Immunotherapy in Prostate Cancer: From a “Cold” Tumor to a “Hot” Prospect



Simple Summary
Abstract
1. Introduction
2. TME and the Immunological Landscape of Prostate Cancer
2.1. Molecular Pathophysiology
2.1.1. Androgen Receptor (AR) Signaling and Historical Context
2.1.2. Transition to CRPC
2.2. Prostate Cancer as a “Cold Tumor”: Challenges in Immunotherapy
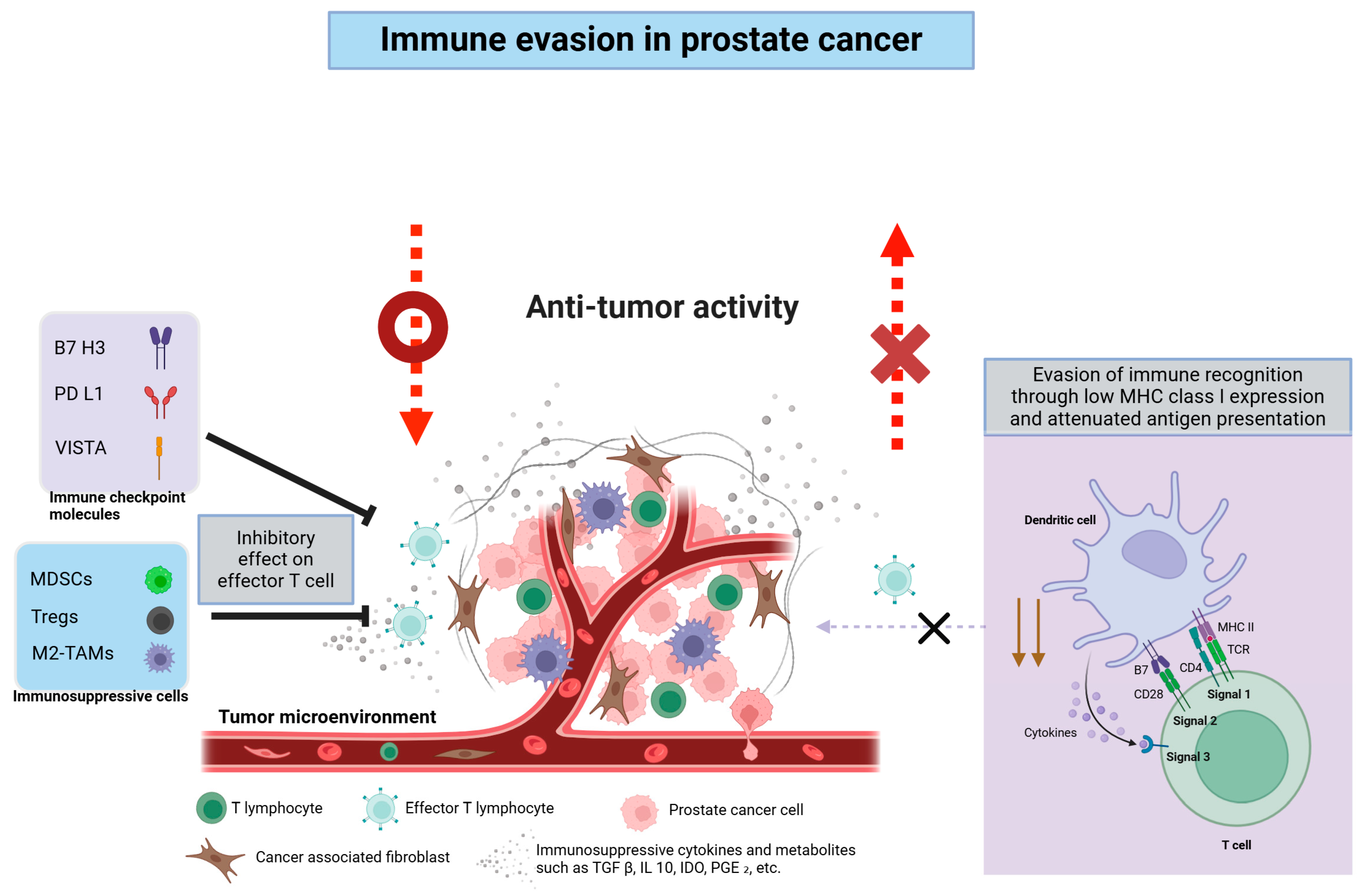
2.2.1. Lack of Tumor-Infiltrating Lymphocytes
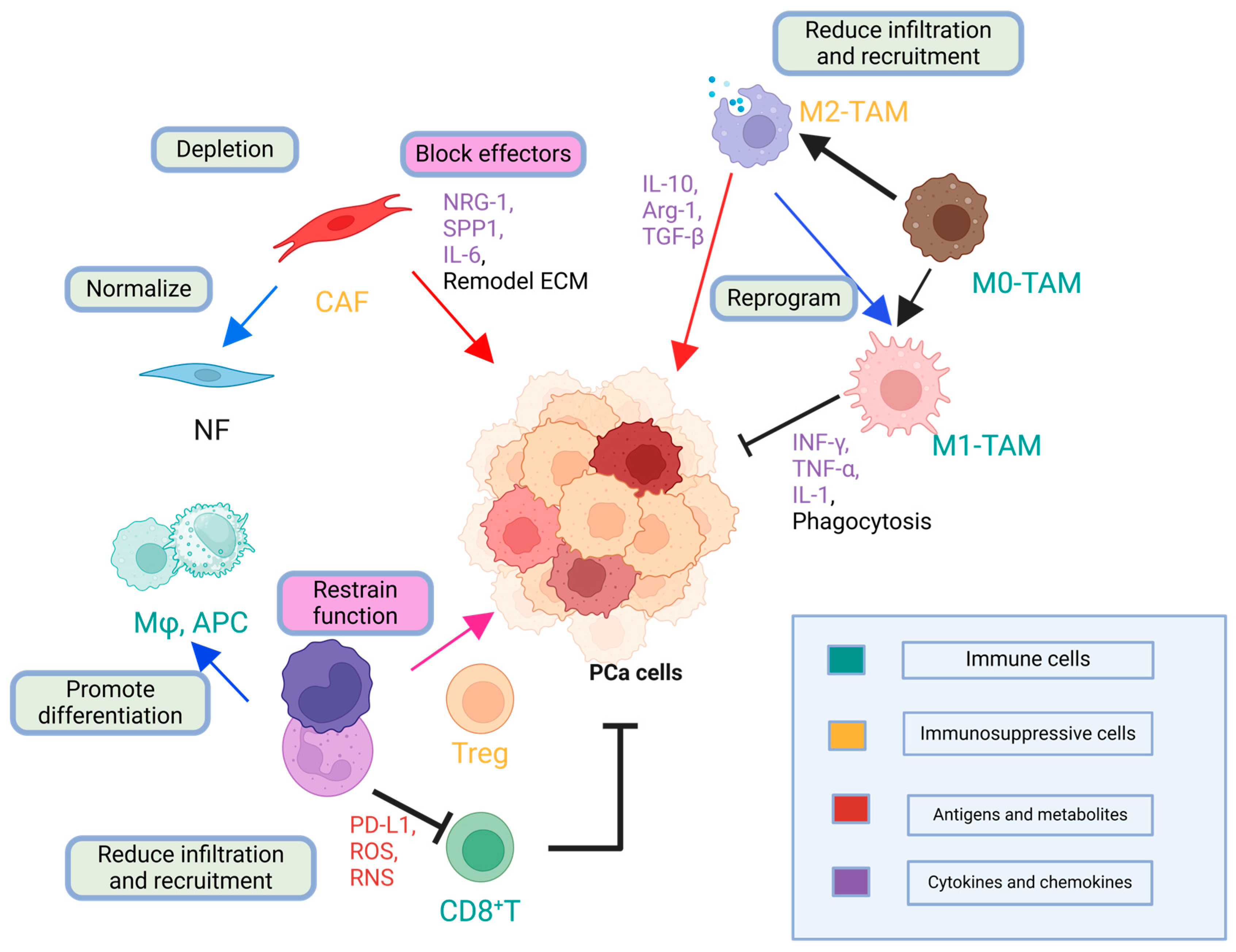
2.2.2. Abundant Immunosuppressive Cells
2.2.3. Soluble Factors in the TME
2.2.4. Tumor Neoantigens and Low Immunogenicity
2.3. Key Immune Evasion Mechanisms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunosuppressive Cell/Molecule | Key Factors | Immunosuppressive Mechanisms | Clinical Significance | References |
|---|---|---|---|---|
| Tregs | TGF-β, IL-10 | Suppress T-cell function | Poor prognosis | [4,34] |
| MDSCs | Arginase-1, IDO | Impair antigen presentation | Therapeutic target | [31,33] |
| TAMs (M2 type) | CCL5, TGF-β | Promote tumor growth | High density = worse outcomes | [32] |
| PD-L1/PD-L2 | PD-L1, PD-L2 | Induce T-cell exhaustion | Resistance marker | [38,40] |
| IDO | Tryptophan | Inhibit T-cell growth | Potential combo target | [35] |
| TGF-β | TGF-β cytokine | Promote Treg development | Potential therapeutic target | [2,4] |
| Adenosine | CD39/CD73 | Suppress immune activity | Novel therapeutic pathway | [2] |
2.4. Prostate Cancer-Specific Antigens and Therapeutic Targets
3. Past and Present of Immunotherapy Approaches for Prostate Cancer
3.1. Early Attempts and Key Lessons
3.2. Current Strategies and Clinical Experience: Immunotherapeutic Approaches
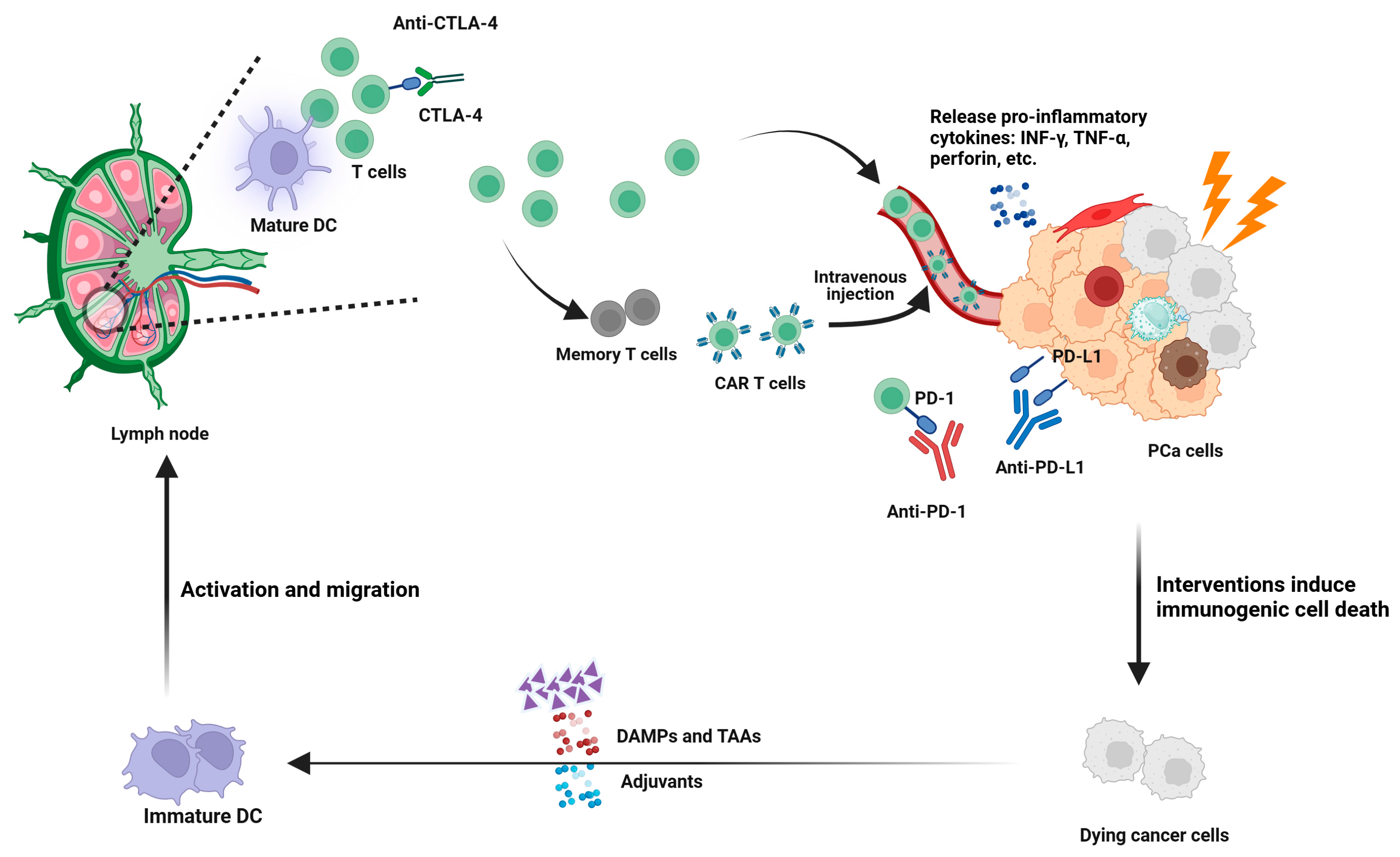
3.2.1. Immune Checkpoint Blockade
3.2.2. Induction of ICD
3.2.3. TME Reversal (Immunologically “Hot” Conversion)
3.2.4. Tumor Vaccines
3.2.5. Bispecific T-Cell Engagement
3.2.6. Immune Adjuvants
3.2.7. CAR T-Cell Therapy
3.2.8. Interventions Targeting Barriers to TME Infiltration
3.2.9. Clinical Trials of Immunotherapeutic Approaches: Current Evidence and Challenges
3.2.10. Overall Critique and Future Direction
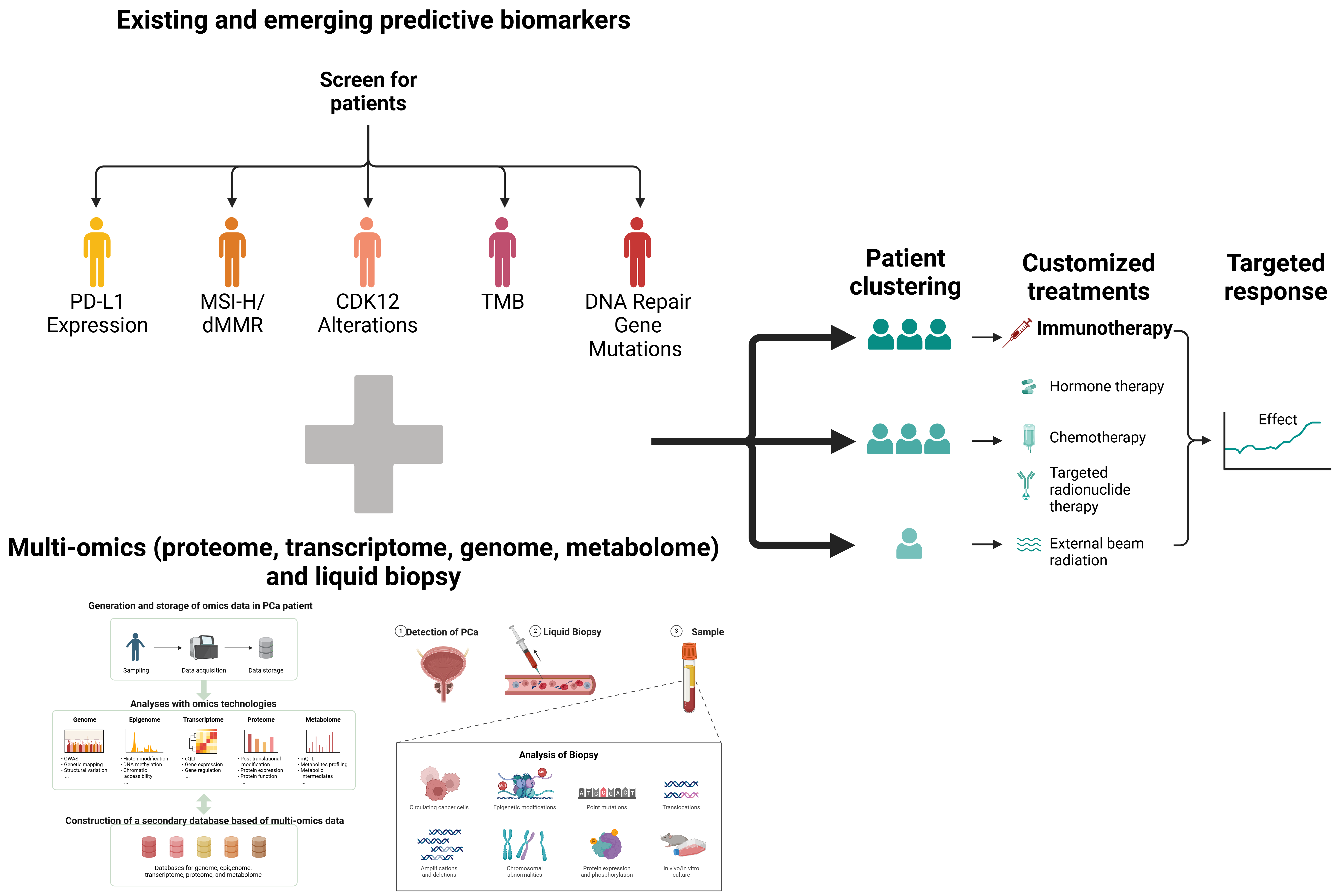
4. Predictive Biomarkers and Patient Stratification
4.1. Biomarkers for Immunotherapy Response
4.2. Prostate Cancer-Specific Markers
4.3. Precision Medicine and Comprehensive Profiling
5. Combination Therapies
5.1. Hormonal Therapy Plus Immunotherapy
5.2. Chemotherapy, Targeted Therapies, and Beyond
5.3. Radiotherapy and Immunotherapy
6. Limitations and Future Directions
6.1. Novel Technologies and Therapeutic Concepts
6.2. Precision Oncology Approaches
6.3. Innovation in Clinical Trial Design
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, L.; Lu, B.; He, M.; Wang, Y.; Wang, Z.; Du, L. Prostate Cancer Incidence and Mortality: Global Status and Temporal Trends in 89 Countries from 2000 to 2019. Front Public Health 2022, 10, 811044. [Google Scholar] [CrossRef] [PubMed]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.R.; Lee, J.H.; Ponnazhagan, S. Revisiting Immunotherapy: A Focus on Prostate Cancer. Cancer Res. 2020, 80, 1615–1623. [Google Scholar] [CrossRef]
- Wang, I.; Song, L.; Wang, B.Y.; Rezazadeh Kalebasty, A.; Uchio, E.; Zi, X. Prostate cancer immunotherapy: A review of recent advancements with novel treatment methods and efficacy. Am. J. Clin. Exp. Urol. 2022, 10, 210–233. [Google Scholar] [PubMed]
- Liang, H.; Liu, Y.; Guo, J.; Dou, M.; Zhang, X.; Hu, L.; Chen, J. Progression in immunotherapy for advanced prostate cancer. Front. Oncol. 2023, 13, 1126752. [Google Scholar] [CrossRef]
- Liu, Y.T.; Sun, Z.J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Desai, N.; Hasan, U.; Mani, R.; Chauhan, M.; Basu, S.M.; Giri, J. Biomaterial-based platforms for modulating immune components against cancer and cancer stem cells. Acta Biomater. 2023, 161, 1–36. [Google Scholar] [CrossRef]
- Lenis, A.T.; Ravichandran, V.; Brown, S.; Alam, S.M.; Katims, A.; Truong, H.; Reisz, P.A.; Vasselman, S.; Nweji, B.; Autio, K.A.; et al. Microsatellite Instability, Tumor Mutational Burden, and Response to Immune Checkpoint Blockade in Patients with Prostate Cancer. Clin. Cancer Res. 2024, 30, 3894–3903. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hsu, J.M.; Sun, L.; Wang, S.C.; Hung, M.C. Advances and prospects of biomarkers for immune checkpoint inhibitors. Cell Rep. Med. 2024, 5, 101621. [Google Scholar] [CrossRef]
- Galsky, M.D.; Autio, K.A.; Cabanski, C.R.; Wentzel, K.; Graff, J.N.; Friedlander, T.W.; Howes, T.R.; Shotts, K.M.; Densmore, J.; Spasic, M.; et al. Clinical and Translational Results from PORTER, a Multi-cohort Phase 1 Platform Trial of Combination Immunotherapy in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2025, OF1–OF13. [Google Scholar] [CrossRef]
- Schütz, V.; Nessler, C.-L.; Duensing, A.; Zschäbitz, S.; Jäger, D.; Debus, J.; Hohenfellner, M.; Duensing, S. Improved survival of patients with newly diagnosed oligometastatic prostate cancer through intensified multimodal treatment. Front. Oncol. 2024, 14, 1475914. [Google Scholar] [CrossRef] [PubMed]
- Le, T.K.; Duong, Q.H.; Baylot, V.; Fargette, C.; Baboudjian, M.; Colleaux, L.; Taïeb, D.; Rocchi, P. Castration-Resistant Prostate Cancer: From Uncovered Resistance Mechanisms to Current Treatments. Cancers 2023, 15, 5047. [Google Scholar] [CrossRef]
- DuHadaway, J.B.; Muller, A.J.; Laury-Kleintop, L.D.; Wallon, U.M.; Gilmour, S.K.; Webster, M.; Williams, J.R.; Rossi, G.R.; Mautino, M.R.; Lewis, J.; et al. Abstract LB355: Cryo-immune vaccination (CIV) by SYNC-T therapy: Preclinical modeling of a novel device-multidrug immunotherapeutic approach to eradicate advanced metastatic cancers. Cancer Res. 2024, 84, LB355. [Google Scholar] [CrossRef]
- Mukherjee, D.; Romano, E.; Walshaw, R.; Zeef, L.A.H.; Banyard, A.; Kitcatt, S.J.; Cheadle, E.J.; Tuomela, K.; Pendharkar, S.; Al-Deka, A.; et al. Reprogramming the immunosuppressive tumor microenvironment results in successful clearance of tumors resistant to radiation therapy and anti-PD-1/PD-L1. Oncoimmunology 2023, 12, 2223094. [Google Scholar] [CrossRef]
- Khosravi, G.R.; Mostafavi, S.; Bastan, S.; Ebrahimi, N.; Gharibvand, R.S.; Eskandari, N. Immunologic tumor microenvironment modulators for turning cold tumors hot. Cancer Commun. 2024, 44, 521–553. [Google Scholar] [CrossRef]
- Dai, C.; Dehm, S.M.; Sharifi, N. Targeting the Androgen Signaling Axis in Prostate Cancer. J. Clin. Oncol. 2023, 41, 4267–4278. [Google Scholar] [CrossRef] [PubMed]
- Lumahan, L.E.V.; Arif, M.; Whitener, A.E.; Yi, P. Regulating Androgen Receptor Function in Prostate Cancer: Exploring the Diversity of Post-Translational Modifications. Cells 2024, 13, 191. [Google Scholar] [CrossRef]
- Mercader, M.; Bodner, B.K.; Moser, M.T.; Kwon, P.S.; Park, E.S.Y.; Manecke, R.G.; Ellis, T.M.; Wojcik, E.M.; Yang, D.; Flanigan, R.C.; et al. T cell infiltration of the prostate induced by androgen withdrawal in patients with prostate cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 14565–14570. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, A.Z.; Dallos, M.C.; Zahurak, M.L.; Partin, A.W.; Schaeffer, E.M.; Ross, A.E.; Allaf, M.E.; Nirschl, T.R.; Liu, D.; Chapman, C.G.; et al. T-Cell Infiltration and Adaptive Treg Resistance in Response to Androgen Deprivation With or Without Vaccination in Localized Prostate Cancer. Clin. Cancer Res. 2020, 26, 3182–3192. [Google Scholar] [CrossRef]
- Muralidhar, A.; Gamat-Huber, M.; Vakkalanka, S.; McNeel, D.G. Sequence of androgen receptor-targeted vaccination with androgen deprivation therapy affects anti-prostate tumor efficacy. J. ImmunoTherapy Cancer 2024, 12, e008848. [Google Scholar] [CrossRef]
- Cheng, B.; Huang, H. Expanding horizons in overcoming therapeutic resistance in castration-resistant prostate cancer: Targeting the androgen receptor-regulated tumor immune microenvironment. Cancer Biol. Med. 2023, 20, 568–574. [Google Scholar] [CrossRef]
- Chesner, L.N.; Polesso, F.; Graff, J.N.; Hawley, J.E.; Smith, A.K.; Lundberg, A.; Das, R.; Shenoy, T.; Sjöström, M.; Zhao, F.; et al. Androgen Receptor Inhibition Increases MHC Class I Expression and Improves Immune Response in Prostate Cancer. Cancer Discov. 2025, 15, OF1–OF14. [Google Scholar] [CrossRef]
- Palmbos, P.L.; Daignault-Newton, S.; Tomlins, S.A.; Agarwal, N.; Twardowski, P.; Morgans, A.K.; Kelly, W.K.; Arora, V.K.; Antonarakis, E.S.; Siddiqui, J.; et al. A Randomized Phase II Study of Androgen Deprivation Therapy with or without Palbociclib in RB-positive Metastatic Hormone-Sensitive Prostate Cancer. Clin. Cancer Res. 2021, 27, 3017–3027. [Google Scholar] [CrossRef]
- Brea, L.; Yu, J. Tumor-intrinsic regulators of the immune-cold microenvironment of prostate cancer. Trends Endocrinol. Metab. 2025; Online ahead of print. [Google Scholar] [CrossRef]
- Lyu, A.; Fan, Z.; Clark, M.; Lea, A.; Luong, D.; Setayesh, A.; Starzinski, A.; Wolters, R.; Arias-Badia, M.; Allaire, K.; et al. Evolution of myeloid-mediated immunotherapy resistance in prostate cancer. Nature 2025, 637, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Novysedlak, R.; Guney, M.; Al Khouri, M.; Bartolini, R.; Koumbas Foley, L.; Benesova, I.; Ozaniak, A.; Novak, V.; Vesely, S.; Pacas, P.; et al. The Immune Microenvironment in Prostate Cancer: A Comprehensive Review. Oncology, 2024; 1–25, Online ahead of print. [Google Scholar] [CrossRef]
- Xu, P.; Wasielewski, L.J.; Yang, J.C.; Cai, D.; Evans, C.P.; Murphy, W.J.; Liu, C. The Immunotherapy and Immunosuppressive Signaling in Therapy-Resistant Prostate Cancer. Biomedicines 2022, 10, 1778. [Google Scholar] [CrossRef]
- Jiang, W.; He, Y.; He, W.; Wu, G.; Zhou, X.; Sheng, Q.; Zhong, W.; Lu, Y.; Ding, Y.; Lu, Q.; et al. Exhausted CD8+ T Cells in the Tumor Immune Microenvironment: New Pathways to Therapy. Front. Immunol. 2020, 11, 622509. [Google Scholar] [CrossRef]
- Hirz, T.; Mei, S.; Sarkar, H.; Kfoury, Y.; Wu, S.; Verhoeven, B.M.; Subtelny, A.O.; Zlatev, D.V.; Wszolek, M.W.; Salari, K.; et al. Dissecting the immune suppressive human prostate tumor microenvironment via integrated single-cell and spatial transcriptomic analyses. Nat. Commun. 2023, 14, 663. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wang, X.; Huang, Y.; Zhang, X.; Sun, W.; Du, Y.; Xu, Z.; Kou, H.; Zhu, S.; Liu, C.; et al. Prostate cancer cell-derived exosomal IL-8 fosters immune evasion by disturbing glucolipid metabolism of CD8(+) T cell. Cell Rep. 2023, 42, 113424. [Google Scholar] [CrossRef]
- Koinis, F.; Xagara, A.; Chantzara, E.; Leontopoulou, V.; Aidarinis, C.; Kotsakis, A. Myeloid-Derived Suppressor Cells in Prostate Cancer: Present Knowledge and Future Perspectives. Cells 2021, 11, 20. [Google Scholar] [CrossRef]
- Han, C.; Deng, Y.; Xu, W.; Liu, Z.; Wang, T.; Wang, S.; Liu, J.; Liu, X. The Roles of Tumor-Associated Macrophages in Prostate Cancer. J. Oncol. 2022, 2022, 8580043. [Google Scholar] [CrossRef]
- Kobayashi, T.; Nagata, M.; Hachiya, T.; Wakita, H.; Ikehata, Y.; Takahashi, K.; China, T.; Shimizu, F.; Lu, J.; Jin, Y.; et al. Increased circulating polymorphonuclear myeloid-derived suppressor cells are associated with prognosis of metastatic castration-resistant prostate cancer. Front. Immunol. 2024, 15, 1372771. [Google Scholar] [CrossRef]
- Ju, M.; Fan, J.; Zou, Y.; Yu, M.; Jiang, L.; Wei, Q.; Bi, J.; Hu, B.; Guan, Q.; Song, X.; et al. Computational Recognition of a Regulatory T-cell-specific Signature With Potential Implications in Prognosis, Immunotherapy, and Therapeutic Resistance of Prostate Cancer. Front. Immunol. 2022, 13, 807840. [Google Scholar] [CrossRef]
- Mao, C.; Ding, Y.; Xu, N. A Double-Edged Sword Role of Cytokines in Prostate Cancer Immunotherapy. Front. Oncol. 2021, 11, 688489. [Google Scholar] [CrossRef]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef]
- Bou-Dargham, M.J.; Sha, L.; Sang, Q.A.; Zhang, J. Immune landscape of human prostate cancer: Immune evasion mechanisms and biomarkers for personalized immunotherapy. BMC Cancer 2020, 20, 572. [Google Scholar] [CrossRef]
- Noori, M.; Azizi, S.; Mahjoubfar, A.; Abbasi Varaki, F.; Fayyaz, F.; Mousavian, A.-H.; Bashash, D.; Kardoust Parizi, M.; Kasaeian, A. Efficacy and safety of immune checkpoint inhibitors for patients with prostate cancer: A systematic review and meta-analysis. Front. Immunol. 2023, 14, 1181051. [Google Scholar] [CrossRef]
- Rebuzzi, S.E.; Rescigno, P.; Catalano, F.; Mollica, V.; Vogl, U.M.; Marandino, L.; Massari, F.; Pereira Mestre, R.; Zanardi, E.; Signori, A.; et al. Immune Checkpoint Inhibitors in Advanced Prostate Cancer: Current Data and Future Perspectives. Cancers 2022, 14, 1245. [Google Scholar] [CrossRef]
- Venkatachalam, S.; McFarland, T.R.; Agarwal, N.; Swami, U. Immune Checkpoint Inhibitors in Prostate Cancer. Cancers 2021, 13, 2187. [Google Scholar] [CrossRef]
- Taylor, B.C.; Balko, J.M. Mechanisms of MHC-I Downregulation and Role in Immunotherapy Response. Front. Immunol. 2022, 13, 844866. [Google Scholar] [CrossRef]
- Bhasin, A.; Mille, P.; Eturi, A.; Iskander, A.; Tester, W.; Zarrabi, K. Tumor-associated antigen targets for novel immune-based strategies in prostate cancer. J. Transl. Genet. Genom. 2024, 8, 55–76. [Google Scholar]
- Bilusic, M.; Madan, R.A.; Gulley, J.L. Immunotherapy of Prostate Cancer: Facts and Hopes. Clin. Cancer Res. 2017, 23, 6764–6770. [Google Scholar] [CrossRef]
- Sutherland, S.I.M.; Ju, X.; Horvath, L.G.; Clark, G.J. Moving on From Sipuleucel-T: New Dendritic Cell Vaccine Strategies for Prostate Cancer. Front. Immunol. 2021, 12, 641307. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [PubMed]
- Almeida, D.V.P.d.; Fong, L.; Rettig, M.B.; Autio, K.A. Immune Checkpoint Blockade for Prostate Cancer: Niche Role or Next Breakthrough? Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e89–e106. [Google Scholar] [CrossRef] [PubMed]
- Barnett, R.M.; Jang, A.; Lanka, S.; Fu, P.; Bucheit, L.A.; Babiker, H.; Bryce, A.; Meyer, H.M.; Choi, Y.; Moore, C.; et al. Blood-based tumor mutational burden impacts clinical outcomes of immune checkpoint inhibitor treated breast and prostate cancers. Commun. Med. 2024, 4, 256. [Google Scholar] [CrossRef]
- Autio, K.A.; Higano, C.S.; Nordquist, L.; Appleman, L.J.; Zhang, T.; Zhu, X.-H.; Babiker, H.; Vogelzang, N.J.; Prasad, S.M.; Schweizer, M.T.; et al. First-in-human, phase 1 study of PF-06753512, a vaccine-based immunotherapy regimen (VBIR), in non-metastatic hormone-sensitive biochemical recurrence and metastatic castration-resistant prostate cancer (mCRPC). J. ImmunoTherapy Cancer 2023, 11, e005702. [Google Scholar] [CrossRef]
- Nguyen, C.B.; Reimers, M.A.; Perera, C.; Abida, W.; Chou, J.; Feng, F.Y.; Antonarakis, E.S.; McKay, R.R.; Pachynski, R.K.; Zhang, J.; et al. Evaluating Immune Checkpoint Blockade in Metastatic Castration-Resistant Prostate Cancers with Deleterious CDK12 Alterations in the Phase 2 IMPACT Trial. Clin. Cancer Res. 2024, 30, 3200–3210. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Azad, A.; Carles, J.; Matsubara, N.; Oudard, S.; Saad, F.; Merseburger, A.S.; Soares, A.; McGregor, B.A.; Zurawski, B. CONTACT-02: Phase 3 study of cabozantinib (C) plus atezolizumab (A) vs second novel hormonal therapy (NHT) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2024, 42, 18. [Google Scholar] [CrossRef]
- Powles, T.; Yuen, K.C.; Gillessen, S.; Kadel, E.E., 3rd; Rathkopf, D.; Matsubara, N.; Drake, C.G.; Fizazi, K.; Piulats, J.M.; Wysocki, P.J.; et al. Atezolizumab with enzalutamide versus enzalutamide alone in metastatic castration-resistant prostate cancer: A randomized phase 3 trial. Nat. Med. 2022, 28, 144–153. [Google Scholar] [CrossRef]
- Jafari, S.; Ardakan, A.K.; Aghdam, E.M.; Mesbahi, A.; Montazersaheb, S.; Molavi, O. Induction of immunogenic cell death and enhancement of the radiation-induced immunogenicity by chrysin in melanoma cancer cells. Sci. Rep. 2024, 14, 23231. [Google Scholar] [CrossRef]
- Soler-Agesta, R.; Moreno-Loshuertos, R.; Yim, C.Y.; Congenie, M.T.; Ames, T.D.; Johnson, H.L.; Stossi, F.; Mancini, M.G.; Mancini, M.A.; Ripollés-Yuba, C.; et al. Cancer cell-selective induction of mitochondrial stress and immunogenic cell death by PT-112 in human prostate cell lines. J. Transl. Med. 2024, 22, 927. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, E.; Beltrán-Visiedo, M.; Galluzzi, L.; Krysko, D.V. Immunogenicity of cell death and cancer immunotherapy with immune checkpoint inhibitors. Cell. Mol. Immunol. 2025, 22, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Hu, Y. Approaches for boosting antitumor immunity in prostate cancer therapy: A comprehensive review on drugs, products, and nanoparticles. J. Drug Deliv. Sci. Technol. 2023, 89, 105048. [Google Scholar] [CrossRef]
- Wu, Q.; Xiao, Q.; Tang, X.; Li, L.; Song, D.; Zhou, Y.; Li, B.; Ren, G.; Luo, F. DAMPs prognostic signature predicts tumor immunotherapy, and identifies immunosuppressive mechanism of pannexin 1 channels in pancreatic ductal adenocarcinoma. Front. Immunol. 2025, 15, 641307. [Google Scholar] [CrossRef]
- Hansen, S.B.; Unal, B.; Kuzu, O.F.; Saatcioglu, F. Immunological facets of prostate cancer and the potential of immune checkpoint inhibition in disease management. Theranostics 2024, 14, 6913–6934. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, B.; Li, B.; Wu, H.; Jiang, M. Cold and hot tumors: From molecular mechanisms to targeted therapy. Signal Transduct. Target. Ther. 2024, 9, 274. [Google Scholar] [CrossRef]
- Tomassetti, C.; Insinga, G.; Gimigliano, F.; Morrione, A.; Giordano, A.; Giurisato, E. Insights into CSF-1R Expression in the Tumor Microenvironment. Biomedicines 2024, 12, 2381. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. ImmunoTherapy Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Escamilla, J.; Schokrpur, S.; Liu, C.; Priceman, S.J.; Moughon, D.; Jiang, Z.; Pouliot, F.; Magyar, C.; Sung, J.L.; Xu, J.; et al. CSF1 receptor targeting in prostate cancer reverses macrophage-mediated resistance to androgen blockade therapy. Cancer Res. 2015, 75, 950–962. [Google Scholar] [CrossRef]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 127. [Google Scholar] [CrossRef]
- Jumaniyazova, E.; Lokhonina, A.; Dzhalilova, D.; Miroshnichenko, E.; Kosyreva, A.; Fatkhudinov, T. The Role of Macrophages in Various Types of Tumors and the Possibility of Their Use as Targets for Antitumor Therapy. Cancers 2025, 17, 342. [Google Scholar] [CrossRef] [PubMed]
- Nesslinger, N.J.; Ng, A.; Tsang, K.Y.; Ferrara, T.; Schlom, J.; Gulley, J.L.; Nelson, B.H. A viral vaccine encoding prostate-specific antigen induces antigen spreading to a common set of self-proteins in prostate cancer patients. Clin. Cancer Res. 2010, 16, 4046–4056. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Lu, X.; Tian, F.; Luo, Q.; Zhou, W.; Yang, S.; Li, W.; Yang, Y.; Shi, M.; Zhou, T. Vaccine Therapies for Prostate Cancer: Current Status and Future Outlook. Vaccines 2024, 12, 1384. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Zhang, M.; Yang, J.; Zhu, Z.; Cao, W.; Dong, C. Therapeutic cancer vaccines: Advancements, challenges and prospects. Signal Transduct. Target. Ther. 2023, 8, 450. [Google Scholar] [CrossRef]
- Handa, S.; Hans, B.; Goel, S.; Bashorun, H.O.; Dovey, Z.; Tewari, A. Immunotherapy in prostate cancer: Current state and future perspectives. Ther. Adv. Urol. 2020, 12, 1756287220951404. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O.; Li, C.; Liang, L.; Zhan, J.; Estrada, J.; Osgood, T.; Li, F.; Zhang, H.; Case, R.; Murawsky, C.M.; et al. AMG 509 (Xaluritamig), an Anti-STEAP1 XmAb 2+1 T-cell Redirecting Immune Therapy with Avidity-Dependent Activity against Prostate Cancer. Cancer Discov. 2024, 14, 90–103. [Google Scholar] [CrossRef]
- Kelly, W.K.; Danila, D.C.; Lin, C.C.; Lee, J.L.; Matsubara, N.; Ward, P.J.; Armstrong, A.J.; Pook, D.; Kim, M.; Dorff, T.B.; et al. Xaluritamig, a STEAP1 × CD3 XmAb 2+1 Immune Therapy for Metastatic Castration-Resistant Prostate Cancer: Results from Dose Exploration in a First-in-Human Study. Cancer Discov. 2024, 14, 76–89. [Google Scholar] [CrossRef]
- Hushmandi, K.; Einollahi, B.; Lee, E.; Sakaizawa, R.; Glaviano, A.; Reiter, R.J.; Saadat, S.H.; Farani, M.R.; Huh, Y.S.; Aref, A.R. Bispecific antibodies as powerful immunotherapeutic agents for urological cancers: Recent innovations based on preclinical and clinical evidence. Int. J. Biol. Sci. 2025, 21, 1410–1435. [Google Scholar]
- Lampe, H.; Tam, L.; Hansen, A.R. Bi-specific T-cell engagers (BiTEs) in prostate cancer and strategies to enhance development: Hope for a BiTE-r future. Front. Pharmacol. 2024, 15, 1399802. [Google Scholar] [CrossRef]
- Xu, M.; Wummer, B.; Kelly, W.K.; Bashir, B. Association between bispecific T cell engager (BiTE) therapy and inflammatory adverse effects (AEs) in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2023, 41, e14534. [Google Scholar] [CrossRef]
- Temizoz, B.; Kuroda, E.; Ishii, K.J. Vaccine adjuvants as potential cancer immunotherapeutics. Int. Immunol. 2016, 28, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Makker, S.; Galley, C.; Bennett, C.L. Cancer vaccines: From an immunology perspective. Immunother. Adv. 2023, 4, ltad030. [Google Scholar] [CrossRef]
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; McArdle, S.E.B. Cancer Vaccines: Adjuvant Potency, Importance of Age, Lifestyle, and Treatments. Front. Immunol. 2021, 11, 615240. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Wahi, A.; Sharma, P.; Nagpal, R.; Raina, N.; Kaurav, M.; Bhattacharya, J.; Rodrigues Oliveira, S.M.; Dolma, K.G.; Paul, A.K.; et al. Recent Advances in Cancer Vaccines: Challenges, Achievements, and Futuristic Prospects. Vaccines 2022, 10, 2011. [Google Scholar] [CrossRef]
- Chung, S.W.; Xie, Y.; Suk, J.S. Overcoming physical stromal barriers to cancer immunotherapy. Drug Deliv. Transl. Res. 2021, 11, 2430–2447. [Google Scholar] [CrossRef] [PubMed]
- Hossain, D.M.; Pal, S.K.; Moreira, D.; Duttagupta, P.; Zhang, Q.; Won, H.; Jones, J.; D’Apuzzo, M.; Forman, S.; Kortylewski, M. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin. Cancer Res. 2015, 21, 3771–3782. [Google Scholar] [CrossRef]
- Sharma, P.; Krainer, M.; Saad, F.; Castellano, D.; Bedke, J.; Kwiatkowski, M.; Patnaik, A.; Procopio, G.; Wiechno, P.; Kochuparambil, S.T.; et al. Nivolumab plus ipilimumab for the treatment of post-chemotherapy metastatic castration-resistant prostate cancer (mCRPC): Additional results from the randomized phase 2 CheckMate 650 trial. J. Clin. Oncol. 2023, 41, 22. [Google Scholar] [CrossRef]
- Graff, J.N.; Hoimes, C.J.; Gerritsen, W.R.; Vaishampayan, U.N.; Elliott, T.; Hwang, C.; Ten Tije, A.J.; Omlin, A.; McDermott, R.S.; Fradet, Y.; et al. Pembrolizumab plus enzalutamide for metastatic castration-resistant prostate cancer progressing on enzalutamide: Cohorts 4 and 5 of the phase 2 KEYNOTE-199 study. Prostate Cancer Prostatic Dis. 2024; Online ahead of print. [Google Scholar] [CrossRef]
- Elyas, S.; Vaishampayan, U.N. Redefining Treatment Paradigms in Metastatic Castration-resistant Prostate Cancer: A Comprehensive Analysis of Cabozantinib and Atezolizumab Based on the CONTACT-02 Study Results. Oncol. Haematol. 2024, 20, 45–50. [Google Scholar] [CrossRef]
- Gulley, J.L.; Borre, M.; Vogelzang, N.J.; Ng, S.; Agarwal, N.; Parker, C.C.; Pook, D.W.; Rathenborg, P.; Flaig, T.W.; Carles, J.; et al. Results of PROSPECT: A randomized phase 3 trial of PROSTVAC-V/F (PRO) in men with asymptomatic or minimally symptomatic metastatic, castration-resistant prostate cancer. J. Clin. Oncol. 2018, 36, 5006. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Chen, C.-L.; Jian, J.-Y.; Wei, M.; Hsieh, W.-Y.; Huang, C.-W.; Chen, Y.-C.; Liao, T.-H.; Wang, S.-J. Abstract 818: Preclinical studies of the cabazitaxel nanoformulation MPB-1734 support therapeutic application in chemo-resistant solid tumors and synergistic benefit in combination with immune checkpoint inhibitor. Cancer Res. 2023, 83, 818. [Google Scholar] [CrossRef]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef]
- Shariati, A.; Khani, P.; Nasri, F.; Afkhami, H.; Khezrpour, A.; Kamrani, S.; Shariati, F.; Alavimanesh, S.; Modarressi, M.H. mRNA cancer vaccines from bench to bedside: A new era in cancer immunotherapy. Biomark. Res. 2024, 12, 157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, X.; Xie, J.; Cheng, X.; Chen, J.; Shen, M.; Ding, W.; Wang, S.; Zhang, Z.; Wang, C.; et al. Clinicopathological and molecular analysis of microsatellite instability in prostate cancer: A multi-institutional study in China. Front. Oncol. 2023, 13, 1277233. [Google Scholar] [CrossRef]
- Isaacsson Velho, P.; Antonarakis, E.S. PD-1/PD-L1 pathway inhibitors in advanced prostate cancer. Expert. Rev. Clin. Pharmacol. 2018, 11, 475–486. [Google Scholar] [CrossRef]
- Wang, L.; Yao, Y.; Xu, C.; Wang, X.; Wu, D.; Hong, Z. Exploration of the Tumor Mutational Burden as a Prognostic Biomarker and Related Hub Gene Identification in Prostate Cancer. Technol. Cancer Res. Treat. 2021, 20, 15330338211052154. [Google Scholar] [CrossRef]
- Zvirble, M.; Survila, Z.; Bosas, P.; Dobrovolskiene, N.; Mlynska, A.; Zaleskis, G.; Jursenaite, J.; Characiejus, D.; Pasukoniene, V. Prognostic significance of soluble PD-L1 in prostate cancer. Front. Immunol. 2024, 15, 1401097. [Google Scholar] [CrossRef]
- Alva, A.S.; Li, J.; Chou, J.; Reimers, M.A.; McKay, R.R.; Zhang, J.; Daignault-Newton, S.; Palmbos, P.L.; Reichert, Z.R.; Cieslik, M.; et al. Phase 2 trial of immunotherapy in tumors with CDK12 inactivation (IMPACT): Results from cohort A of patients (pts) with metastatic castration resistant prostate cancer (mCRPC) receiving dual immune checkpoint inhibition (ICI). J. Clin. Oncol. 2022, 40, 103. [Google Scholar] [CrossRef]
- Palicelli, A.; Bonacini, M.; Croci, S.; Magi-Galluzzi, C.; Cañete-Portillo, S.; Chaux, A.; Bisagni, A.; Zanetti, E.; De Biase, D.; Melli, B.; et al. What Do We Have to Know about PD-L1 Expression in Prostate Cancer? A Systematic Literature Review. Part 2: Clinic–Pathologic Correlations. Cells 2021, 10, 3165. [Google Scholar] [CrossRef]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Hu, Q.; Rizvi, A.A.; Schau, G.; Ingale, K.; Muller, Y.; Baits, R.; Pretzer, S.; BenTaieb, A.; Gordhamer, A.; Nussenzveig, R.; et al. Development and validation of a deep learning-based microsatellite instability predictor from prostate cancer whole-slide images. npj Precis. Oncol. 2024, 8, 88. [Google Scholar] [CrossRef]
- Kavun, A.; Veselovsky, E.; Lebedeva, A.; Belova, E.; Kuznetsova, O.; Yakushina, V.; Grigoreva, T.; Mileyko, V.; Fedyanin, M.; Ivanov, M. Microsatellite Instability: A Review of Molecular Epidemiology and Implications for Immune Checkpoint Inhibitor Therapy. Cancers 2023, 15, 2288. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Fizazi, K.; Laird, D.; Matsubara, N.; Azad, A.; Joung, J.Y.; Fong, P.C.C.; Bruwaene, S.V.; Liu, G.; Voog, E.; et al. Exploration of circulating tumor cell (CTC) conversion and CTC0 as prognostic biomarkers for efficacy in TALAPRO-2: Phase 3 study of talazoparib (TALA) + enzalutamide (ENZA) vs placebo (PBO) + ENZA as first-line (1L) treatment in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2024, 42, 5023. [Google Scholar] [CrossRef]
- Kwan, E.M.; Hofman, M.S.; Ng, S.W.; Emmett, L.; Sandhu, S.; Buteau, J.P.; Iravani, A.; Joshua, A.M.; Francis, R.J.; Subhash, V.; et al. Circulating tumour DNA fraction as a predictor of treatment efficacy in a randomized phase 2 trial of [177Lu]Lu-PSMA-617 (LuPSMA) versus cabazitaxel in metastatic castration-resistant prostate cancer (mCRPC) progressing after docetaxel (TheraP ANZUP 1603). J. Clin. Oncol. 2024, 42, 5055. [Google Scholar] [CrossRef]
- Bono, J.S.D.; Morris, M.J.; Sartor, A.O.; Wei, X.X.; Fizazi, K.; Herrmann, K.; Piulats, J.M.; Mahammedi, H.; Logothetis, C.; George, D.J.; et al. Association of baseline and on-treatment ctDNA fraction with clinical outcomes in patients with mCRPC in the PSMAfore study of 177Lu-PSMA-617. J. Clin. Oncol. 2025, 43, 16. [Google Scholar] [CrossRef]
- Alarcón-Zendejas, A.P.; Scavuzzo, A.; Jiménez-Ríos, M.A.; Álvarez-Gómez, R.M.; Montiel-Manríquez, R.; Castro-Hernández, C.; Jiménez-Dávila, M.A.; Pérez-Montiel, D.; González-Barrios, R.; Jiménez-Trejo, F.; et al. The promising role of new molecular biomarkers in prostate cancer: From coding and non-coding genes to artificial intelligence approaches. Prostate Cancer Prostatic Dis. 2022, 25, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.S.; Lawrentschuk, N.; Francis, R.J.; Tang, C.; Vela, I.; Thomas, P.; Rutherford, N.; Martin, J.M.; Frydenberg, M.; Shakher, R.; et al. Prostate-specific membrane antigen PET-CT in patients with high-risk prostate cancer before curative-intent surgery or radiotherapy (proPSMA): A prospective, randomised, multicentre study. Lancet 2020, 395, 1208–1216. [Google Scholar] [CrossRef]
- Karan, D.; Holzbeierlein, J.M.; Van Veldhuizen, P.; Thrasher, J.B. Cancer immunotherapy: A paradigm shift for prostate cancer treatment. Nat. Rev. Urol. 2012, 9, 376–385. [Google Scholar] [CrossRef]
- Heo, Y.J.; Hwa, C.; Lee, G.-H.; Park, J.-M.; An, J.-Y. Integrative Multi-Omics Approaches in Cancer Research: From Biological Networks to Clinical Subtypes. Mol. Cells 2021, 44, 433–443. [Google Scholar] [CrossRef]
- Alqualo, N.O.; Campos-Fernandez, E.; Picolo, B.U.; Ferreira, E.L.; Henriques, L.M.; Lorenti, S.; Moreira, D.C.; Simião, M.P.S.; Oliveira, L.B.T.; Alonso-Goulart, V. Molecular biomarkers in prostate cancer tumorigenesis and clinical relevance. Crit. Rev. Oncol. Hematol. 2024, 194, 104232. [Google Scholar] [CrossRef]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.-S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate cancer and PARP inhibitors: Progress and challenges. J. Hematol. Oncol. 2021, 14, 51. [Google Scholar] [CrossRef]
- Kwon, W.A. PARP Inhibitors in the Treatment of Prostate Cancer: From Scientific Rationale to Clinical Development. World J. Mens. Health 2024, 42, 290–303. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, W.; Xiao, Y.-T.; Huang, H.; Gu, D.; Ren, S. Targeting signaling pathways in prostate cancer: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2022, 7, 198. [Google Scholar] [CrossRef]
- Feng, F.Y.; Smith, M.R.; Saad, F.; Mobadersany, P.; Tian, S.K.; Yip, S.S.F.; Greshock, J.; Khan, N.; Yu, M.K.; McCarthy, S.; et al. Digital Pathology-Based Multimodal Artificial Intelligence Scores and Outcomes in a Randomized Phase III Trial in Men With Nonmetastatic Castration-Resistant Prostate Cancer. JCO Precis. Oncol. 2025, 9, e2400653. [Google Scholar] [CrossRef] [PubMed]
- Marques, L.; Costa, B.; Pereira, M.; Silva, A.; Santos, J.; Saldanha, L.; Silva, I.; Magalhães, P.; Schmidt, S.; Vale, N. Advancing Precision Medicine: A Review of Innovative In Silico Approaches for Drug Development, Clinical Pharmacology and Personalized Healthcare. Pharmaceutics 2024, 16, 332. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Yang, Y.; Mortazavi, A.; Zhang, J. Emerging Immunotherapy Approaches for Treating Prostate Cancer. Int. J. Mol. Sci. 2023, 24, 14347. [Google Scholar] [CrossRef]
- Gamat, M.; McNeel, D.G. Androgen deprivation and immunotherapy for the treatment of prostate cancer. Endocr. Relat. Cancer 2017, 24, T297–T310. [Google Scholar] [CrossRef]
- Calvillo-Rodríguez, K.M.; Lorenzo-Anota, H.Y.; Rodríguez-Padilla, C.; Martínez-Torres, A.C.; Scott-Algara, D. Immunotherapies inducing immunogenic cell death in cancer: Insight of the innate immune system. Front. Immunol. 2023, 14, 1294434. [Google Scholar] [CrossRef]
- Bailly, C.; Thuru, X.; Quesnel, B. Combined cytotoxic chemotherapy and immunotherapy of cancer: Modern times. NAR Cancer 2020, 2, zcaa002. [Google Scholar] [CrossRef]
- Li, Z.; Lai, X.; Fu, S.; Ren, L.; Cai, H.; Zhang, H.; Gu, Z.; Ma, X.; Luo, K. Immunogenic Cell Death Activates the Tumor Immune Microenvironment to Boost the Immunotherapy Efficiency. Adv. Sci. 2022, 9, 2201734. [Google Scholar] [CrossRef]
- Nelson, B.E.; Adashek, J.J.; Lin, S.H.; Subbiah, V. The abscopal effect in patients with cancer receiving immunotherapy. Med 2023, 4, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; Liu, J.; Subudhi, S.K.; Poon, C.; Gant, K.L.; et al. Immune checkpoint therapy—Current perspectives and future directions. Cell 2023, 186, 1652–1669. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Jang, A.; Kendi, A.T.; Sartor, O. Status of PSMA-targeted radioligand therapy in prostate cancer: Current data and future trials. Ther. Adv. Med. Oncol. 2023, 15, 17588359231157632. [Google Scholar] [CrossRef]
- Ramnaraign, B.; Sartor, O. PSMA-Targeted Radiopharmaceuticals in Prostate Cancer: Current Data and New Trials. Oncologist 2023, 28, 392–401. [Google Scholar] [CrossRef]
- Yin, Q.; Wu, L.; Han, L.; Zheng, X.; Tong, R.; Li, L.; Bai, L.; Bian, Y. Immune-related adverse events of immune checkpoint inhibitors: A review. Front. Immunol. 2023, 14, 1167975. [Google Scholar] [CrossRef]
- Dorff, T.B.; Blanchard, M.S.; Adkins, L.N.; Luebbert, L.; Leggett, N.; Shishido, S.N.; Macias, A.; Del Real, M.M.; Dhapola, G.; Egelston, C.; et al. PSCA-CAR T cell therapy in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2024, 30, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Chehelgerdi, M.; Chehelgerdi, M.; Khorramian-Ghahfarokhi, M.; Shafieizadeh, M.; Mahmoudi, E.; Eskandari, F.; Rashidi, M.; Arshi, A.; Mokhtari-Farsani, A. Comprehensive review of CRISPR-based gene editing: Mechanisms, challenges, and applications in cancer therapy. Mol. Cancer 2024, 23, 9. [Google Scholar] [CrossRef]
- Xu, Z.; Xiao, Z.X.; Wang, J.; Qiu, H.W.; Cao, F.; Zhang, S.Q.; Xu, Y.D.; Lei, H.Q.; Xia, H.; He, Y.R.; et al. Novel mRNA adjuvant ImmunER enhances prostate cancer tumor-associated antigen mRNA therapy via augmenting T cell activity. Oncoimmunology 2024, 13, 2373526. [Google Scholar] [CrossRef]
- Riaz, I.B.; Harmon, S.; Chen, Z.; Naqvi, S.A.A.; Cheng, L. Applications of Artificial Intelligence in Prostate Cancer Care: A Path to Enhanced Efficiency and Outcomes. Am. Soc. Clin. Oncol. Educ. Book. 2024, 44, e438516. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Santos, M.J.; García-Martín, S.; Fustero-Torre, C.; Di Domenico, T.; Gómez-López, G.; Al-Shahrour, F. Bioinformatics roadmap for therapy selection in cancer genomics. Mol. Oncol. 2022, 16, 3881–3908. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.-P.; Qin, B.-D.; Jiao, X.-D.; Liu, K.; Wang, Z.; Zang, Y.-S. New clinical trial design in precision medicine: Discovery, development and direction. Signal Transduct. Target. Ther. 2024, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, S.; Zaheer, S. Advancements in pathology: Digital transformation, precision medicine, and beyond. J. Pathol. Inform. 2025, 16, 100408. [Google Scholar] [CrossRef]
- Galustian, C.; Dalgleish, A.; Bodman-Smith, M.; Kusmartsev, S.; Dasgupta, P. Editorial: Immunotherapy for Prostate Cancer—Turning the immunological desert into an oasis of hope. Front. Oncol. 2022, 12, 1021870. [Google Scholar] [CrossRef]
| Strategy | Selected Clinical Trials | Phase | Population (Patients) | Status/Key Findings |
|---|---|---|---|---|
| Immune checkpoint blockade | ||||
| CheckMate 650 (NCT02985957) | II | mCRPC | Limited efficacy; subgroup activity in selected biomarkers | |
| KEYNOTE-199 (NCT02787005) | II | mCRPC | Modest response (~5–10%); better in MSI-H/dMMR patients | |
| IMbassador250 (NCT03016312) | II | mCRPC (post abiraterone) | No significant OS benefit; better PFS in PD-L1/CD8-high subset | |
| CONTACT-02 (NCT04446117) | III | mCRPC (after novel hormonal therapy) | Improved PFS; no OS benefit overall; better results in liver/bone metastases | |
| Induction of ICD | ||||
| Docetaxel + vaccine combos (e.g., NCT02649855) | I/II | mHSPC | Mixed immunological outcomes; larger trials needed | |
| TME reversal (“cold-to-hot”) | ||||
| CSF-1R inhibitors (pexidartinib/NCT02472275) | I | Intermediate/high-risk PCa | Improved T-cell infiltration; combination trials ongoing | |
| Tumor vaccines | ||||
| IMPACT (sipuleucel-T/NCT00065442) | III | mCRPC | Improved survival; delayed responses typical | |
| PROSPECT (PROSTVAC/NCT01322490) | III | mCRPC | No significant survival benefit | |
| mRNA vaccine (BNT112/NCT04382898) | I/II | mCRPC and high-risk localized | Promising immunogenicity; ongoing | |
| Bispecific T-cell engagers (BiTEs) | ||||
| Xaluritamig (AMG 509/NCT04221542) | I→III | mCRPC (heavily pretreated) | Early promising PSA responses; notable CRS events; phase III planned | |
| Immune adjuvants | ||||
| SD-101 + pembrolizumab + RT + ADT/NCT03007732) | II | Hormone-naïve oligometastatic PCa | Enhanced immune response; moderate efficacy observed | |
| CAR T-cell therapy | ||||
| PSCA-directed CAR T/NCT0387380 | I | mCRPC | Early phase; promising but limited responses; ongoing | |
| Interventions targeting stromal barriers | ||||
| COSMIC-021 (cabozantinib + atezolizumab/NCT03170960) | I/II/III | mCRPC | Improved immune infiltration; transient clinical benefits |
| Approach | Mechanism | Representative Agents or Examples | Key Clinical Findings | Limitations/ Challenges | References |
|---|---|---|---|---|---|
| Checkpoint inhibitors | Block inhibitory receptors (PD-1, CTLA-4) on T cells | Nivolumab, ipilimumab | Effective in MSI-H subset | Limited overall response | [38,39] |
| Induction of ICD | Promote release of DAMPs; enhance antigen presentation and DC activation | Docetaxel, SBRT | May synergize with ICIs | Inconsistent biomarkers | [52,53] |
| TME reversal | Modify the immunosuppressive milieu; shift M2-like TAMs to an M1 phenotype; reduce Treg and MDSC infiltration | CSF-1R inhibitors | Improved T-cell infiltration | Multiple targets needed | [59,60] |
| Tumor vaccines | Present tumor-specific antigens (e.g., PSA, PAP) to prime T cells; elicit long-term adaptive immunity | Sipuleucel-T | Improved survival | Antigen escape possible | [65,66,67] |
| CAR T-cell therapy | Genetically engineer T cells to target prostate-specific antigens (e.g., PSMA, PAP); potential for high specificity | PSMA-targeted CAR | Early responses observed | Toxicity and high cost | [68] |
| Interventions targeting stromal barriers | Alter tumor stroma/fibrosis to allow for T-cell penetration; normalize vasculature for better immune cell and drug delivery | TGF-β inhibitors | Enhanced immune infiltration | Risk of damaging healthy tissue | [78] |
| Biomarker | Detection Method | Clinical Significance | Limitations/Challenges | References |
|---|---|---|---|---|
| PD-L1 expression | IHC | Partial predictive value | Generally low expression | [90,92] |
| MSI-H/dMMR | NGS, IHC | Predicts checkpoint inhibitor response | Rare in prostate cancer | [93,95] |
| CDK12 alterations | NGS panels | Associated with neoantigen load | Requires validation | [91] |
| TMB | Whole-exome sequencing | Indicates tumor immunogenicity | Lack of standardized cutoffs | [89] |
| DNA repair mutations | NGS | Facilitates targeted therapy | Variability in response | [104,105] |
| Liquid biopsy (ctDNA, CTC) | Blood-based analysis | Tracks resistance in real time | Sensitivity/specificity issues | [97,98] |
| Combination Approach | Mechanism/Rationale | Representative Trials or Evidence | Key Outcomes/ Observations | Challenges | References |
|---|---|---|---|---|---|
| Hormonal + immunotherapy | Transiently enhances T-cell infiltration by modulating AR signaling; may upregulate tumor antigens | ADT + ICIs | Improved T-cell infiltration | Optimal timing unclear | [39,110] |
| Chemo + immunotherapy | Certain chemotherapeutics (e.g., docetaxel) induce immunogenic cell death; potentially increase antigen release | Docetaxel + ICIs | Enhanced immune responses | Variable ICD induction | [111,112] |
| Radiotherapy + immunotherapy | Radiation can induce local tumor cell death and promote abscopal effects; releases tumor antigens for DC activation | SBRT + ICIs | Potential abscopal effect | Inconsistent results | [113,114] |
| PARP inhibitors + immunotherapy | DNA repair inhibition elevates tumor mutational load; may increase neoantigen presentation | Olaparib + ICIs | Efficacy in DNA repair-deficient patients | Limited to certain mutations | [104] |
| TME-targeting agents + immunotherapy | Disrupt immunosuppressive networks (MDSCs, Tregs, TAMs) to facilitate T-cell infiltration | IDO, TGF-β inhibitors + ICIs | Improved T-cell activity | Complexity of pathways | [60,61,67] |
| Multi-target regimens | Simultaneously address multiple resistance mechanisms (e.g., PD-1 + CTLA-4 + hormonal + radiotherapy) | PD-1 + CTLA-4 + ADT | Potential increased response | High cumulative toxicity | [67,115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, W.-A.; Joung, J.Y. Immunotherapy in Prostate Cancer: From a “Cold” Tumor to a “Hot” Prospect. Cancers 2025, 17, 1064. https://doi.org/10.3390/cancers17071064
Kwon W-A, Joung JY. Immunotherapy in Prostate Cancer: From a “Cold” Tumor to a “Hot” Prospect. Cancers. 2025; 17(7):1064. https://doi.org/10.3390/cancers17071064
Chicago/Turabian StyleKwon, Whi-An, and Jae Young Joung. 2025. "Immunotherapy in Prostate Cancer: From a “Cold” Tumor to a “Hot” Prospect" Cancers 17, no. 7: 1064. https://doi.org/10.3390/cancers17071064
APA StyleKwon, W.-A., & Joung, J. Y. (2025). Immunotherapy in Prostate Cancer: From a “Cold” Tumor to a “Hot” Prospect. Cancers, 17(7), 1064. https://doi.org/10.3390/cancers17071064