
Advancing Immunotherapy in Pancreatic Cancer: A Brief Review of Emerging Adoptive Cell Therapies


Simple Summary
Abstract

1. Introduction
2. Current Management of Pancreatic Ductal Adenocarcinoma
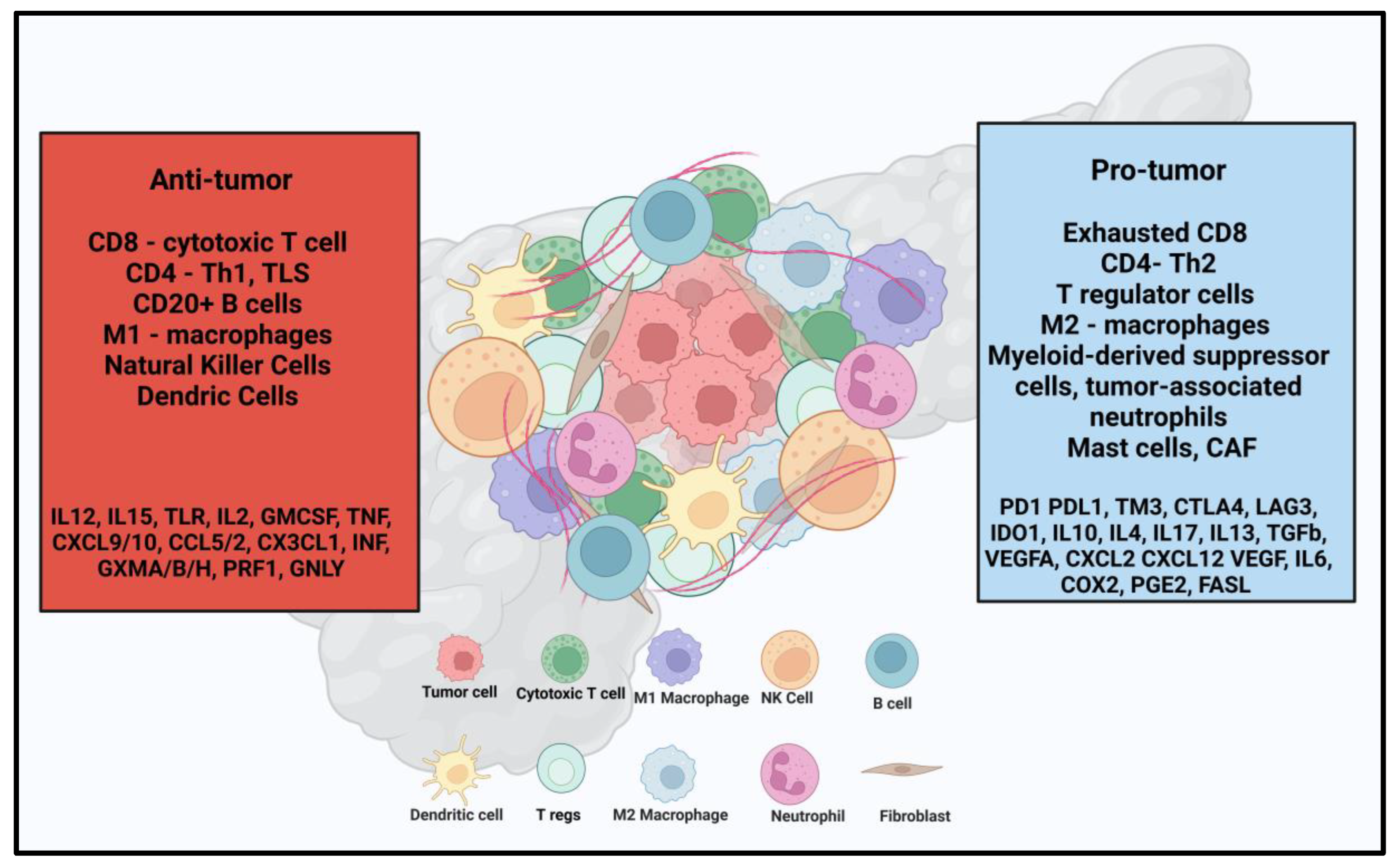
3. Immunotherapy in PDAC
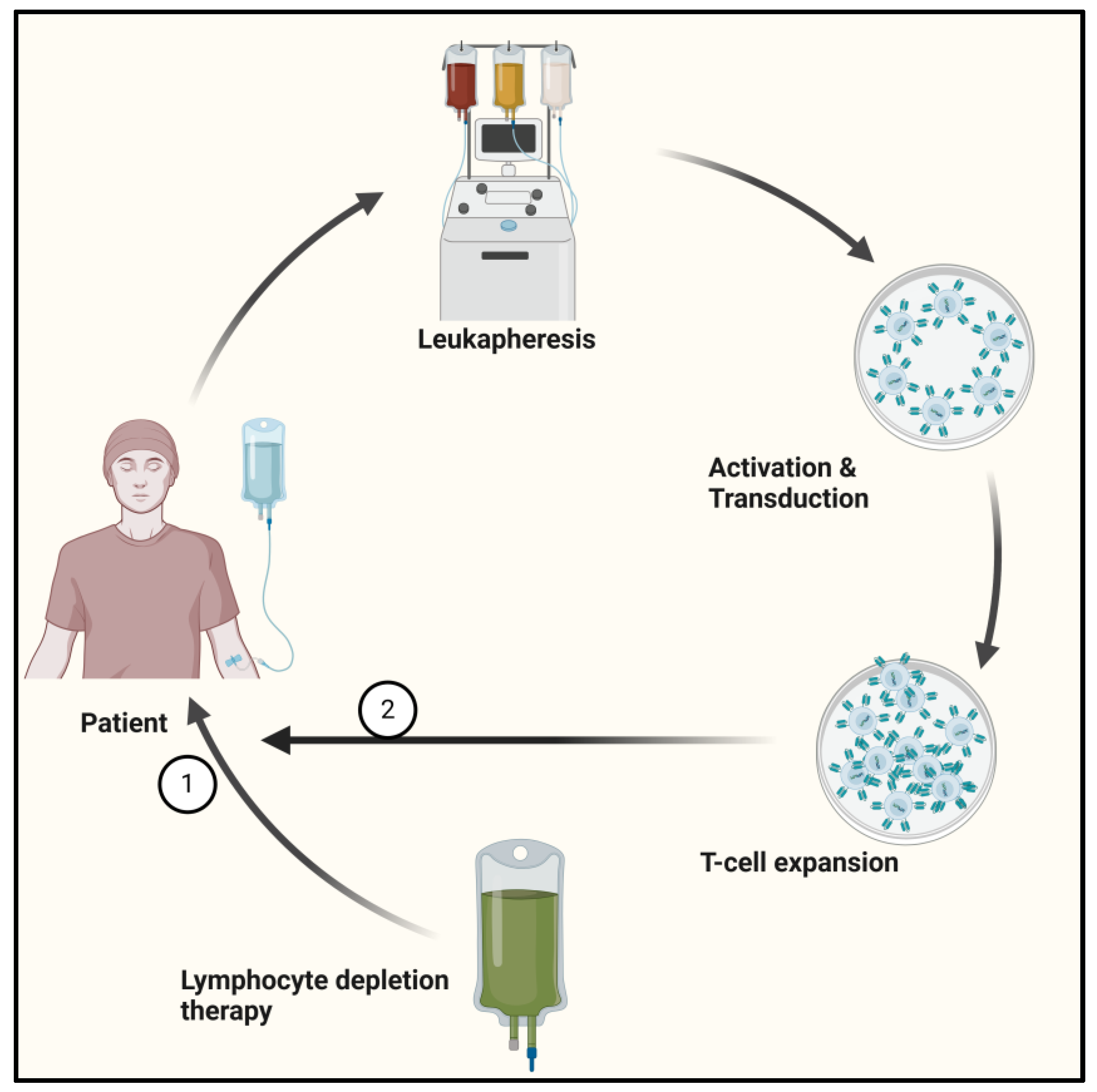
3.1. Adoptive Cell Therapy in PDAC
3.1.1. CAR-T in PDAC
3.1.2. Tumor-Infiltrating Lymphocyte Therapy (TIL) in PDAC
3.1.3. CAR-NK Cell Therapy
3.1.4. TCR-Engineered T-Cell Therapy
3.1.5. Cytokine-Induced Killer (CIK) Cells
4. Discussion
4.1. Challenges in ACT
4.2. Future Directions in ACT
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology 2022, 163, 386–402.e381. [Google Scholar] [CrossRef] [PubMed]
- Ilic, I.; Ilic, M. International patterns in incidence and mortality trends of pancreatic cancer in the last three decades: A joinpoint regression analysis. World J. Gastroenterol. 2022, 28, 4698–4715. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.X.; Zhao, C.F.; Chen, W.B.; Liu, Q.C.; Li, Q.W.; Lin, Y.Y.; Gao, F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Roalsø, M.; Aunan, J.R.; Søreide, K. Refined TNM-staging for pancreatic adenocarcinoma—Real progress or much ado about nothing? Eur. J. Surg. Oncol. 2020, 46, 1554–1557. [Google Scholar] [CrossRef]
- Al-Hawary, M.M.; Francis, I.R.; Chari, S.T.; Fishman, E.K.; Hough, D.M.; Lu, D.S.; Macari, M.; Megibow, A.J.; Miller, F.H.; Mortele, K.J.; et al. Pancreatic ductal adenocarcinoma radiology reporting template: Consensus statement of the Society of Abdominal Radiology and the American Pancreatic Association. Radiology 2014, 270, 248–260. [Google Scholar] [CrossRef]
- Evans, D.B. What Makes a Pancreatic Cancer Resectable? Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 300–305. [Google Scholar] [CrossRef]
- Vernuccio, F.; Messina, C.; Merz, V.; Cannella, R.; Midiri, M. Resectable and Borderline Resectable Pancreatic Ductal Adenocarcinoma: Role of the Radiologist and Oncologist in the Era of Precision Medicine. Diagnostics 2021, 11, 2166. [Google Scholar] [CrossRef]
- Isaji, S.; Mizuno, S.; Windsor, J.A.; Bassi, C.; Fernández-Del Castillo, C.; Hackert, T.; Hayasaki, A.; Katz, M.H.G.; Kim, S.W.; Kishiwada, M.; et al. International consensus on definition and criteria of borderline resectable pancreatic ductal adenocarcinoma 2017. Pancreatology 2018, 18, 2–11. [Google Scholar] [CrossRef]
- Barcellini, A.; Peloso, A.; Pugliese, L.; Vitolo, V.; Cobianchi, L. Locally Advanced Pancreatic Ductal Adenocarcinoma: Challenges and Progress. OncoTargets Ther. 2020, 13, 12705–12720. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Melisi, D.; Macarulla, T.; Pazo-Cid, R.; Chandana, S.R.; De La Fouchardiere, C.; Dean, A.P.; Kiss, I.; Lee, W.; Goetze, T.O.; et al. NAPOLI-3: A randomized, open-label phase 3 study of liposomal irinotecan + 5-fluorouracil/leucovorin + oxaliplatin (NALIRIFOX) versus nab-paclitaxel + gemcitabine in treatment-naïve patients with metastatic pancreatic ductal adenocarcinoma (mPDAC). J. Clin. Oncol. 2023, 41, LBA661. [Google Scholar] [CrossRef]
- Dayyani, F.; Macarulla, T.; Johnson, A.; Wainberg, Z.A. Second-line treatment options for patients with metastatic pancreatic ductal adenocarcinoma: A systematic literature review. Cancer Treat. Rev. 2023, 113, 102502. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Dillman, R.O. Cancer immunotherapy. Cancer Biother. Radiopharm. 2011, 26, 1–64. [Google Scholar] [CrossRef]
- Nasar, N.; Eikenboom, E.; Seier, K.; Gonen, M.; Wagner, A.; Jarnagin, W.R.; Drebin, J.A.; D’Angelica, M.I.; Kingham, T.P.; Balachandran, V.P.; et al. Survival of patients with microsatellite instability-high and Lynch syndrome-associated pancreatic ductal adenocarcinomas. J. Clin. Oncol. 2024, 42, 640. [Google Scholar] [CrossRef]
- Velcheti, V.; Schalper, K. Basic Overview of Current Immunotherapy Approaches in Cancer. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 298–308. [Google Scholar] [CrossRef]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef]
- Sangro, B.; Chan, S.L.; Kelley, R.K.; Lau, G.; Kudo, M.; Sukeepaisarnjaroen, W.; Yarchoan, M.; De Toni, E.N.; Furuse, J.; Kang, Y.K.; et al. Four-year overall survival update from the phase III HIMALAYA study of tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. Ann. Oncol. 2024, 35, 448–457. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.V.; Merle, P.; et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). J. Clin. Oncol. 2021, 39, 267. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Ruth He, A.; Qin, S.; Chen, L.-T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Ah Lee, M.; Kitano, M.; et al. Durvalumab plus Gemcitabine and Cisplatin in Advanced Biliary Tract Cancer. N. Engl. J. Med. Evid. 2022, 1, EVIDoa2200015. [Google Scholar] [CrossRef] [PubMed]
- Farhangnia, P.; Khorramdelazad, H.; Nickho, H.; Delbandi, A.-A. Current and future immunotherapeutic approaches in pancreatic cancer treatment. J. Hematol. Oncol. 2024, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.; Amin, A.; Kaye, F.J.; Morse, M.A.; Taylor, M.H.; Peltola, K.J.; Sharma, P.; O’Reilly, E.M.; Meadows Shropshire, S.; O’Brien, S.; et al. Nivolumab monotherapy or combination with ipilimumab with or without cobimetinib in previously treated patients with pancreatic adenocarcinoma (CheckMate 032). J. Immunother. Cancer 2024, 12, e007883. [Google Scholar] [CrossRef]
- Renouf, D.J.; Loree, J.M.; Knox, J.J.; Topham, J.T.; Kavan, P.; Jonker, D.; Welch, S.; Couture, F.; Lemay, F.; Tehfe, M.; et al. The CCTG PA.7 phase II trial of gemcitabine and nab-paclitaxel with or without durvalumab and tremelimumab as initial therapy in metastatic pancreatic ductal adenocarcinoma. Nat. Commun. 2022, 13, 5020. [Google Scholar] [CrossRef]
- Padrón, L.J.; Maurer, D.M.; O’Hara, M.H.; O’Reilly, E.M.; Wolff, R.A.; Wainberg, Z.A.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: Clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat. Med. 2022, 28, 1167–1177. [Google Scholar] [CrossRef]
- Bockorny, B.; Macarulla, T.; Semenisty, V.; Borazanci, E.; Feliu, J.; Ponz-Sarvise, M.; Abad, D.G.; Oberstein, P.; Alistar, A.; Muñoz, A.; et al. Motixafortide and Pembrolizumab Combined to Nanoliposomal Irinotecan, Fluorouracil, and Folinic Acid in Metastatic Pancreatic Cancer: The COMBAT/KEYNOTE-202 Trial. Clin. Cancer Res. 2021, 27, 5020–5027. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.-Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.-C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Haldar, S.D.; Huff, A.; Diwan, E.A.; Ferguson, A.; Judkins, C.; Lu, J.; Wang, H.; Sinan, H.; Thoburn, C.; Bever, K.M.; et al. Abstract CT022: Mutant KRAS peptide-based vaccine in patients at high risk of developing pancreatic cancer: Preliminary analysis from a phase I study. Cancer Res. 2024, 84, CT022. [Google Scholar] [CrossRef]
- Hanahan, D.; Michielin, O.; Pittet, M.J. Convergent inducers and effectors of T cell paralysis in the tumour microenvironment. Nat. Rev. Cancer 2025, 25, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Liu, J.; Qian, H.; Zhuang, Q. Cancer-associated fibroblasts: From basic science to anticancer therapy. Exp. Mol. Med. 2023, 55, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, J.; Jacobs, K.A.; Rizzollo, F.; Lodi, F.; Hua, Y.; Poźniak, J.; Narayanan Srinivasan, A.; Houbaert, D.; Shankar, G.; More, S.; et al. Tumor endothelial cell autophagy is a key vascular-immune checkpoint in melanoma. EMBO Mol. Med. 2023, 15, e18028. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Andreeva, E.R.; Eremin, I.I.; Markin, A.M.; Nadelyaeva, I.I.; Orekhov, A.N.; Melnichenko, A.A. The Role of Pericytes in Regulation of Innate and Adaptive Immunity. Biomedicines 2023, 11, 600. [Google Scholar] [CrossRef]
- Miksch, R.C.; Schoenberg, M.B.; Weniger, M.; Bösch, F.; Ormanns, S.; Mayer, B.; Werner, J.; Bazhin, A.V.; D’Haese, J.G. Prognostic Impact of Tumor-Infiltrating Lymphocytes and Neutrophils on Survival of Patients with Upfront Resection of Pancreatic Cancer. Cancers 2019, 11, 39. [Google Scholar] [CrossRef]
- Panahi, M.; Rezagholizadeh, F.; Mollazadehghomi, S.; Farhangnia, P.; Niya, M.H.K.; Ajdarkosh, H.; Tameshkel, F.S.; Heshmati, S.M. The association between CD3+ and CD8+ tumor-infiltrating lymphocytes (TILs) and prognosis in patients with pancreatic adenocarcinoma. Cancer Treat. Res. Commun. 2023, 35, 100699. [Google Scholar] [CrossRef]
- Orhan, A.; Vogelsang, R.P.; Andersen, M.B.; Madsen, M.T.; Hölmich, E.R.; Raskov, H.; Gögenur, I. The prognostic value of tumour-infiltrating lymphocytes in pancreatic cancer: A systematic review and meta-analysis. Eur. J. Cancer 2020, 132, 71–84. [Google Scholar] [CrossRef]
- Guo, J.; Wu, M.; Guo, L.; Zuo, Q. Pretreatment blood neutrophil/lymphocyte ratio is associated with metastasis and predicts survival in patients with pancreatic cancer. Bull. Cancer 2018, 105, 146–154. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Guo, J.; Wang, S.; Gao, Q. An integrated overview of the immunosuppression features in the tumor microenvironment of pancreatic cancer. Front. Immunol. 2023, 14, 1258538. [Google Scholar] [CrossRef]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Tumor Immunology and Tumor Evolution: Intertwined Histories. Immunity 2020, 52, 55–81. [Google Scholar] [CrossRef] [PubMed]
- Hartupee, C.; Nagalo, B.M.; Chabu, C.Y.; Tesfay, M.Z.; Coleman-Barnett, J.; West, J.T.; Moaven, O. Pancreatic cancer tumor microenvironment is a major therapeutic barrier and target. Front. Immunol. 2024, 15, 1287459. [Google Scholar] [CrossRef]
- Kirtane, K.; Elmariah, H.; Chung, C.H.; Abate-Daga, D. Adoptive cellular therapy in solid tumor malignancies: Review of the literature and challenges ahead. J. Immunother. Cancer 2021, 9, e002723. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ’Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- DeSelm, C.J.; Tano, Z.E.; Varghese, A.M.; Adusumilli, P.S. CAR T-cell therapy for pancreatic cancer. J. Surg. Oncol. 2017, 116, 63–74. [Google Scholar] [CrossRef]
- Asmamaw Dejenie, T.; Tiruneh G/Medhin, M.; Dessie Terefe, G.; Tadele Admasu, F.; Wale Tesega, W.; Chekol Abebe, E. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum. Vaccines Immunother. 2022, 18, 2114254. [Google Scholar] [CrossRef]
- Papathanasiou, M.M.; Stamatis, C.; Lakelin, M.; Farid, S.; Titchener-Hooker, N.; Shah, N. Autologous CAR T-cell therapies supply chain: Challenges and opportunities? Cancer Gene Ther. 2020, 27, 799–809. [Google Scholar] [CrossRef]
- Watanabe, N.; Mo, F.; McKenna, M.K. Impact of Manufacturing Procedures on CAR T Cell Functionality. Front. Immunol. 2022, 13, 876339. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Miyazaki, Y.; Tsukasa, K.; Matsubara, S.; Yoshimitsu, M.; Takao, S. CD133 facilitates epithelial-mesenchymal transition through interaction with the ERK pathway in pancreatic cancer metastasis. Mol. Cancer 2014, 13, 15. [Google Scholar] [CrossRef]
- Maeda, S.; Shinchi, H.; Kurahara, H.; Mataki, Y.; Maemura, K.; Sato, M.; Natsugoe, S.; Aikou, T.; Takao, S. CD133 expression is correlated with lymph node metastasis and vascular endothelial growth factor-C expression in pancreatic cancer. Br. J. Cancer 2008, 98, 1389–1397. [Google Scholar] [CrossRef]
- Katz, S.C.; Moody, A.E.; Guha, P.; Hardaway, J.C.; Prince, E.; LaPorte, J.; Stancu, M.; Slansky, J.E.; Jordan, K.R.; Schulick, R.D.; et al. HITM-SURE: Hepatic immunotherapy for metastases phase Ib anti-CEA CAR-T study utilizing pressure enabled drug delivery. J. Immunother. Cancer 2020, 8, e001097. [Google Scholar] [CrossRef]
- Meng, Q.; Shi, S.; Liang, C.; Liang, D.; Xu, W.; Ji, S.; Zhang, B.; Ni, Q.; Xu, J.; Yu, X. Diagnostic and prognostic value of carcinoembryonic antigen in pancreatic cancer: A systematic review and meta-analysis. OncoTargets Ther. 2017, 10, 4591–4598. [Google Scholar] [CrossRef]
- Hall, C.; Clarke, L.; Pal, A.; Buchwald, P.; Eglinton, T.; Wakeman, C.; Frizelle, F. A Review of the Role of Carcinoembryonic Antigen in Clinical Practice. Ann. Coloproctol. 2019, 35, 294–305. [Google Scholar] [CrossRef]
- van Manen, L.; Groen, J.V.; Putter, H.; Vahrmeijer, A.L.; Swijnenburg, R.-J.; Bonsing, B.A.; Mieog, J.S.D. Elevated CEA and CA19-9 serum levels independently predict advanced pancreatic cancer at diagnosis. Biomarkers 2020, 25, 186–193. [Google Scholar] [CrossRef]
- Kato, H.; Kishiwada, M.; Hayasaki, A.; Chipaila, J.; Maeda, K.; Noguchi, D.; Gyoten, K.; Fujii, T.; Iizawa, Y.; Tanemura, A.; et al. Role of Serum Carcinoma Embryonic Antigen (CEA) Level in Localized Pancreatic Adenocarcinoma: CEA Level Before Operation is a Significant Prognostic Indicator in Patients With Locally Advanced Pancreatic Cancer Treated With Neoadjuvant Therapy Followed by Surgical Resection: A Retrospective Analysis. Ann. Surg. 2022, 275, e698–e707. [Google Scholar] [CrossRef]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3, e99573. [Google Scholar] [CrossRef]
- Hassan, R.; Thomas, A.; Alewine, C.; Le, D.T.; Jaffee, E.M.; Pastan, I. Mesothelin Immunotherapy for Cancer: Ready for Prime Time? J. Clin. Oncol. 2016, 34, 4171–4179. [Google Scholar] [CrossRef] [PubMed]
- Klampatsa, A.; Dimou, V.; Albelda, S.M. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert Opin. Biol. Ther. 2021, 21, 473–486. [Google Scholar] [CrossRef]
- Johnston, F.M.; Tan, M.C.B.; Tan, B.R., Jr.; Porembka, M.R.; Brunt, E.M.; Linehan, D.C.; Simon, P.O., Jr.; Plambeck-Suess, S.; Eberlein, T.J.; Hellstrom, K.E.; et al. Circulating Mesothelin Protein and Cellular Antimesothelin Immunity in Patients with Pancreatic Cancer. Clin. Cancer Res. 2009, 15, 6511–6518. [Google Scholar] [CrossRef]
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef]
- QI, C.; Liu, C.; Gong, J.; Li, J.; Liu, D.; Wang, X.; Zhang, P.; Qin, Y.; Zhang, M.; Peng, Z.; et al. Claudin18.2-targeted chimeric antigen receptor T cell-therapy for patients with gastrointestinal cancers: Final results of CT041-CG4006 phase 1 trial. J. Clin. Oncol. 2024, 42, 2501. [Google Scholar] [CrossRef]
- Qin, S.; Tian, W.; Li, M.; Wei, H.; Sun, L.; Xie, Q.; Lin, E.; Xu, D.; Tian, J.; Chen, J.; et al. 1054P A phase Ia study to evaluate the safety, tolerability, pharmacokinetics and preliminary efficacy of a modular CLDN18.2-targeting PG CAR-T therapy (IBI345) in patients with CLDN18.2+ solid tumors. Ann. Oncol. 2023, 34, S638. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, Y.; Wu, Z.; Feng, K.; Tong, C.; Wang, Y.; Dai, H.; Shi, F.; Yang, Q.; Han, W. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy 2020, 22, 573–580. [Google Scholar] [CrossRef]
- Fitzgerald, T.L.; Lertpiriyapong, K.; Cocco, L.; Martelli, A.M.; Libra, M.; Candido, S.; Montalto, G.; Cervello, M.; Steelman, L.; Abrams, S.L.; et al. Roles of EGFR and KRAS and their downstream signaling pathways in pancreatic cancer and pancreatic cancer stem cells. Adv. Biol. Regul. 2015, 59, 65–81. [Google Scholar] [CrossRef]
- Oliveira-Cunha, M.; Newman, W.G.; Siriwardena, A.K. Epidermal Growth Factor Receptor in Pancreatic Cancer. Cancers 2011, 3, 1513–1526. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2018, 9, 838–847. [Google Scholar] [CrossRef]
- Kumar, R.; Yarmand-Bagheri, R. The role of HER2 in angiogenesis. Semin. Oncol. 2001, 28, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Schmidts, A.; Wehrli, M.; Maus, M.V. Toward Better Understanding and Management of CAR-T Cell–Associated Toxicity. Annu. Rev. Med. 2021, 72, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Deng, J.; Rao, S.; Guo, S.; Shen, J.; Du, F.; Wu, X.; Chen, Y.; Li, M.; Chen, M.; et al. Tumor Infiltrating Lymphocyte (TIL) Therapy for Solid Tumor Treatment: Progressions and Challenges. Cancers 2022, 14, 4160. [Google Scholar] [CrossRef] [PubMed]
- Betof Warner, A.; Corrie, P.G.; Hamid, O. Tumor-Infiltrating Lymphocyte Therapy in Melanoma: Facts to the Future. Clin. Cancer Res. 2023, 29, 1835–1854. [Google Scholar] [CrossRef]
- Kazemi, M.H.; Sadri, M.; Najafi, A.; Rahimi, A.; Baghernejadan, Z.; Khorramdelazad, H.; Falak, R. Tumor-infiltrating lymphocytes for treatment of solid tumors: It takes two to tango? Front. Immunol. 2022, 13, 1018962. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of Patients With Metastatic Melanoma With Autologous Tumor-Infiltrating Lymphocytes and Interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Dafni, U.; Michielin, O.; Lluesma, S.M.; Tsourti, Z.; Polydoropoulou, V.; Karlis, D.; Besser, M.J.; Haanen, J.; Svane, I.M.; Ohashi, P.S.; et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: A systematic review and meta-analysis. Ann. Oncol. 2019, 30, 1902–1913. [Google Scholar] [CrossRef]
- Ellebaek, E.; Iversen, T.Z.; Junker, N.; Donia, M.; Engell-Noerregaard, L.; Met, Ö.; Hölmich, L.R.; Andersen, R.S.; Hadrup, S.R.; Andersen, M.H.; et al. Adoptive cell therapy with autologous tumor infiltrating lymphocytes and low-dose Interleukin-2 in metastatic melanoma patients. J. Transl. Med. 2012, 10, 169. [Google Scholar] [CrossRef]
- Ben-Avi, R.; Farhi, R.; Ben-Nun, A.; Gorodner, M.; Greenberg, E.; Markel, G.; Schachter, J.; Itzhaki, O.; Besser, M.J. Establishment of adoptive cell therapy with tumor infiltrating lymphocytes for non-small cell lung cancer patients. Cancer Immunol. Immunother. 2018, 67, 1221–1230. [Google Scholar] [CrossRef]
- Kradin, R.L.; Boyle, L.A.; Preffer, F.I.; Callahan, R.J.; Barlai-Kovach, M.; Strauss, H.W.; Dubinett, S.; Kurnick, J.T. Tumor-derived interleukin-2-dependent lymphocytes in adoptive immunotherapy of lung cancer. Cancer Immunol. Immunother. 1987, 24, 76–85. [Google Scholar] [CrossRef]
- Freedman, R.S.; Kudelka, A.P.; Kavanagh, J.J.; Verschraegen, C.; Edwards, C.L.; Nash, M.; Levy, L.; Atkinson, E.N.; Zhang, H.Z.; Melichar, B.; et al. Clinical and biological effects of intraperitoneal injections of recombinant interferon-gamma and recombinant interleukin 2 with or without tumor-infiltrating lymphocytes in patients with ovarian or peritoneal carcinoma. Clin. Cancer Res. 2000, 6, 2268–2278. [Google Scholar] [PubMed]
- Fujita, K.; Ikarashi, H.; Takakuwa, K.; Kodama, S.; Tokunaga, A.; Takahashi, T.; Tanaka, K. Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin. Cancer Res. 1995, 1, 501–507. [Google Scholar] [PubMed]
- Pedersen, M.; Westergaard, M.C.W.; Milne, K.; Nielsen, M.; Borch, T.H.; Poulsen, L.G.; Hendel, H.W.; Kennedy, M.; Briggs, G.; Ledoux, S.; et al. Adoptive cell therapy with tumor-infiltrating lymphocytes in patients with metastatic ovarian cancer: A pilot study. Oncoimmunology 2018, 7, e1502905. [Google Scholar] [CrossRef] [PubMed]
- Kverneland, A.H.; Pedersen, M.; Westergaard, M.C.W.; Nielsen, M.; Borch, T.H.; Olsen, L.R.; Aasbjerg, G.; Santegoets, S.J.; van der Burg, S.H.; Milne, K.; et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget 2020, 11, 2092–2105. [Google Scholar] [CrossRef]
- O’Malley, D.; Lee, S.; Psyrri, A.; Sukari, A.; Thomas, S.; Wenham, R.; Gogas, H.; Jazaeri, A.; Monk, B.; Rose, P.; et al. 492 Phase 2 efficacy and safety of autologous tumor-infiltrating lymphocyte (TIL) cell therapy in combination with pembrolizumab in immune checkpoint inhibitor-naïve patients with advanced cancers. J. ImmunoTherapy Cancer 2021, 9, A523–A524. [Google Scholar] [CrossRef]
- Li, J.; Chen, Q.Y.; He, J.; Li, Z.L.; Tang, X.F.; Chen, S.P.; Xie, C.M.; Li, Y.Q.; Huang, L.X.; Ye, S.B.; et al. Phase I trial of adoptively transferred tumor-infiltrating lymphocyte immunotherapy following concurrent chemoradiotherapy in patients with locoregionally advanced nasopharyngeal carcinoma. Oncoimmunology 2015, 4, e976507. [Google Scholar] [CrossRef]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef]
- Amaria, R.; Knisely, A.; Vining, D.; Kopetz, S.; Overman, M.J.; Javle, M.; Antonoff, M.B.; Tzeng, C.D.; Wolff, R.A.; Pant, S.; et al. Efficacy and safety of autologous tumor-infiltrating lymphocytes in recurrent or refractory ovarian cancer, colorectal cancer, and pancreatic ductal adenocarcinoma. J. Immunother. Cancer 2024, 12, e006822. [Google Scholar] [CrossRef]
- Granhøj, J.S.; Witness Præst Jensen, A.; Presti, M.; Met, Ö.; Svane, I.M.; Donia, M. Tumor-infiltrating lymphocytes for adoptive cell therapy: Recent advances, challenges, and future directions. Expert Opin. Biol. Ther. 2022, 22, 627–641. [Google Scholar] [CrossRef]
- Betof Warner, A.; Hamid, O.; Komanduri, K.; Amaria, R.; Butler, M.O.; Haanen, J.; Nikiforow, S.; Puzanov, I.; Sarnaik, A.; Bishop, M.R.; et al. Expert consensus guidelines on management and best practices for tumor-infiltrating lymphocyte cell therapy. J. ImmunoTherapy Cancer 2024, 12, e008735. [Google Scholar] [CrossRef]
- Froelich, W. CAR NK Cell Therapy Directed Against Pancreatic Cancer. Oncol. Times 2021, 43, 46. [Google Scholar] [CrossRef]
- Fang, F.; Xie, S.; Chen, M.; Li, Y.; Yue, J.; Ma, J.; Shu, X.; He, Y.; Xiao, W.; Tian, Z. Advances in NK cell production. Cell. Mol. Immunol. 2022, 19, 460–481. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. eBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Teng, K.Y.; Mansour, A.G.; Zhu, Z.; Li, Z.; Tian, L.; Ma, S.; Xu, B.; Lu, T.; Chen, H.; Hou, D.; et al. Off-the-Shelf Prostate Stem Cell Antigen-Directed Chimeric Antigen Receptor Natural Killer Cell Therapy to Treat Pancreatic Cancer. Gastroenterology 2022, 162, 1319–1333. [Google Scholar] [CrossRef] [PubMed]
- Da, Y.; Liu, Y.; Hu, Y.; Liu, W.; Ma, J.; Lu, N.; Zhang, C.; Zhang, C. STING agonist cGAMP enhances anti-tumor activity of CAR-NK cells against pancreatic cancer. Oncoimmunology 2022, 11, 2054105. [Google Scholar] [CrossRef]
- Pan, K.; Farrukh, H.; Chittepu, V.; Xu, H.; Pan, C.X.; Zhu, Z. CAR race to cancer immunotherapy: From CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 2022, 41, 119. [Google Scholar] [CrossRef]
- Wang, K.; Wang, L.; Wang, Y.; Xiao, L.; Wei, J.; Hu, Y.; Wang, D.; Huang, H. Reprogramming natural killer cells for cancer therapy. Mol. Ther. 2024, 32, 2835–2855. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Harrison, C. TCR cell therapies vanquish solid tumors—Finally. Nat. Biotechnol. 2024, 42, 1477–1479. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, T.; Li, P.; Gai, J.; Chen, S.; Espinoza, G.; Kung, H.-C.; Zhang, R.; Fujiwara, K.; Fu, J.; et al. Engineered TCR T-cell therapy targeting mass spectrometry-identified natural epitope in PDAC. Cancer Lett. 2023, 573, 216366. [Google Scholar] [CrossRef]
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer Therapy With TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef] [PubMed]
- Hiltensperger, M.; Krackhardt, A.M. Current and future concepts for the generation and application of genetically engineered CAR-T and TCR-T cells. Front. Immunol. 2023, 14, 1121030. [Google Scholar] [CrossRef] [PubMed]
- Gerdemann, U.; Katari, U.; Christin, A.S.; Cruz, C.R.; Tripic, T.; Rousseau, A.; Gottschalk, S.M.; Savoldo, B.; Vera, J.F.; Heslop, H.E.; et al. Cytotoxic T Lymphocytes Simultaneously Targeting Multiple Tumor-associated Antigens to Treat EBV Negative Lymphoma. Mol. Ther. 2011, 19, 2258–2268. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Yossef, R.; Cafri, G.; Paria, B.C.; Lowery, F.J.; Jafferji, M.; Good, M.L.; Sachs, A.; Copeland, A.R.; Kim, S.P.; et al. Antigen Experienced T Cells from Peripheral Blood Recognize p53 Neoantigens. Clin. Cancer Res. 2020, 26, 1267–1276. [Google Scholar] [CrossRef]
- Cai, Y.; Prochazkova, M.; Jiang, C.; Song, H.W.; Jin, J.; Moses, L.; Gkitsas, N.; Somerville, R.P.; Highfill, S.L.; Panch, S.; et al. Establishment and validation of in-house cryopreserved CAR/TCR-T cell flow cytometry quality control. J. Transl. Med. 2021, 19, 523. [Google Scholar] [CrossRef]
- Leidner, R.; Sanjuan Silva, N.; Huang, H.; Sprott, D.; Zheng, C.; Shih, Y.P.; Leung, A.; Payne, R.; Sutcliffe, K.; Cramer, J.; et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N. Engl. J. Med. 2022, 386, 2112–2119. [Google Scholar] [CrossRef]
- Zhang, Y.; Schmidt-Wolf, I.G.H. Ten-year update of the international registry on cytokine-induced killer cells in cancer immunotherapy. J. Cell. Physiol. 2020, 235, 9291–9303. [Google Scholar] [CrossRef]
- Choi, J.H.; Nam, G.H.; Hong, J.-m.; Cho, I.R.; Paik, W.H.; Ryu, J.K.; Kim, Y.-T.; Lee, S.H. Cytokine-Induced Killer Cell Immunotherapy Combined With Gemcitabine Reduces Systemic Metastasis in Pancreatic Cancer: An Analysis Using Preclinical Adjuvant Therapy-Mimicking Pancreatic Cancer Xenograft Model. Pancreas 2022, 51, 1251–1257. [Google Scholar] [CrossRef]
- Wang, M.; Shi, S.B.; Qi, J.L.; Tang, X.Y.; Tian, J. S-1 plus CIK as second-line treatment for advanced pancreatic cancer. Med. Oncol. 2013, 30, 747. [Google Scholar] [CrossRef]
- Gao, X.; Mi, Y.; Guo, N.; Xu, H.; Xu, L.; Gou, X.; Jin, W. Cytokine-Induced Killer Cells As Pharmacological Tools for Cancer Immunotherapy. Front. Immunol. 2017, 8, 774. [Google Scholar] [CrossRef]
- Cappuzzello, E.; Vigolo, E.; D’Accardio, G.; Astori, G.; Rosato, A.; Sommaggio, R. How can Cytokine-induced killer cells overcome CAR-T cell limits. Front. Immunol. 2023, 14, 1229540. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Qiao, G.; Wang, X.; Morse, M.A.; Gwin, W.R.; Zhou, L.; Song, Y.; Zhao, Y.; Chen, F.; Zhou, X.; et al. Dendritic Cell/Cytokine-Induced Killer Cell Immunotherapy Combined with S-1 in Patients with Advanced Pancreatic Cancer: A Prospective Study. Clin. Cancer Res. 2017, 23, 5066–5073. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, G.; Fraietta, J.A.; Gerson, J.N.; Van Deerlin, V.M.; Morrissette, J.J.D.; Caponetti, G.C.; Paruzzo, L.; Harris, J.C.; Chong, E.A.; Susanibar Adaniya, S.P.; et al. T cell lymphoma and secondary primary malignancy risk after commercial CAR T cell therapy. Nat. Med. 2024, 30, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Malviya, M.; Aretz, Z.E.H.; Molvi, Z.; Lee, J.; Pierre, S.; Wallisch, P.; Dao, T.; Scheinberg, D.A. Challenges and solutions for therapeutic TCR-based agents. Immunol. Rev. 2023, 320, 58–82. [Google Scholar] [CrossRef]
- Ebrahimiyan, H.; Tamimi, A.; Shokoohian, B.; Minaei, N.; Memarnejadian, A.; Hossein-Khannazer, N.; Hassan, M.; Vosough, M. Novel insights in CAR-NK cells beyond CAR-T cell technology; promising advantages. Int. Immunopharmacol. 2022, 106, 108587. [Google Scholar] [CrossRef]
- Wang, S.; Sun, J.; Chen, K.; Ma, P.; Lei, Q.; Xing, S.; Cao, Z.; Sun, S.; Yu, Z.; Liu, Y.; et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021, 19, 140. [Google Scholar] [CrossRef]
- Monberg, T.J.; Borch, T.H.; Svane, I.M.; Donia, M. TIL Therapy: Facts and Hopes. Clin. Cancer Res. 2023, 29, 3275–3283. [Google Scholar] [CrossRef]
- Liu, S.; Meng, Y.; Liu, L.; Lv, Y.; Yu, W.; Liu, T.; Wang, L.; Mu, D.; Zhou, Q.; Liu, M.; et al. CD4+ T cells are required to improve the efficacy of CIK therapy in non-small cell lung cancer. Cell Death Dis. 2022, 13, 441. [Google Scholar] [CrossRef]
- Ghanbari Sevari, F.; Mehdizadeh, A.; Abbasi, K.; Hejazian, S.S.; Raeisi, M. Cytokine-induced killer cells: New insights for therapy of hematologic malignancies. Stem Cell Res. Ther. 2024, 15, 254. [Google Scholar] [CrossRef]
- Hamzah, J.; Jugold, M.; Kiessling, F.; Rigby, P.; Manzur, M.; Marti, H.H.; Rabie, T.; Kaden, S.; Gröne, H.J.; Hämmerling, G.J.; et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 2008, 453, 410–414. [Google Scholar] [CrossRef] [PubMed]
- de Aguiar, R.B.; de Moraes, J.Z. Exploring the Immunological Mechanisms Underlying the Anti-vascular Endothelial Growth Factor Activity in Tumors. Front. Immunol. 2019, 10, 1023. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- Kakarla, S.; Chow, K.K.; Mata, M.; Shaffer, D.R.; Song, X.T.; Wu, M.F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassará, A.; Wyser Rmili, C.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9, eaak9670. [Google Scholar] [CrossRef]
- Ferreira, C.S.; Babitzki, G.; Klaman, I.; Krieter, O.; Lechner, K.; Bendell, J.; Vega Harring, S.; Heil, F. Predictive potential of angiopoietin-2 in a mCRC subpopulation treated with vanucizumab in the McCAVE trial. Front. Oncol. 2023, 13, 1157596. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Binnewies, M.; Pollack, J.L.; Rudolph, J.; Dash, S.; Abushawish, M.; Lee, T.; Jahchan, N.S.; Canaday, P.; Lu, E.; Norng, M.; et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 2021, 37, 109844. [Google Scholar] [CrossRef]
- Gao, Y.; Lin, H.; Guo, D.; Cheng, S.; Zhou, Y.; Zhang, L.; Yao, J.; Farooq, M.A.; Ajmal, I.; Duan, Y.; et al. Suppression of 4.1R enhances the potency of NKG2D-CAR T cells against pancreatic carcinoma via activating ERK signaling pathway. Oncogenesis 2021, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, D.; Tomiuk, S.; Küster, L.N.; Rawashdeh, W.A.; Henze, J.; Tischler-Höhle, G.; Agorku, D.J.; Brauner, J.; Linnartz, C.; Lock, D.; et al. Identification of CD318, TSPAN8 and CD66c as target candidates for CAR T cell based immunotherapy of pancreatic adenocarcinoma. Nat. Commun. 2021, 12, 1453. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Wang, L.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef] [PubMed]
- Waaga-Gasser, A.M.; Böldicke, T. Genetically Engineered T Cells and Recombinant Antibodies to Target Intracellular Neoantigens: Current Status and Future Directions. Int. J. Mol. Sci. 2024, 25, 13504. [Google Scholar] [CrossRef]
- He, Q.; Liu, Z.; Liu, Z.; Lai, Y.; Zhou, X.; Weng, J. TCR-like antibodies in cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 99. [Google Scholar] [CrossRef]
- Walseng, E.; Köksal, H.; Sektioglu, I.M.; Fåne, A.; Skorstad, G.; Kvalheim, G.; Gaudernack, G.; Inderberg, E.M.; Wälchli, S. A TCR-based Chimeric Antigen Receptor. Sci. Rep. 2017, 7, 10713. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Li, K.; Liu, Y.; Xu, J.; Ma, J.; An, L.; Wang, H.; Chu, X. A novel dominant-negative PD-1 armored anti-CD19 CAR T cell is safe and effective against refractory/relapsed B cell lymphoma. Transl. Oncol. 2021, 14, 101085. [Google Scholar] [CrossRef]
- Lin, G.; Zhang, Y.; Yu, L.; Wu, D. Cytotoxic effect of CLL-1 CAR-T cell immunotherapy with PD-1 silencing on relapsed/refractory acute myeloid leukemia. Mol. Med. Rep. 2021, 23, 208. [Google Scholar] [CrossRef]
- Serganova, I.; Moroz, E.; Cohen, I.; Moroz, M.; Mane, M.; Zurita, J.; Shenker, L.; Ponomarev, V.; Blasberg, R. Enhancement of PSMA-Directed CAR Adoptive Immunotherapy by PD-1/PD-L1 Blockade. Mol. Ther. Oncolytics 2017, 4, 41–54. [Google Scholar] [CrossRef]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [PubMed]
- Pang, N.; Shi, J.; Qin, L.; Chen, A.; Tang, Y.; Yang, H.; Huang, Y.; Wu, Q.; Li, X.; He, B.; et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J. Hematol. Oncol. 2021, 14, 118. [Google Scholar] [CrossRef] [PubMed]
- Hou, A.J.; Chang, Z.L.; Lorenzini, M.H.; Zah, E.; Chen, Y.Y. TGF-β-responsive CAR-T cells promote anti-tumor immune function. Bioeng. Transl. Med. 2018, 3, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, S.; Li, S.; Qu, Y.; Wang, H.Y.; Liu, J.; Dunn, Z.S.; Cinay, G.E.; MacMullan, M.A.; Hu, F.; et al. Secretion of bispecific protein of anti-PD-1 fused with TGF-β trap enhances antitumor efficacy of CAR-T cell therapy. Mol. Ther. Oncolytics 2021, 21, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.; Abken, H. Chimeric antigen receptors designed to overcome transforming growth factor-β-mediated repression in the adoptive T-cell therapy of solid tumors. Clin. Transl. Immunol. 2019, 8, e1064. [Google Scholar] [CrossRef]
- Al-Haideri, M.; Tondok, S.B.; Safa, S.H.; Maleki, A.H.; Rostami, S.; Jalil, A.T.; Al-Gazally, M.E.; Alsaikhan, F.; Rizaev, J.A.; Mohammad, T.A.M.; et al. CAR-T cell combination therapy: The next revolution in cancer treatment. Cancer Cell Int. 2022, 22, 365. [Google Scholar] [CrossRef]
- Gumber, D.; Wang, L.D. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine 2022, 77, 103941. [Google Scholar] [CrossRef]
- Zhang, R.Y.; Wei, D.; Liu, Z.K.; Yong, Y.L.; Wei, W.; Zhang, Z.Y.; Lv, J.J.; Zhang, Z.; Chen, Z.N.; Bian, H. Doxycycline Inducible Chimeric Antigen Receptor T Cells Targeting CD147 for Hepatocellular Carcinoma Therapy. Front. Cell Dev. Biol. 2019, 7, 233. [Google Scholar] [CrossRef]
- Webster, B.; Xiong, Y.; Hu, P.; Wu, D.; Alabanza, L.; Orentas, R.J.; Dropulic, B.; Schneider, D. Self-driving armored CAR-T cells overcome a suppressive milieu and eradicate CD19+ Raji lymphoma in preclinical models. Mol. Ther. 2021, 29, 2691–2706. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wen, J.; Yi, H.; Hou, X.; Yin, Y.; Ye, G.; Wu, X.; Jiang, X. Split chimeric antigen receptor-modified T cells targeting glypican-3 suppress hepatocellular carcinoma growth with reduced cytokine release. Ther. Adv. Med. Oncol. 2020, 12, 1758835920910347. [Google Scholar] [CrossRef]
- Zajc, C.U.; Dobersberger, M.; Schaffner, I.; Mlynek, G.; Pühringer, D.; Salzer, B.; Djinović-Carugo, K.; Steinberger, P.; De Sousa Linhares, A.; Yang, N.J.; et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc. Natl. Acad. Sci. USA 2020, 117, 14926–14935. [Google Scholar] [CrossRef]
- Yu, S.; Yi, M.; Qin, S.; Wu, K. Next generation chimeric antigen receptor T cells: Safety strategies to overcome toxicity. Mol. Cancer 2019, 18, 125. [Google Scholar] [CrossRef]
- Lee, Y.G.; Marks, I.; Srinivasarao, M.; Kanduluru, A.K.; Mahalingam, S.M.; Liu, X.; Chu, H.; Low, P.S. Use of a Single CAR T Cell and Several Bispecific Adapters Facilitates Eradication of Multiple Antigenically Different Solid Tumors. Cancer Res. 2019, 79, 387–396. [Google Scholar] [CrossRef]
- Baeuerle, P.A.; Ding, J.; Patel, E.; Thorausch, N.; Horton, H.; Gierut, J.; Scarfo, I.; Choudhary, R.; Kiner, O.; Krishnamurthy, J.; et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat. Commun. 2019, 10, 2087. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, G.; Wang, J.; Zheng, Z.Y.; Jia, L.; Rui, W.; Huang, D.; Zhou, Z.X.; Zhou, L.; Wu, X.; et al. Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci. Transl. Med. 2021, 13, eabb5191. [Google Scholar] [CrossRef]
- Wang, C.; Li, M.; Wei, R.; Wu, J. Adoptive transfer of TILs plus anti-PD1 therapy: An alternative combination therapy for treating metastatic osteosarcoma. J. Bone Oncol. 2020, 25, 100332. [Google Scholar] [CrossRef]
- Jimeno, A.; Papa, S.; Haigentz, M.; Rodríguez-Moreno, J.; Schardt, J.; Fardis, M.; Finckenstein, F.G.; Fiaz, R.; Chen, G.; Cacovean, A.; et al. 353 Safety and efficacy of tumor infiltrating lymphocytes (TIL, LN-145) in combination with pembrolizumab for advanced, recurrent or metastatic HNSCC. J. ImmunoTherapy Cancer 2020, 8, A215–A216. [Google Scholar] [CrossRef]
- Stevanović, S.; Helman, S.R.; Wunderlich, J.R.; Langhan, M.M.; Doran, S.L.; Kwong, M.L.M.; Somerville, R.P.T.; Klebanoff, C.A.; Kammula, U.S.; Sherry, R.M.; et al. A Phase II Study of Tumor-infiltrating Lymphocyte Therapy for Human Papillomavirus-associated Epithelial Cancers. Clin. Cancer Res. 2019, 25, 1486–1493. [Google Scholar] [CrossRef]
- Zacharakis, N.; Huq, L.M.; Seitter, S.J.; Kim, S.P.; Gartner, J.J.; Sindiri, S.; Hill, V.K.; Li, Y.F.; Paria, B.C.; Ray, S.; et al. Breast Cancers Are Immunogenic: Immunologic Analyses and a Phase II Pilot Clinical Trial Using Mutation-Reactive Autologous Lymphocytes. J. Clin. Oncol. 2022, 40, 1741–1754. [Google Scholar] [CrossRef] [PubMed]
- Kverneland, A.H.; Chamberlain, C.A.; Borch, T.H.; Nielsen, M.; Mørk, S.K.; Kjeldsen, J.W.; Lorentzen, C.L.; Jørgensen, L.P.; Riis, L.B.; Yde, C.W.; et al. Adoptive cell therapy with tumor-infiltrating lymphocytes supported by checkpoint inhibition across multiple solid cancer types. J. Immunother. Cancer 2021, 9, e003499. [Google Scholar] [CrossRef] [PubMed]
- Laumont, C.M.; Banville, A.C.; Gilardi, M.; Hollern, D.P.; Nelson, B.H. Tumour-infiltrating B cells: Immunological mechanisms, clinical impact and therapeutic opportunities. Nat. Rev. Cancer 2022, 22, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Rokade, S.; Damani, A.M.; Oft, M.; Emmerich, J. IL-2 based cancer immunotherapies: An evolving paradigm. Front. Immunol. 2024, 15, 1433989. [Google Scholar] [CrossRef]
- Waldmann, T.A.; Dubois, S.; Miljkovic, M.D.; Conlon, K.C. IL-15 in the Combination Immunotherapy of Cancer. Front. Immunol. 2020, 11, 868. [Google Scholar] [CrossRef]
- Nikita, S.; Mishra, S.; Gupta, K.; Runkana, V.; Gomes, J.; Rathore, A.S. Advances in bioreactor control for production of biotherapeutic products. Biotechnol. Bioeng. 2023, 120, 1189–1214. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Liu, D.; Naing, M.W. Development of a closed and automated bioreactor technology for cell therapy manufacturing—A sharing of our journey. Regen. Med. 2020, 15, 2335–2340. [Google Scholar] [CrossRef]
- Laskowski, T.J.; Biederstädt, A.; Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 2022, 22, 557–575. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Li, R.e.; Shang, Y.; Zhang, Y.; Zhao, L.; Li, W.; Yang, Y.; Zhang, X.; Yang, T.; et al. Autologous cytokine-induced killer cell transfusion increases overall survival in advanced pancreatic cancer. J. Hematol. Oncol. 2016, 9, 6. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, C.L.; Jiang, S.W.; Feng, Y.; Yuan, L.; Chen, P.; Zhang, L.; Huang, S.; Li, J.; Xia, J.C.; et al. Retrospective analysis of the efficacy of adjuvant CIK cell therapy in epithelial ovarian cancer patients who received postoperative chemotherapy. Oncoimmunology 2019, 8, e1528411. [Google Scholar] [CrossRef]
- Kong, D.S.; Nam, D.H.; Kang, S.H.; Lee, J.W.; Chang, J.H.; Kim, J.H.; Lim, Y.J.; Koh, Y.C.; Chung, Y.G.; Kim, J.M.; et al. Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in Korea. Oncotarget 2017, 8, 7003–7013. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xiong, Y.; Wang, Y.; Meng, Y.; Zhang, W.; Shen, M.; Zhang, X.; Li, S.; Ren, B.; Li, R.; et al. A Phase IB Trial of Autologous Cytokine-Induced Killer Cells in Combination with Sintilimab, Monoclonal Antibody Against Programmed Cell Death-1, plus Chemotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2022, 23, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, T.; Song, Y.; Yang, Y.; Ma, B.; Zhang, Y.; Shang, Y.; Xu, B.; Guo, J.; Qin, P.; et al. High Complete Response Rate in Patients With Metastatic Renal Cell Carcinoma Receiving Autologous Cytokine-Induced Killer Cell Therapy Plus Anti-Programmed Death-1 Agent: A Single-Center Study. Front. Immunol. 2022, 12, 779248. [Google Scholar] [CrossRef]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, S.; Zhan, Y.; Zhang, X. CRISPR-Cas9 applications in T cells and adoptive T cell therapies. Cell Mol. Biol. Lett. 2024, 29, 52. [Google Scholar] [CrossRef]
- De Castro, V.; Galaine, J.; Loyon, R.; Godet, Y. CRISPR-Cas gene knockouts to optimize engineered T cells for cancer immunotherapy. Cancer Gene Ther. 2024, 31, 1124–1134. [Google Scholar] [CrossRef]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Trial | Target | Outcomes | Adverse Effects | Notes on the Target |
|---|---|---|---|---|
| NCT02541370 * [52] (n = 23) | CD-133 (B) PDAC—7/23 | PR—2 SD—3 PD—2 | Hyperbilirubinemia, Anemia, Leucopenia, Thrombocytopenia, Anorexia, and Mucosal hyperemia | It is a transmembrane protein and the most commonly expressed cancer stem cell marker in several cancer types [53]. Correlates with histologic type, lymphatic invasion, and metastasis in pancreatic cancer [54]. |
| NCT02850536 [55] (n = 5) | CEA | OS—23.2 m DOR—13 m | Fever, Electrolyte abnormalities, Hypertension | It can be elevated in PDAC and a level >7.2 ng/mL in LA-PDAC is often associated with systemic disease [56,57,58,59]. |
| NCT01897415 [60] (n = 6) | Mesothelin | SD—2 PD—4 | Abdominal pain, Back pain Dysgeusia, Gastritis | It is an important factor in pancreatic growth by promoting proliferation and inhibiting apoptosis through p53-dependent and p53-independent pathways [61,62]. Mesothelin-specific T cells were generated in 50% of pancreatic cancer patients in a study [63]. |
| NCT02159716 [64] (n = 15) | Mesothelin (B) PDAC—5/15 | PD—3 SD—2 | Anemia, Lymphopenia, Fatigue, Dysgeusia, DIC | |
| NCT03874897 [65] (n = 37) | Claudin 18.2 (B) PDAC—5/37 | PD—1 SD—3 PR—1 | Lymphopenia, Neutropenia, Anemia, Thrombocytopenia, Elevated conjugated bilirubin, Elevated aminotransferase, Hypokalemia, Pyrexia | It is a transmembrane protein that controls the paracellular space through which molecules pass in the epithelial and endothelial tissues and is essential for normal membrane barrier function [46]. It is overexpressed in various cancers and plays an important role in the progression of pancreatic neoplasms. Claudin types could be tumor-specific. |
| NCT05199519 [66] (n = 7) | Claudin 18.2 (B) PDAC—2/5 | PR—1 SD—1 | Neutropenia, Anorexia | |
| NCT01869166 [67] (n = 14) | EGFR | PR—4 SD—8 PD—2 | Lymphocytopenia, Pleural effusion, Pulmonary interstitial exudation, Dermatitis Herpetiformis, Gastrointestinal hemorrhage | It plays a crucial role in normal cellular growth, prevention of apoptosis, and development of metastasis in many types of cancer [68]. There are four receptors in the EGF family: HER1, HER2, HER3, and HER4 [69]. |
| NCT01935843 [70] (n = 11) | HER2 (B) PDAC—2/11 | SD—2 | Anemia, Lymphopenia, Fever, Fatigue, Transaminase elevation, Gastrointestinal hemorrhage | It is a cell-membrane protein involved in promoting cell division and differentiation and contributes to tumor progression by triggering angiogenesis [71]. |
| Trial | Phase | Size | Target | Primary Outcome | Secondary Outcomes |
|---|---|---|---|---|---|
| NCT06464965 | I | 30 | Claudin 18.2 | MTD, DLT | ORR, DCR, DOR, PFS, OS |
| NCT05472857 | I | 30 | MTD, AE | ORR, DCR, DOR, PFS | |
| NCT04404595 | Ib | 110 | MTD, DLT, AE, ORR | ORR, DCR, DOR, PFS, OS, HRQoL | |
| NCT04581473 | I/II | 192 | MTD, AE, PFS | ORR, DCR, DOR, PFS, OS | |
| NCT05393986 | I | 63 | MTD, DLT | ORR, DCR, DOR, PFS, OS, AE, PK | |
| NCT05275062 | I | 6 | AE | ORR, DCR, PFS, OS, CAR-T %, Tumor marker, RR, IM92 Ab | |
| NCT06126406 | I | 60 | CEA | DLT, AE | DCR, AUCS, CMAX, TMAX, CEA content |
| NCT06043466 | I | 30 | MTD, DLT, Dose range | DCR, AUCS, CMAX, TMAX, CEA content | |
| NCT06010862 | I | 36 | MTD, AE | ORR, DCR, DOR, PFS, OS, AUCS, CMAX, TMAX, CEA content | |
| NCT05736731 | I/II | 160 | DLT, ORR, RP2D | A2B530%, Cytokine analysis | |
| NCT04660929 | I | 48 | HER 2 | AE, Feasibility of manufacturing, CT—0508 | ORR, PFS |
| NCT03740256 | I | 45 | DLT | ORR, DCR, PFS, OS AEs grade 3 | |
| NCT06051695 | I/II | 230 | Mesothelin | DLT, ORR, RP2D | A2B694 persistence, Cytokine analysis |
| NCT05239143 | I | 180 | MUC1—C | MTD, ORR, R2PD | - |
| NCT06158139 | I | 27 | B7-H3 | AE, CRS, Neurotoxicity | ORR, DCR, PFS, OS, B7-H3 expression, DLT |
| NCT02830724 | I/II | 124 | CD 70 | AE within 2 weeks, RR | AE (within 6 weeks) |
| Trial | Phase | Size | Target | Outcomes | |
|---|---|---|---|---|---|
| TIL therapy | NCT05098197 | I | 50 | - | ORR, DCR, DOR, PFS, OS, TRAE |
| NCT03935893 | II | 240 | - | ORR, DCR, DOR, PFS, OS | |
| NCT04426669 | I/II | 20 | - | MTD, PE, AE PFS, OS | |
| NCT01174121 | II | 332 | - | ORR, TRAE Efficacy | |
| NCT05098197 | I | 50 | - | ORR, DCR, DOR, PFS, OS, TRAE | |
| CAR-NK | NCT03941457 | I/II | 9 | ROBO1 | TRAE |
| NCT02839954 | I/II | 10 | MUC1 | TRAE ORR | |
| NCT03841110 | I | 64 | NK cell + ICI | DLT ORR, DOR | |
| NCT06464965 | I | 30 | Claudin18.2 | MTD, DLT ORR, DCR, DOR, PFS, OS | |
| NCT05922930 | I/II | TROP2 | ORR, PFS, DLT | ||
| Cytokine-induced killer (CIK) cells | NCT03509298 | II | 90 | CIK with anti-CD3-MUC1 bispecific antibody | ORR, DCR, PFS, OS, SSR, TTP |
| NCT05955157 | II/III randomized | 52 | DC-CIK_S-1 vs. S-1 | TRAE, Hematological CBR Efficacy | |
| T-cell receptor-engineered T-cells | NCT04809766 | I | 15 | Mesothelin | TRAE ORR, PFS, OS |
| NCT05438667 | I | 18 | KRAS (G12V or G12D) | PFS, OS, AUC, CMAX, TMAX, AE, TTP, EFS, DFS, DoE TCR-T cell number, peak value of cytokines | |
| NCT06487377 | I | 12 | KRAS (G12V or G12D) | DLT, TRAE, SAE ORR, DCR, DOR, PFS, OS, TTR, TCR-T cell counts, TCR gene copies, anti-drug antibodies, changes in tumor markers | |
| NCT04146298 | I/II | 30 | KRAS (G12V) | ORR, TRAE OS, TCR transduced T cell % | |
| NCT06054984 | I | 18 | RAS/TP 53 | AUC, CMAX, TMAX, TRAE ORR, DCR, PFS, OS Change in tumor size, biomarker | |
| NCT06043713 | I | 24 | KRAS (G12V) | MTD, DLT, AE ORR, PFS, OS, CBR, SD, changes in TME | |
| NCT05877599 | I | 162 | TP53 | ORR, DOR, PFS, DLT, AE, SAE, TRAE, CBR, TTR, BOR | |
| NCT06218914 | I | 24 | KRAS (G12D) | DLT, AE, SAE ORR, DOR, PFS, OS, CBR, TTR, BOR | |
| NCT06105021 | I/II | 100 | KRAS (G12V) | DLT, SAE, TEAE, OBD ORR, DOR, PFS, OS, CBR, TTR, | |
| NCT04622423 | Observational | 475 | PFS, OS, tumor mutational burden, gene expression profile, antigenic landscape, T-cell repertoire, change in tumor marker | ||
| NCT05964361 | I/II | 10 | WT-1 | DOR, BOR, leukapheresis %, SAE, ORR, DCR, PFS, OS, QoLA | |
| NCT03190941 | I/II | 110 | KRAS (G12V) | TRAE, RR | |
| NCT03745326 | I/II | 70 | KRAS (G12D) | TRAE, RR | |
| NCT04810910 | I | 20 | Personalized Neo-antigen vaccine | OS, TRAE, RFS, CD4/CD8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sherpally, D.; Manne, A. Advancing Immunotherapy in Pancreatic Cancer: A Brief Review of Emerging Adoptive Cell Therapies. Cancers 2025, 17, 589. https://doi.org/10.3390/cancers17040589
Sherpally D, Manne A. Advancing Immunotherapy in Pancreatic Cancer: A Brief Review of Emerging Adoptive Cell Therapies. Cancers. 2025; 17(4):589. https://doi.org/10.3390/cancers17040589
Chicago/Turabian StyleSherpally, Deepak, and Ashish Manne. 2025. "Advancing Immunotherapy in Pancreatic Cancer: A Brief Review of Emerging Adoptive Cell Therapies" Cancers 17, no. 4: 589. https://doi.org/10.3390/cancers17040589
APA StyleSherpally, D., & Manne, A. (2025). Advancing Immunotherapy in Pancreatic Cancer: A Brief Review of Emerging Adoptive Cell Therapies. Cancers, 17(4), 589. https://doi.org/10.3390/cancers17040589