
Mutational Profile of Blood and Tumor Tissue and Biomarkers of Response to PD-1 Inhibitors in Patients with Cutaneous Squamous Cell Carcinoma

 , , , and
, , , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Clinical Management and Treatment Response
2.3. Tumor Molecular Profiling
2.4. Statistical Analysis
3. Results
3.1. Demographics and Tumor Characteristics
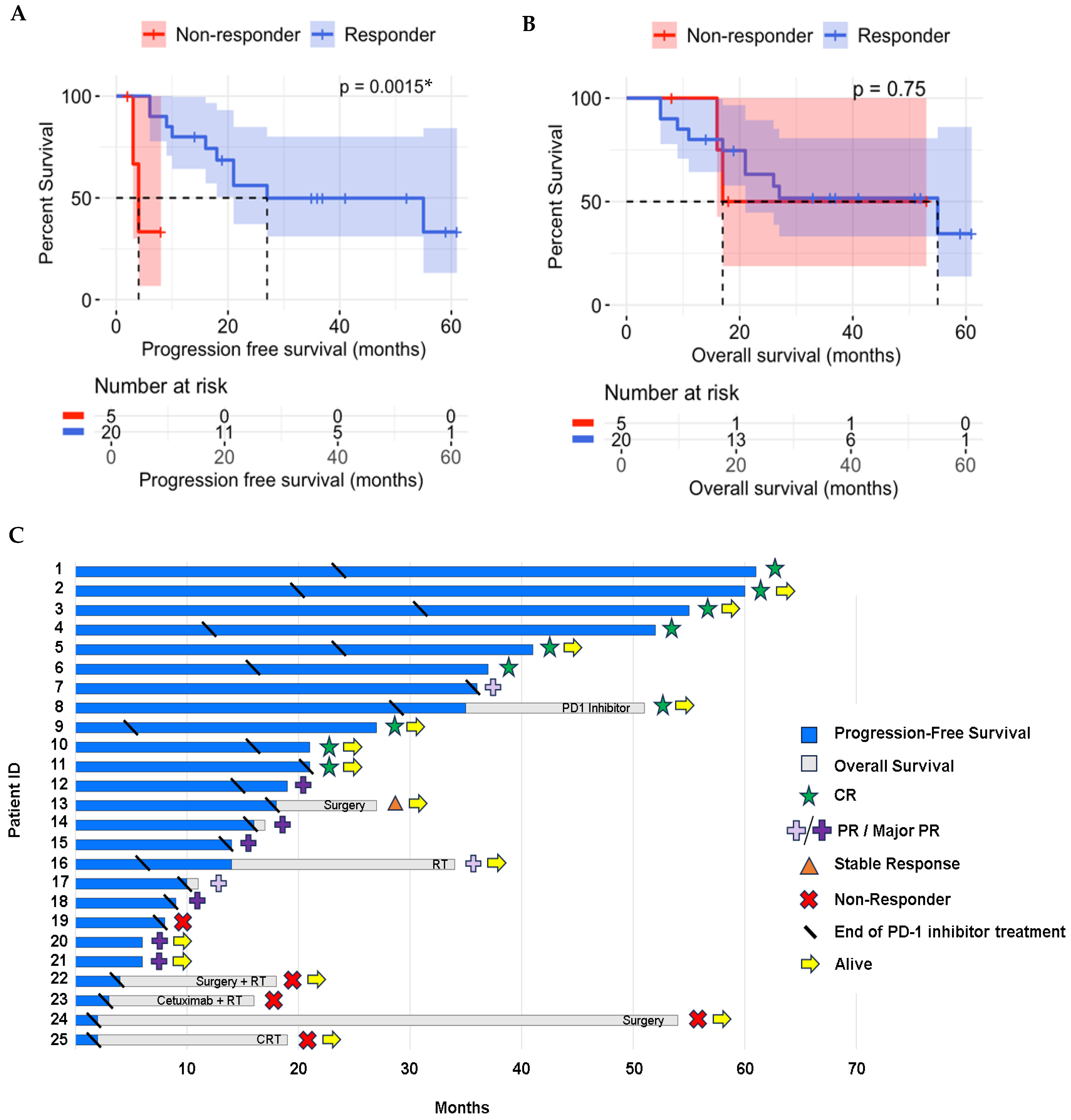
3.2. Treatment Results
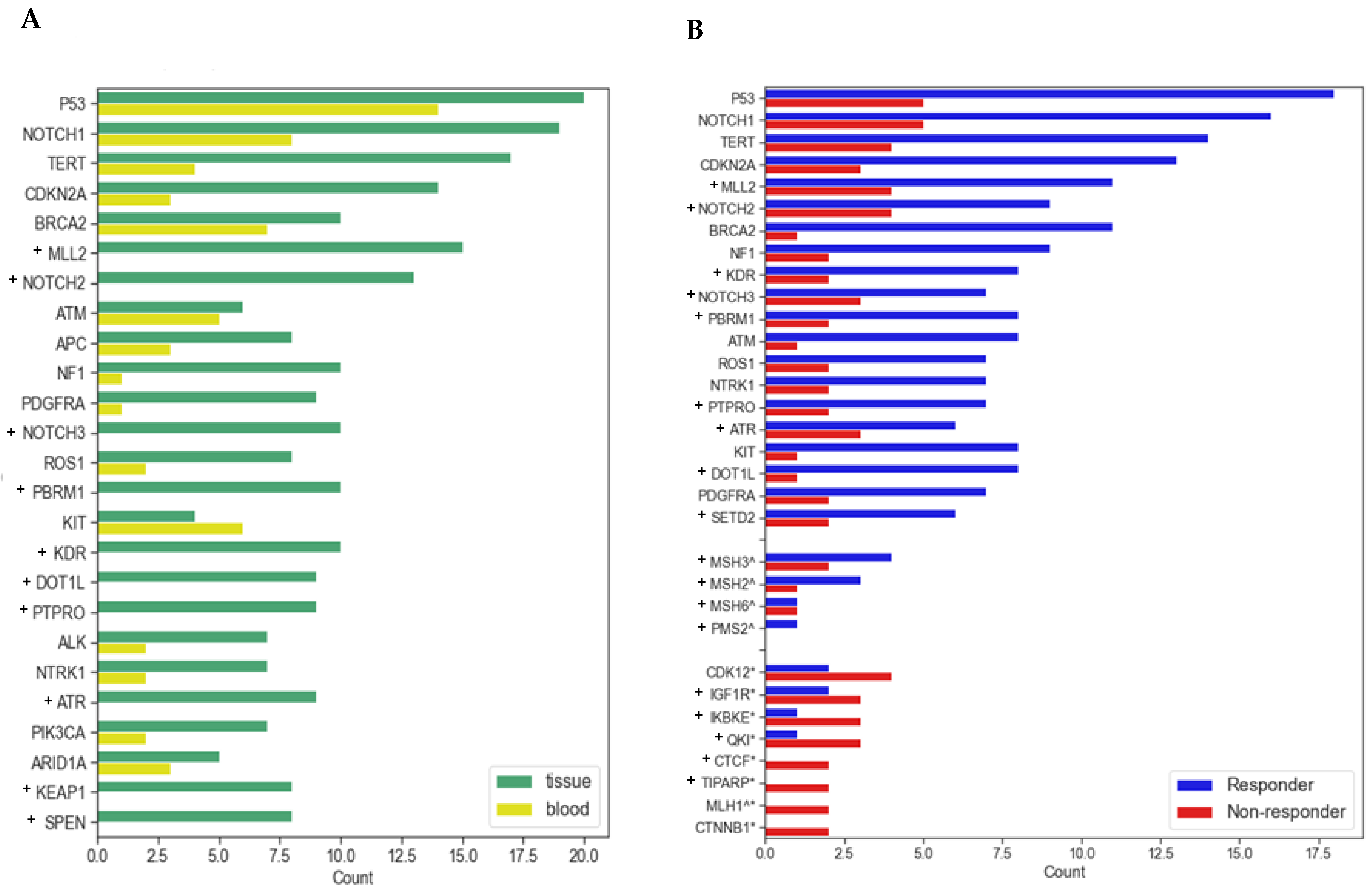
3.3. Molecular and Genomic Results
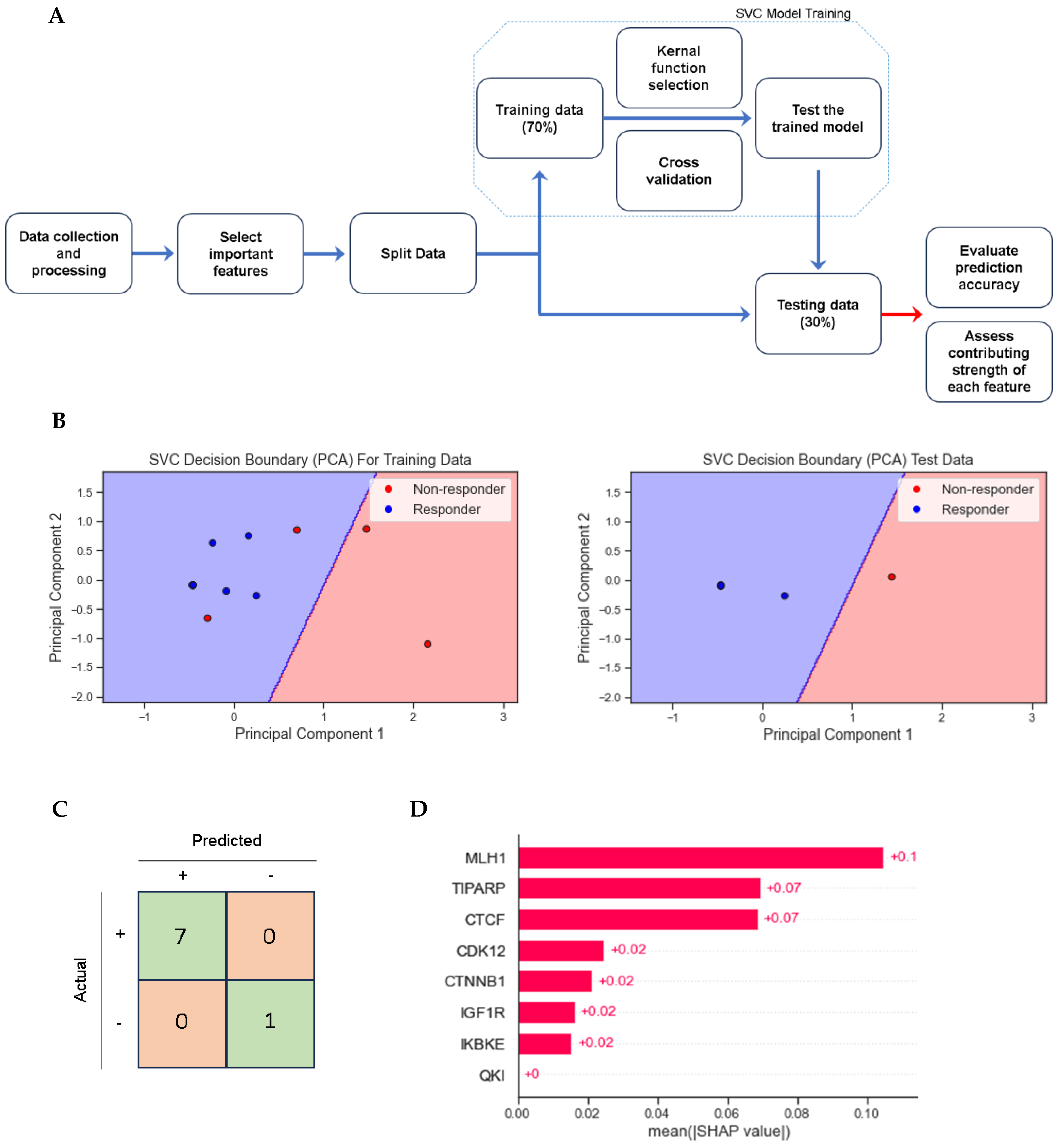
3.4. Multivariate and Support Vector Classifier Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Circulating tumor DNA | ctDNA |
| Combined positive score | CPS |
| Cutaneous squamous cell carcinoma | cSCC |
| Circulating tumor DNA | ctDNA |
| Cross-validation score | CV score |
| DNA damage repair | DDR |
| Eastern Cooperative Oncology group | ECOG |
| Immune-related Response Evaluation Criteria in Solid Tumors | iRECIST |
| Leave-one-out cross-validation | LOO-CV |
| Multivariate analysis of variance | MANOVA |
| Microsatellite instability | MSI |
| Mismatch repair | MMR |
| National Comprehensive Cancer Network | NCCN |
| Next generation sequencing | NGS |
| Overall survival | OS |
| Principal component analysis | PCA |
| Programmed cell death protein 1 | PD-1 |
| Programmed death-ligand 1 | PD-L1 |
| Progression-free survival | PFS |
| Radiation therapy | RT |
| Tumor tissue DNA | tDNA |
| Tumor microenvironment | TME |
| Tumor mutational burden | TMB |
| Tumor proportion score | TPS |
| Support vector classifier | SVC |
| SWItch/Sucrose Non-Fermentable | SWI/SNF |
References
- Wysong, A. Squamous-Cell Carcinoma of the Skin. N. Engl. J. Med. 2023, 388, 2262–2273. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Fritz, M.; Que, S.K.T. Cutaneous Squamous Cell Carcinoma: An Updated Review. Cancers 2024, 16, 1800. [Google Scholar] [CrossRef] [PubMed]
- Winge, M.C.G.; Kellman, L.N.; Guo, K.; Tang, J.Y.; Swetter, S.M.; Aasi, S.Z.; Sarin, K.Y.; Chang, A.L.S.; Khavari, P.A. Advances in cutaneous squamous cell carcinoma. Nat. Rev. Cancer 2023, 23, 430–449. [Google Scholar] [CrossRef] [PubMed]
- Brunner, M.; Veness, M.J.; Ch’ng, S.; Elliott, M.; Clark, J.R. Distant metastases from cutaneous squamous cell carcinoma—Analysis of AJCC stage IV. Head Neck 2013, 35, 72–75. [Google Scholar] [CrossRef]
- Brougham, N.D.; Dennett, E.R.; Cameron, R.; Tan, S.T. The incidence of metastasis from cutaneous squamous cell carcinoma and the impact of its risk factors. J. Surg. Oncol. 2012, 106, 811–815. [Google Scholar] [CrossRef]
- Thompson, A.K.; Kelley, B.F.; Prokop, L.J.; Murad, M.H.; Baum, C.L. Risk Factors for Cutaneous Squamous Cell Carcinoma Recurrence, Metastasis, and Disease-Specific Death: A Systematic Review and Meta-analysis. JAMA Dermatol. 2016, 152, 419–428. [Google Scholar] [CrossRef]
- Chong, C.Y.; Goh, M.S.; Porceddu, S.V.; Rischin, D.; Lim, A.M. The Current Treatment Landscape of Cutaneous Squamous Cell Carcinoma. Am. J. Clin. Dermatol. 2023, 24, 25–40. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Kyriakos, P.; Papadopoulos, M.; Johnson, M.L.; Martin, M.G.; Moreno, V.; Salama, A.K.S.; Calvo, E.; Yee, N.S.; Safran, H.; et al. Phase 1 study of cemiplimab, a human monoclonal anti-PD-1, in patients with unresectable locally advanced or metastatic cutaneous squamous cell carcinoma (CSCC): Final efficacy and safety data. J. Clin. Oncol. 2018, 36, 9557. [Google Scholar] [CrossRef]
- Rischin, D.; Migden, M.R.; Lim, A.M.; Schmults, C.D.; Khushalani, N.I.; Hughes BG, M.; Schadendorf, D.; Dunn, L.A.; Hernandez-Aya, L.; Chang, A.L.S.; et al. Phase 2 study of cemiplimab, a human monoclonal anti-PD-1, in patients (pts) with metastatic cutaneous squamous cell carcinoma (mCSCC.; Group 1): 12-month follow-up. J. Clin. Oncol. 2019, 37, 9526. [Google Scholar] [CrossRef]
- Grob, J.J.; Gonzalez, R.; Basset-Seguin, N.; Vornicova, O.; Schachter, J.; Joshi, A.; Meyer, N.; Grange, F.; Piulats, J.M.; Bauman, J.R.; et al. Pembrolizumab Monotherapy for Recurrent or Metastatic Cutaneous Squamous Cell Carcinoma: A Single-Arm Phase II Trial (KEYNOTE-629). J. Clin. Oncol. 2020, 38, 2916–2925. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B.G.M.; Munoz-Couselo, E.; Mortier, L.; Bratland, Å.; Gutzmer, R.; Roshdy, O.; González Mendoza, R.; Schachter, J.; Arance, A.; Grange, F.; et al. Pembrolizumab for locally advanced and recurrent/metastatic cutaneous squamous cell carcinoma (KEYNOTE-629 study): An open-label, nonrandomized, multicenter, phase II trial. Ann. Oncol. 2021, 32, 1276–1285, Erratum in Ann. Oncol. 2022, 33, 853. https://doi.org/10.1016/j.annonc.2022.05.517. [Google Scholar] [CrossRef] [PubMed]
- Silk, A.W.; Barker, C.A.; Bhatia, S.; Bollin, K.B.; Chandra, S.; Eroglu, Z.; Gastman, B.R.; Kendra, K.L.; Kluger, H.; Lipson, E.J.; et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immunotherapy for the treatment of nonmelanoma skin cancer. J. Immunother. Cancer 2022, 10, e004434. [Google Scholar] [CrossRef]
- Hober, C.; Fredeau, L.; Pham-Ledard, A.; Boubaya, M.; Herms, F.; Celerier, P.; Aubin, F.; Beneton, N.; Dinulescu, M.; Jannic, A.; et al. Cemiplimab for Locally Advanced and Metastatic Cutaneous Squamous-Cell Carcinomas: Real-Life Experience from the French CAREPI Study Group. Cancers 2021, 13, 3547. [Google Scholar] [CrossRef]
- Guillaume, T.; Puzenat, E.; Popescu, D.; Aubin, F.; Nardin, C. Cemiplimab-rwlc in advanced cutaneous squamous cell carcinoma: Real-world experience in a French dermatology department. Br. J. Dermatol. 2021, 185, 1056–1058. [Google Scholar] [CrossRef]
- Baggi, A.; Quaglino, P.; Rubatto, M.; Depenni, R.; Guida, M.; Ascierto, P.A.; Trojaniello, C.; Queirolo, P.; Saponara, M.; Peris, K.; et al. Real world data of cemiplimab in locally advanced and metastatic cutaneous squamous cell carcinoma. Eur. J. Cancer 2021, 157, 250–258, Erratum in Eur. J. Cancer 2022, 166, 309–310. https://doi.org/10.1016/j.ejca.2022.02.027. [Google Scholar] [CrossRef]
- Salzmann, M.; Leiter, U.; Loquai, C.; Zimmer, L.; Ugurel, S.; Gutzmer, R.; Thoms, K.M.; Enk, A.H.; Hassel, J.C. Programmed cell death protein 1 inhibitors in advanced cutaneous squamous cell carcinoma: Real-world data of a retrospective, multicenter study. Eur. J. Cancer 2020, 138, 125–132. [Google Scholar] [CrossRef]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Primary analysis of phase 2 results of cemiplimab, a human monoclonal anti-PD-1, in patients (pts) with locally advanced cutaneous squamous cell carcinoma (laCSCC). J. Clin. Oncol. 2019, 37, 6015. [Google Scholar] [CrossRef]
- Maubec, E.; Boubaya, M.; Petrow, P.; Beylot-Barry, M.; Basset-Seguin, N.; Deschamps, L.; Grob, J.J.; Dréno, B.; Scheer-Senyarich, I.; Bloch-Queyrat, C.; et al. Phase II Study of Pembrolizumab as First-Line, Single-Drug Therapy for Patients with Unresectable Cutaneous Squamous Cell Carcinomas. J. Clin. Oncol. 2020, 38, 3051–3061. [Google Scholar] [CrossRef]
- Hanna, G.J.; Ruiz, E.S.; LeBoeuf, N.R.; Thakuria, M.; Schmults, C.D.; Decaprio, J.A.; Silk, A.W. Real-world outcomes treating patients with advanced cutaneous squamous cell carcinoma with immune checkpoint inhibitors (CPI). Br. J. Cancer 2020, 123, 1535–1542. [Google Scholar] [CrossRef] [PubMed]
- In, G.K.; Vaidya, P.; Filkins, A.; Hermel, D.J.; King, K.G.; Ragab, O.; Tseng, W.W.; Swanson, M.; Kokot, N.; Lang, J.E.; et al. PD-1 inhibition therapy for advanced cutaneous squamous cell carcinoma: A retrospective analysis from the University of Southern California. J. Cancer Res. Clin. Oncol. 2021, 147, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Kacew, A.J.; Harris, E.J.; Lorch, J.H.; Haddad, R.I.; Chau, N.G.; Rabinowits, G.; LeBoeuf, N.R.; Schmults, C.D.; Thakuria, M.; MacConaill, L.E.; et al. Chromosome 3q arm gain linked to immunotherapy response in advanced cutaneous squamous cell carcinoma. Eur. J. Cancer 2019, 113, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotto, R.; Amit, M.; Nagarajan, P.; Rubin, M.L.; Yuan, Y.; Bell, D.; El-Naggar, A.K.; Johnson, J.M.; Morrison, W.H.; Rosenthal, D.I.; et al. Pilot Phase II Trial of Neoadjuvant Immunotherapy in Locoregionally Advanced, Resectable Cutaneous Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2021, 27, 4557–4565, Erratum in Clin. Cancer Res. 2022, 28, 1735. https://doi.org/10.1158/1078-0432.CCR-22-0468. [Google Scholar] [CrossRef]
- Wang, D.; Elenbaas, B.; Murugesan, K.; Shah, K.; Montesion, M.; Gounaris, I.; Scheuenpflug, J.; Locatelli, G.; Feng, Z. Relationship among DDR gene mutations, TMB and PD-L1 in solid tumour genomes identified using clinically actionable biomarker assays. NPJ Precis. Oncol. 2023, 7, 103. [Google Scholar] [CrossRef]
- Assam, J.H.; Powell, S.; Spanos, W.C. Unresectable cutaneous squamous cell carcinoma of the forehead with MLH1 mutation showing dramatic response to Programmed Cell Death Protein 1 Inhibitor Therapy. Clin. Skin. Cancer 2016, 1, 26–29. [Google Scholar] [CrossRef]
- Zelin, E.; Maronese, C.A.; Dri, A.; Toffoli, L.; Di Meo, N.; Nazzaro, G.; Zalaudek, I. Identifying Candidates for Immunotherapy among Patients with Non-Melanoma Skin Cancer: A Review of the Potential Predictors of Response. J. Clin. Med. 2022, 11, 3364. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593, Erratum in Science 2019, 363, eaax1384. https://doi.org/10.1126/science.aax1384. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Lu, K.Q.; Li, Z.L.; Zhang, Q.; Yin, Q.; Zhang, Y.L.; Ni, W.J.; Jiang, L.Z.; He, W.; Wang, B. CDK12 is a potential biomarker for diagnosis, prognosis and immunomodulation in pan-cancer. Sci. Rep. 2024, 14, 6574. [Google Scholar] [CrossRef]
- Gao, C.; Wang, Y.; Broaddus, R.; Sun, L.; Xue, F.; Zhang, W. Exon 3 mutations of CTNNB1 drive tumorigenesis: A review. Oncotarget 2017, 9, 5492–5508. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, Q.; Liu, J.; Zhang, W. CTNNB1 Alternation Is a Potential Biomarker for Immunotherapy Prognosis in Patients with Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 759565. [Google Scholar] [CrossRef]
- Shan, Q.; Zhu, S.; Chen, X.; Liu, J.; Yuan, S.; Li, X.; Peng, W.; Xue, H.H. Tcf1-CTCF cooperativity shapes genomic architecture to promote CD8+ T cell homeostasis. Nat. Immunol. 2022, 23, 1222–1235. [Google Scholar] [CrossRef]
- Wang, W.; Chandra, A.; Goldman, N.; Yoon, S.; Ferrari, E.K.; Nguyen, S.C.; Joyce, E.F.; Vahedi, G. TCF-1 promotes chromatin interactions across topologically associating domains in T cell progenitors. Nat. Immunol. 2022, 23, 1052–1062. [Google Scholar] [CrossRef]
- Ajona, D.; Ortiz-Espinosa, S.; Lozano, T.; Exposito, F.; Calvo, A.; Valencia, K.; Redrado, M.; Remírez, A.; Lecanda, F.; Alignani, D.; et al. Short-term starvation reduces IGF-1 levels to sensitize lung tumors to PD-1 immune checkpoint blockade. Nat. Cancer 2020, 1, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Alfaro-Arnedo, E.; López, I.P.; Piñeiro-Hermida, S.; Canalejo, M.; Gotera, C.; Sola, J.J.; Roncero, A.; Peces-Barba, G.; Ruíz-Martínez, C.; Pichel, J.G. IGF1R acts as a cancer-promoting factor in the tumor microenvironment facilitating lung metastasis implantation and progression. Oncogene 2022, 41, 3625–3639. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Revach, O.Y.; Anderson, S.; Kessler, E.A.; Wolfe, C.H.; Jenney, A.; Mills, C.E.; Robitschek, E.J.; Davis TG, R.; Kim, S.; et al. Targeting TBK1 to overcome resistance to cancer immunotherapy. Nature 2023, 615, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Wang, X.; Lu, J. Advances in IKBKE as a potential target for cancer therapy. Cancer Med. 2020, 9, 247–258. [Google Scholar] [CrossRef]
- Gozgit, J.M.; Vasbinder, M.M.; Abo, R.P.; Kunii, K.; Kuplast-Barr, K.G.; Gui, B.; Lu, A.Z.; Molina, J.R.; Minissale, E.; Swinger, K.K.; et al. PARP7 negatively regulates the type I interferon response in cancer cells and its inhibition triggers antitumor immunity. Cancer Cell 2021, 39, 1214–1226.e10. [Google Scholar] [CrossRef]
- Wolde, T.; Huang, J.; Huang, P.; Pandey, V.; Qin, P. Depleted-MLH1 Expression Predicts Prognosis and Immunotherapeutic Efficacy in Uterine Corpus Endometrial Cancer: An In Silico Approach. BioMedInformatics 2024, 4, 326–346. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar]
- de Bruijn, I.; Kundra, R.; Mastrogiacomo, B.; Tran, T.N.; Sikina, L.; Mazor, T.; Li, X.; Ochoa, A.; Zhao, G.; Lai, B.; et al. Analysis and Visualization of Longitudinal Genomic and Clinical Data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 2023, 83, 3861–3867. [Google Scholar]
- Jee, J.; Fong, C.; Pichotta, K.; Tran, T.N.; Luthra, A.; Waters, M.; Fu, C.; Altoe, M.; Liu, S.Y.; Maron, S.B.; et al. Automated real-world data integration improves cancer outcome prediction. Nature 2024, 636, 728–736. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Margolis, C.A.; Vokes, N.I.; Liu, D.; Taylor-Weiner, A.; Wankowicz, S.M.; Adeegbe, D.; Keliher, D.; Schilling, B.; Tracy, A.; et al. Genomic correlates of response to immune checkpoint blockade in microsatellite-stable solid tumors. Nat. Genet. 2018, 50, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Piha-Paul, S.A.; Won, H.; Rodon, J.; Saura, C.; Shapiro, G.I.; Juric, D.; Quinn, D.I.; Moreno, V.; Doger, B.; et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018, 554, 189–194. [Google Scholar] [CrossRef]
- Rosen, E.Y.; Goldman, D.A.; Hechtman, J.F.; Benayed, R.; Schram, A.M.; Cocco, E.; Shifman, S.; Gong, Y.; Kundra, R.; Solomon, J.P.; et al. TRK Fusions Are Enriched in Cancers with Uncommon Histologies and the Absence of Canonical Driver Mutations. Clin. Cancer Res. 2020, 26, 1624–1632. [Google Scholar] [CrossRef]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef]
- Wu, L.; Yao, H.; Chen, H.; Wang, A.; Guo, K.; Gou, W.; Yu, Y.; Li, X.; Yao, M.; Yuan, S.; et al. Landscape of somatic alterations in large-scale solid tumors from an Asian population. Nat. Commun. 2022, 13, 4264. [Google Scholar] [CrossRef]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 2022, 185, 563–575.e11. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Patients | Responders | Non-Responders | (p-Value *) | ||||
|---|---|---|---|---|---|---|---|
| (n = 25) | (n = 20) | (n = 5) | |||||
| Age (range) | 77 | (58–97) | 78 | (59–97) | 75 | (58–92) | 0.73 |
| Gender | 0.53 | ||||||
| Male | 20 | 80% | 17 | 85% | 3 | 60% | |
| Female | 5 | 20% | 3 | 15% | 2 | 40% | |
| ECOG performance status | 0.29 | ||||||
| 0 | 2 | 8% | 1 | 5% | 1 | 20% | |
| 1 | 15 | 60% | 12 | 60% | 3 | 60% | |
| 2 | 6 | 24% | 6 | 30% | 0 | 0% | |
| 3 | 2 | 8% | 1 | 5% | 1 | 20% | |
| Smoking history | 0.48 | ||||||
| Never smoker | 12 | 48% | 10 | 50% | 2 | 40% | |
| Former smoker | 10 | 40% | 7 | 35% | 3 | 60% | |
| Current smoker | 3 | 12% | 3 | 15% | 0 | 0% | |
| Immunosuppression | 0.65 | ||||||
| None | 22 | 88% | 17 | 85% | 17 | 85% | |
| Heme malignancy | 2 | 8% | 2 | 10% | 2 | 10% | |
| Autoimmune disorder | 1 | 4% | 1 | 5% | 1 | 0% | |
| Primary site of disease | 0.33 | ||||||
| Face | 5 | 20% | 5 | 25% | 0 | 0% | |
| Scalp | 9 | 36% | 7 | 35% | 2 | 40% | |
| Eye orbit | 1 | 4% | 1 | 5% | 0 | 0% | |
| Ear | 3 | 12% | 2 | 10% | 1 | 20% | |
| Nose | 2 | 8% | 2 | 10% | 0 | 0% | |
| Neck | 3 | 12% | 3 | 15% | 0 | 0% | |
| Extremity | 1 | 4% | 0 | 0% | 1 | 20% | |
| Cancer status | 0.39 | ||||||
| LRA stage III or IV | 21 | 84% | 16 | 80% | 5 | 100% | |
| Recurrent | 16 | 64% | 14 | 70% | 2 | 40% | |
| Distant metastasis | 4 | 16% | 4 | 20% | 2 | 0% | |
| Prior treatment | 0.66 | ||||||
| None | 5 | 20% | 5 | 25% | 0 | 0% | |
| Surgery alone | 11 | 44% | 8 | 40% | 3 | 60% | |
| RT alone | 1 | 4% | 1 | 5% | 0 | 0% | |
| Chemo alone | 0 | 0% | 0 | 0% | 0 | 0% | |
| Surgery + RT | 7 | 28% | 5 | 25% | 2 | 40% | |
| RT + chemo | 0 | 0% | 0 | 0% | 0 | 0% | |
| Surgery + chemo | 0 | 0% | 0 | 0% | 0 | 0% | |
| Surgery + RT + chemo | 1 | 4% | 1 | 5% | 0 | 0% | |
| Treatment with PD-1 inhibitor | |||||||
| Average # of treatments | 19 | 22 | 6 | ||||
| Cemiplimab | 11 | 44% | 8 | 40% | 3 | 60% | |
| Nivolumab | 2 | 8% | 2 | 10% | 0 | 0% | |
| Pembrolizumab | 12 | 48% | 10 | 50% | 2 | 40% | |
| Genomic testing availability | |||||||
| ctDNA | 23 | 96% | 18 | 90% | 5 | 100% | |
| tDNA | 20 | 80% | 15 | 65% | 5 | 100% | |
| Both | 18 | 64% | 13 | 55% | 5 | 100% | |
| A | |||
| Gene | Responders | Non-Responders | (p-Value *) |
| CDK12 | 2/20 | 4/5 | (0.005 *) |
| CTCF | 0/20 | 2/5 | (0.033 *) |
| CTNNB1 | 0/20 | 2/5 | (0.033 *) |
| IGF1R | 2/20 | 3/5 | (0.038 *) |
| IKBKE | 1/20 | 3/5 | (0.016 *) |
| MLH1 | 0/20 | 2/5 | (0.033 *) |
| QKI | 1/20 | 3/5 | (0.016 *) |
| TIPARP | 0/20 | 2/5 | (0.033 *) |
| B | |||
| DDR Gene Groups | (p-Value *) | Mismatch Repair | |
| Base excision repair | - | ||
| Damage sensor | (0.5) + | ||
| Fanconi anemia | (0.32) | ||
| Homologous recombination | (0.22) | ||
| Mismatch repair | (0.04 *) | MLH1 | (0.03 *) |
| MSH2 | (1) | ||
| MSH3 | (0.56) | ||
| MSH6 | (0.37) | ||
| MS2 | (1) | ||
| Nucleotide excision | (0.13) | ||
| SWI/SNF chromatin remodeling | (0.32) | ||
| TP53 pathway | (0.15) | ||
| Three-Gene Combinations | OR | 95%CI | (p-Value *) | CV Score |
|---|---|---|---|---|
| CDK12 | 0.05 | (−8.05, −0.91) | 0.003 * | 0.97 |
| TIPARP | 0.003 | (−16.03, −1.44) | 0.009 * | |
| IGF1R | 0.04 | (−11.87, −0.23) | 0.063 | |
| CDK12 | 0.15 | (−4.3, −0.26) | 0.023 * | 0.97 |
| MLH1 | 0.39 | (−6.29, 2.86) | 0.547 | |
| IGF1R | 0.22 | (−4.55, 1.42) | 0.282 | |
| TIPARP | 0.03 | (−8.59, −0.82) | 0.010 * | 0.97 |
| MLH1 | 0.03 | (−10.14, −0.27) | 0.067 | |
| IGF1R | 0.61 | (−3.95, 4.98) | 0.766 | |
| CDK12 | 0.09 | (−7.19, −0.42) | 0.018 * | 0.93 |
| TIPARP | 0.01 | (−13.29, −1.37) | 0.005 * | |
| MLH1 | 0.04 | (−8.42, 0.14) | 0.054 | |
| Two-Gene Combinations | OR | 95%CI | (p-Value *) | CV Score |
| TIPARP | 0.01 | (−9.35, −1.29) | 0.004 * | 0.97 |
| MLH1 | 0.01 | (−9.35, −1.29) | 0.004 * | |
| CDK12 | 0.12 | (−4.67, −0.51) | 0.009 * | 0.97 |
| IGF1R | 0.13 | (−4.87, 0.44) | 0.101 | |
| CDK12 | 0.13 | (−4.74, −0.31) | 0.020 * | 0.93 |
| MLH1 | 0.12 | (−7.22, 1.04) | 0.181 | |
| CDK12 | 0.03 | (−8.54, −0.99) | 0.002 * | 0.82 |
| TIPARP | 0.01 | (−10.49, −0.9) | 0.013 * | |
| MLH1 | 0.12 | (−7.33, 1.12) | 0.203 | 0.8 |
| IGF1R | 0.23 | (−3.97, 1.15) | 0.244 | |
| TIPARP | 0.03 | (−8.73, −0.32) | 0.030 * | 0.77 |
| IGF1R | 0.09 | (−5.2, −0.09) | 0.042 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, M.J.; Stamos, D.B.; Urtis, C.; Bowers, N.L.; Schmalz, L.M.; Deyo, L.J.; Porebski, M.F.; Jabir, A.R.; Bunch, P.M.; Lycan, T.W., Jr.; et al. Mutational Profile of Blood and Tumor Tissue and Biomarkers of Response to PD-1 Inhibitors in Patients with Cutaneous Squamous Cell Carcinoma. Cancers 2025, 17, 1172. https://doi.org/10.3390/cancers17071172
Chang MJ, Stamos DB, Urtis C, Bowers NL, Schmalz LM, Deyo LJ, Porebski MF, Jabir AR, Bunch PM, Lycan TW Jr., et al. Mutational Profile of Blood and Tumor Tissue and Biomarkers of Response to PD-1 Inhibitors in Patients with Cutaneous Squamous Cell Carcinoma. Cancers. 2025; 17(7):1172. https://doi.org/10.3390/cancers17071172
Chicago/Turabian StyleChang, Mark J., Daniel B. Stamos, Cetin Urtis, Nathan L. Bowers, Lauren M. Schmalz, Logan J. Deyo, Martin F. Porebski, Abdur Rahman Jabir, Paul M. Bunch, Thomas W. Lycan, Jr., and et al. 2025. "Mutational Profile of Blood and Tumor Tissue and Biomarkers of Response to PD-1 Inhibitors in Patients with Cutaneous Squamous Cell Carcinoma" Cancers 17, no. 7: 1172. https://doi.org/10.3390/cancers17071172
APA StyleChang, M. J., Stamos, D. B., Urtis, C., Bowers, N. L., Schmalz, L. M., Deyo, L. J., Porebski, M. F., Jabir, A. R., Bunch, P. M., Lycan, T. W., Jr., Buchanan Doerfler, L., Patwa, H. S., Waltonen, J. D., Sullivan, C. A., Browne, J. D., Zhang, W., & Porosnicu, M. (2025). Mutational Profile of Blood and Tumor Tissue and Biomarkers of Response to PD-1 Inhibitors in Patients with Cutaneous Squamous Cell Carcinoma. Cancers, 17(7), 1172. https://doi.org/10.3390/cancers17071172