. Magnitude of and prediction for risk of hepatocellular carcinoma in patients with chronic hepatitis B taking entecavir or tenofovir therapy: A systematic review. J. Gastroenterol. Hepatol. 2020, 35, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, H.J.; Lee, J.; Cho, S.; Ko, M.J.; Lim, Y.S. Risk of Hepatocellular Carcinoma in Patients Treated with Entecavir vs Tenofovir for Chronic Hepatitis B: A Korean Nationwide Cohort Study. JAMA Oncol. 2019, 5, 916–917. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.H.; Li, R.H.; Huo, R.R.; Li, M.J.; Papatheodoridis, G.; Zhong, J.H. Lower risk of hepatocellular carcinoma with tenofovir than entecavir treatment in subsets of chronic hepatitis B patients: An updated meta-analysis. J. Gastroenterol. Hepatol. 2022, 37, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.M.; Yip, T.C.F.; Wong, G.L.H.; Kim, W.R.; Yee, L.J.; Brooks-Rooney, C.; Curteis, T.; Cant, H.; Chen, C.H.; Chen, C.Y.; et al. Hepatocellular carcinoma risk in patients with chronic hepatitis B receiving tenofovir-vs. entecavir-based regimens: Individual patient data meta-analysis. J. Hepatol. 2023, 78, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Pol, S. on the behalf of the ANRS/AFEF study group. Similar 5-year HCC occurrence in Tenofovir- and Entecavir-treated HBV chronic infection in the French AFEF/ANRS CO22 Hepather cohort. Aliment. Pharmacol. Ther. 2021, 53, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Mak, L.Y.; Wong, D.K.H.; Pollicino, T.; Raimondo, G.; Hollinger, F.B.; Yuen, M.F. Occult hepatitis B infection and hepatocellular carcinoma: Epidemiology, virology, hepatocarcinogenesis and clinical significance. J. Hepatol. 2020, 73, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, G.; Locarnini, S.; Pollicino, T.; Levrero, M.; Zoulim, F.; Lok, A.S.; Taormina Workshop on Occult HBV Infection Faculty Members. Update of the statements on biology and clinical impact of occult hepatitis B virus infection. J. Hepatol. 2019, 71, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Squadrito, G.; Cerenzia, G.; Cacciola, I.; Raffa, G.; Craxi, A.; Farinati, F.; Missale, G.; Smedile, A.; Tiribelli, C.; et al. Hepatitis B Virus Maintains Its Pro-oncogenic Properties in the Case of Occult HBV Infection. Gastroenterology 2004, 126, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kobayashi, M.; Someya, T.; Saitoh, S.; Hosaka, T.; Akuta, N.; Suzuki, F.; Suzuki, Y.; Arase, Y.; Kumada, H. Occult hepatitis B virus infection increases hepatocellular carcinogenesis by eight times in patients with non-B, non-C liver cirrhosis: A cohort study. J. Viral Hepat. 2009, 16, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Sohn, W.; Chang, Y.; Cho, Y.K.; Hong, Y.S.; Ryu, S. Isolated Hepatitis B Core Antibody Positivity and Long-Term Liver-Related Mortality in Korea: A Cohort Study. Am. J. Gastroenterol. 2023, 118, 95–104. [Google Scholar] [CrossRef]
- Kamal, H.; Fornes, R.; Simin, J.; Stål, P.; Duberg, A.S.; Brusselaers, N.; Aleman, S. Risk of hepatocellular carcinoma in hepatitis B and D virus co-infected patients: A systematic review and meta-analysis of longitudinal studies. J. Viral Hepat. 2021, 28, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Farci, P.; Niro, G.A. Clinical features of hepatitis D. Semin. Liver Dis. 2012, 32, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Puigvehí, M.; Moctezuma-Velázquez, C.; Villanueva, A.; Llovet, J.M. The Oncogenic Role of Hepatitis Delta Virus in Hepatocellular Carcinoma. JHEP Rep. 2019, 1, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Jeong, S.H.; Hwang, S.B. Large Hepatitis Delta Antigen Modulates Transforming Growth Factor-b Signaling Cascades: Implication of Hepatitis Delta Virus–Induced Liver Fibrosis. Gastroenterology 2007, 132, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Du, D.; Zheng, W.; Liao, M.; Zhang, L.; Liang, G.; Gong, M. Small Hepatitis Delta Antigen Selectively Binds to Target mRNA in Hepatic Cells: A Potential Mechanism by Which Hepatitis D Virus Downregulates Glutathione S-Transferase P1 and Induces Liver Injury and Hepatocarcinogenesis. Biochem. Cell Biol. 2019, 97, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Diaz, G.; Engle, R.E.; Tice, A.; Melis, M.; Montenegro, S.; Rodriguez-Canales, J.; Hanson, J.; Emmert-Buck, M.R.; Bock, K.W.; Moore, I.N.; et al. Molecular Signature and Mechanisms of Hepatitis D Virus-Associated Hepatocellular Carcinoma. Mol. Cancer Res. 2018, 16, 1406–1419. [Google Scholar] [CrossRef] [PubMed]
- Stockdale, A.J.; Kreuels, B.; Henrion, M.Y.R.; Giorgi, E.; Kyomuhangi, I.; de Martel, C.; Hutin, Y.; Geretti, A.M. The global prevalence of hepatitis D virus infection: Systematic review and meta-analysis. J. Hepatol. 2020, 73, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Niro, G.A.; Smedile, A.; Ippolito, A.M.; Ciancio, A.; Fontana, R.; Olivero, A.; Valvano, M.R.; Abate, M.L.; Gioffreda, D.; Caviglia, G.P.; et al. Outcome of chronic delta hepatitis in Italy: A long-term cohort study. J. Hepatol. 2010, 53, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Romeo, R.; Del Ninno, E.; Rumi, M.; Russo, A.; Sangiovanni, A.; de Franchis, R.; Ronchi, G.; Colombo, M. A 28-year study of the course of hepatitis Delta infection: A risk factor for cirrhosis and hepatocellular carcinoma. Gastroenterology 2009, 136, 1629–1638. [Google Scholar] [CrossRef]
- Fattovich, G.; Giustina, G.; Christensen, E.; Pantalena, M.; Zagni, I.; Realdi, G.; Schalm, S.W. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European Concerted Action on Viral Hepatitis (Eurohep). Gut 2000, 46, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Alfaiate, D.; Clément, S.; Gomes, D.; Goossens, N.; Negro, F. Chronic hepatitis D and hepatocellular carcinoma: A systematic review and meta-analysis of observational studies. J. Hepatol. 2020, 73, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, G.; Fasano, M.; Grossi, A.; Santantonio, T.A.; Gaeta, G.B. Clinical outcomes in patients with hepatitis D, cirrhosis and persistent hepatitis B virus replication, and receiving long-term tenofovir or entecavir. Aliment. Pharmacol. Ther. 2019, 49, 1071–1076. [Google Scholar] [CrossRef] [PubMed]


| Author, Journal | Country | Methodology | Patients with Cirrhosis | Findings |
|---|---|---|---|---|
| Calvaruso V, Gastroenterology 2018 [46] | Italy | Prospective | 2249 patients (100%) | HCC occurrence: 3% in SVR vs. 12.8% in non-SVR (p < 0.001) HCC overall cumulative rate at 1 year: 2.9% in SVR vs. 8% in non-SVR |
| Kanwal F, Gastroenterology 2017 [47] | USA | Retrospective | 8766 patients (39%) | HCC incidence: 0.9 per 100 p/y in SVR vs. 3.45 per 100 p/y in non-SVR |
| El-Serag HB, Hepatology 2016 [48] | USA | Retrospective | 1548 patients (14.4%) | HCC incidence: 0.93 per 100 p/y in SVR vs. 3.27 per 100 p/y in non-SVR |
| Morisco F, Cancers 2021 [49] | Italy | Prospective | 706 patients (100%) | Liver-related events: 8.9% in SVR vs. 26.3% in non-SVR HCC incidence in SVR: 1.6 per 100 p/y |
| Kondili L, DLD 2023 [50] | Italy | Retrospective | 2064 patients (100%) | HCC incidence in SVR: 2.45 per 100 p/y |
| Kanwal F, Hepatology 2020 [51] | USA | Retrospective | 6938 patients (38.4%) | HCC incidence in SVR: 1.23 per 100 p/y |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capasso, M.; Cossiga, V.; Guarino, M.; Ranieri, L.; Morisco, F. The Role of Hepatitis Viruses as Drivers of Hepatocancerogenesis. Cancers 2024, 16, 1505. https://doi.org/10.3390/cancers16081505
Capasso M, Cossiga V, Guarino M, Ranieri L, Morisco F. The Role of Hepatitis Viruses as Drivers of Hepatocancerogenesis. Cancers. 2024; 16(8):1505. https://doi.org/10.3390/cancers16081505
Chicago/Turabian StyleCapasso, Mario, Valentina Cossiga, Maria Guarino, Luisa Ranieri, and Filomena Morisco. 2024. "The Role of Hepatitis Viruses as Drivers of Hepatocancerogenesis" Cancers 16, no. 8: 1505. https://doi.org/10.3390/cancers16081505