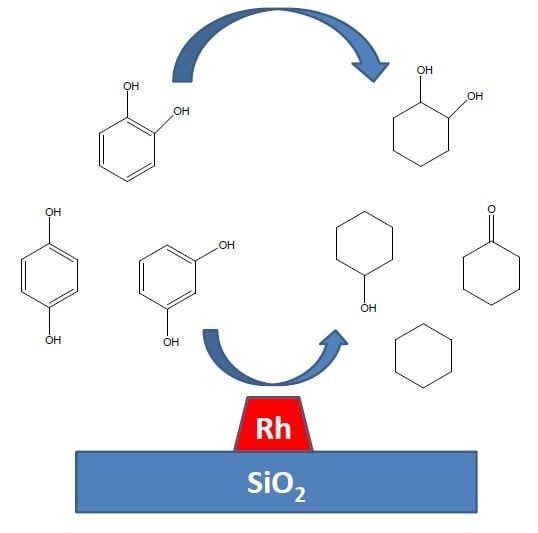
2.1. Hydrogen Reactions
Extra reaction profiles and data analysis are contained in the
Supplementary Data File. To ascertain activation energies and order in reactants, hydrogenation was carried out over a range of temperatures and concentrations (
Table 1). From
Table 1 it can be seen the order of reactivity followed: resorcinol > catechol > hydroquinone and the strength of adsorption based on the order of reaction in organic followed: catechol > hydroquinone > resorcinol. Xylene hydrogenation over a similar catalyst gave an order of reactivity of
para >
ortho >
meta, which is the reverse of that found with dihydroxybenzenes [
12], while the hydrogenation of dimethoxybenzenes over a rhodium catalyst gave an order of activity of
para >
meta >
ortho [
15], indicating that changing the substituents results in a change in the ordering of the reactivity of the isomers. It has been reported that replacing a methyl substituent with a hydroxyl substituent reduces the reactivity of an aromatic species [
16] and, indeed, we have found that dihydroxybenzenes are less reactive than the equivalent xylene [
12], which may be due to the strength of adsorption. With alkylbenzenes, as the inductive effect increases so does the strength of adsorption resulting in a decrease in the rate of hydrogenation [
12]; therefore, as electron donation to the aromatic ring is greater for hydroxyl groups than methyl groups, adsorption of dihydroxybenzenes should be stronger and the rate of hydrogenation slower.
The activation energies for all three substrates were in the range 29–41 kJ mol
−1, in agreement with a previous study over a Rh catalyst [
9]. It was found that resorcinol had the fastest rate of reaction, with the major products (cyclohexanol and cyclohexanone) formed through direct HDO of resorcinol (
Figure 1).
The major product of the reaction was the hydrogenolysis/HDO product, cyclohexanol, at ~24%, with the ring hydrogenated products, isomers of cyclohexanediol and hydroxycyclohexanone, formed to a lesser extent (12%
cis and 8%
trans and 16%, respectively). It was not expected that preferential formation of the HDO product, cyclohexanol, would occur under such mild conditions as the majority of HDO studies operate at elevated temperature and pressure (>473 K and >10 barg hydrogen) in the belief that cleavage of the Ar-OH bond in bio-oil requires harsh conditions. The hydrogenation of resorcinol did not reach completion at 323 K within 180 min, however at 333 K and 343 K reaction did reach completion by 140 and 160 min, respectively (
Supplementary Material, Figure S2 and
Figure 2). From
Figure 2, it is apparent that both cyclohexanone and 3-hydroxycyclohexanone were formed as intermediates, hydrogenating further to cyclohexanol and cyclohexanediol isomers, respectively. It can be seen that the two cyclohexanone intermediates initially had the highest selectivities, being formed directly from resorcinol. Their levels increased steadily over 80 min before decreasing as they were hydrogenated and not replaced from resorcinol.
The hydrogenation/HDO routes are shown in
Figure 3. It has been suggested that cyclohexane is formed from cyclohexanol [
17], however other authors [
18] have suggested direct conversion of phenol to cyclohexane. We have shown that cyclohexanol is stable under these conditions [
14] and does not form cyclohexane, therefore cyclohexane is a direct product from resorcinol. As may be expected, an increase in temperature resulted in higher selectivity to hydrogenolysis products as shown in
Figure 4. Similar behaviour was reported with anisole hydrogenation over rhodium [
14].
The
para isomer, hydroquinone, although exhibiting similar behaviour to resorcinol showed a significantly reduced rate of reaction as can be seen in
Figure 5. This lower rate of reaction for the
para isomer is rarely documented; instead it is generally reported to have the highest reactivity due to its symmetrical arrangement. A study of xylene isomers found
para-xylene to have the highest reactivity [
12], however the presence of two strongly electron donating groups on the dihydroxybenzenes may influence this order. The product distribution for hydroquinone was closely related to that of resorcinol with cyclohexanol the major product at 180 min. At 70% conversion of both resorcinol and hydroquinone, the cumulative yield of hydrogenolysis products was ~40% compared to ~30% for hydrogenated only products. The formation of the hydrogenolysis product cyclohexane (~8% yield) was particularly significant at these conditions, as it required the aromatic to undergo hydrogenation and two bond cleavages to remove both –OH groups. By comparison the hydrogenation of phenol over a similar catalyst gave ~16% yield of cyclohexane at 70% conversion, showing that the process is slower with two hydroxyl substituents in a
para configuration [
14].
From
Figure 6 it is clear that the
ortho-isomer, catechol, shows significantly different behaviour from the other two isomers. In contrast to
meta and
para-isomers, the major products from catechol hydrogenation were the ring hydrogenated
cis-1,2-cyclohexanediol and 2-hydroxycyclohexanone, with selectivity to the
cis-isomer of ~33% compared to ~13% from resorcinol and hydroquinone. As mentioned previously, Song et al. [
7] studied catechol HDO over a Ni catalyst at elevated conditions, and recorded the major product as cyclohexanediol, in agreement with our findings. However, Song et al. detected the presence of phenol (~12% yield) which was not observed in our study. Similarly, they saw no formation of benzene, postulated to be the result of a lower C-O hydrogenolysis barrier for catechol compared to phenol. The rate of phenol hydrogenation [
14] was significantly faster than that of the dihydroxybenzenes in our study (k
phenol 14.7 × 10
−3 min
−1 c.f. k
resorcinol 11.2 × 10
−3 min
−1). It is however, well documented that an increase in the number of substituents on the aromatic ring decreases the rate of reaction [
19,
20].
From
Figure 6 it can be seen that the formation of HDO products from catechol occurred to a lesser extent than observed with hydroquinone and resorcinol, for example, at ~80% conversion of catechol, cyclohexanol yield was ~8% compared to ~19% for resorcinol hydrogenation at a similar conversion. This significant decrease in –OH group cleavage on moving one position on the aromatic ring may be an effect of the close proximity of the two substituents, which can facilitate interaction between the two-hydroxyl groups via hydrogen bonding, resulting in further suppression of the deoxygenation capability of the
ortho- isomer.
Figure 7 clearly demonstrates these differences in levels of hydrogenation and hydrogenolysis product formation between the three isomers with resorcinol and hydroquinone showing a preference to HDO whilst catechol favours hydrogenation. This markedly different behaviour of the
meta and
para-isomers relative to the
ortho-isomer may be due to an adsorption effect. Literature on dihydroxybenzenes adsorption interactions is sparse. Studies by Odebunmi and Ollis on HDO of substituted phenols found similar behaviour with methyl substituted phenols, where the
ortho-methyl phenol exhibited a greater resistance to HDO than the
meta/para-methyl phenols [
21,
22]. Hence, it is possible that the main factor determining the deoxygenation ability of
para/meta-isomers as against
ortho-isomer is the mode of adsorption of the substrate. The assumption made by many is that
para- and
meta-isomers both have a flat mode of adsorption whereas the
ortho isomer adopts an inclined mode as postulated by Bredenberg and Sarbak [
23] in a study of the adsorption of dihydroxybenzenes using chemisorption and infrared spectroscopy. However, a flat mode of adsorption for dihydroxybenzenes is possible in the
ortho position, bonded strongly through the two oxygen atoms, with the hydrogen atoms pointing away from the surface to give a mode of adsorption similar to that of the
meta- and
para-isomers. Rather than mode of adsorption, it may be the proximity and strength of bonding in the
ortho position that significantly suppresses the cleavage of the –OH. However, the substitution of an –OH for a –CH
3 group would result in the inclined mode of adsorption discussed. A study by Furimsky et al. [
13] examined the effect of the addition of a methyl substituent and found a decrease in rate and an increase in the resistance of the ring to undergo hydrogenation. They attributed these findings to a possible steric effect in the transient state between the reactant and catalyst surface.
The rate of hydrogenation for the dihydroxybenzenes gave an order of resorcinol > catechol > hydroquinone (
meta > ortho > para), in contrast to that reported in literature by Smith and Stump over rhodium and platinum catalysts in a sealed reactor, where the order was given as (
para > meta > ortho) [
9]. However, their results do agree that catechol undergoes less hydrogenolysis than hydroquinone or resorcinol. Nevertheless, the higher activity for resorcinol in comparison to the other two isomers found in our study has been documented previously in a study by Maximov et al. [
24] over a ruthenium catalyst, where the favourable arrangement of the two hydroxy groups in the
meta position was postulated as the reason behind the faster rate. Furthermore, a study of HDO of methyl substituted phenols by Furimsky. et al. [
13] also found the
meta-isomer to be the most reactive isomer followed in that instance by the
para-isomer.
A study of phenol hydrogenation under similar reaction conditions recorded a yield of the HDO product, cyclohexane of ~20%, double that found in our study [
20]. The formation of cyclohexane from phenol, however, requires the cleavage of only one –OH group, whereas for dihydroxybenzene cleavage of two –OH groups is necessary. The different bond dissociation energies of aromatic and aliphatic C–O bonds, 468 and 385 kJ·mol
−1, respectively, with the greater energy required to cleave the aromatic-OH bond, used to suggest that initial hydrogenation followed by subsequent hydrogenolysis is the route of –OH bond cleavage [
13]. However, hydrogenation of cyclohexanone found cyclohexanol as the sole product, with no cyclohexane detected [
14] and cyclohexanol was stable under reaction conditions. Both these findings suggest a direct route of cyclohexane formation from the aromatic [
14]. To confirm that HDO was not via a hydrogenated species,
cis-1,2-cyclohexanediol was used as the reactant and subjected to standard reaction conditions for 3 h: no cyclohexane or cyclohexanol was detected (The reaction graph can be found in
Figure S9 of the Supplementary Information Section). Therefore, we propose that the formation of the hydrogenolysis products occurs directly from the aromatic via highly reactive surface intermediates. This idea of reactive surface intermediates being linked to HDO activity was first stated by Smith and Stump during their study on dihydroxybenzene hydrogenation [
9]. The initial hydrogenation of the aromatic results in the formation of highly reactive surface intermediates, containing double bonds, which facilitate the promotion of hydrogenolysis. Examining catechol as an example and considering the intermediates that can be formed (
Figure 8) it is apparent that intermediates A and D contain a double-bond β-γ to a hydroxyl group, rendering this group susceptible to hydrogenolysis. Intermediate B is the most stable configuration for the double bond and is likely the major route for the formation of 2-hydroxcyclohexanone through
keto-enol tautomerism. Intermediate D also has the potential to form 2-hydroxcyclohexanone through
keto-enol tautomerism. Further hydrogenation of 2-hydroxycyclohexanone would result in the formation of
cis/trans-1,2-cyclohexanediol and as such, intermediates B and D are the most likely routes for hydrogenation. It should be noted that although intermediate A can form the HDO products via hydrogenolysis, it can also form the hydrogenated products. Intermediate C is unlikely to contribute significantly to the reaction due to the position of its double bond. When studying intermediate formation for resorcinol and hydroquinone it is apparent that a greater number of those with a double-bond β-γ to the hydroxyl group, as seen above in A, can be potentially formed, which may explain the increased bias towards hydrogenolysis observed with these two isomers.
Further differences in behaviour of the three dihydroxybenzenes substrates was apparent in the
cis:
trans ratio of the cyclohexanediol product. As expected all three preferentially formed the
cis isomer, however, the
cis:
trans ratios of the cyclohexanediols formed from catechol and hydroquinone were significantly greater than that observed with resorcinol as can be seen in
Figure 9. This is slightly surprising as the ring position of substituents dictates that it is thermodynamically more favourable for the
trans-isomer to form with catechol and hydroquinone, whereas our results show that
trans-isomer is being formed more favourably with resorcinol.
This raises the question of how the
trans-isomer is formed. No isomerisation or hydrogenolysis was observed when
cis-1,2-dihydroxycyclohexane was used as reactant indicating that the
trans-isomer is not formed by subsequent isomerisation of the
cis-isomer, thereby necessitating a direct route to the
trans-isomer. The
cis-isomer can be formed through hydrogenation of the enol form of hydroxycyclohexanone (i.e., 1,2-dihydroxycyclohex-1-ene, intermediate B in
Figure 8). Note that this would explain the high yield of cis-1,2-cyclohexanol compared to the 1,3- and 1,4-isomers as only catechol can produce a four-substituted alkene as an intermediate, which will be more stable and hence have a greater chance of hydrogenation relative to tautomerism to the keto-form. In contrast we propose that the
trans-isomer is formed solely via hydrogenation of hydroxycyclohexanone via desorption and subsequent re-adsorption. Hydrogenation of the C=O functionality will be slower than hydrogenation of the C=C functionality hence the
trans-isomer is always formed later in the reaction than the
cis-isomer.
2.2. Deuterium Reactions
To further explore the mechanism of hydrogenation and hydrodeoxygenation, in the following set of reactions, deuterium was used in place of hydrogen for both the reduction and reaction procedure. All other parameters were set as per standard conditions (323 K, 10 mmol substrate, and 3 barg pressure). Comparison of the rate constants from reactions carried out in deuterium and hydrogen was used to calculate the kinetic isotope effect for each substrate. The reaction profiles are shown in
Figure 10,
Figure 11 and
Figure 12. The results suggested significant mechanistic differences between the reactions of the dihydroxybenzenes.
Table 2 show the kinetic isotope effects.
Comparing
Figure 1 with
Figure 10 it is clear that resorcinol showed a faster rate of reaction under deuterium with the conversion after 180 min increasing from 86% under hydrogen to ~96% under deuterium. A marked difference in the production of cyclohexane was observed with a delay of 30 min when using deuterium whereas when using hydrogen, cyclohexane formation occurred immediately.
Comparing
Figure 5 with
Figure 11 it is clear that in, contrast to resorcinol, hydroquinone had a slower rate of reaction under deuterium with the conversion after 180 min decreasing from 70% under hydrogen to ~76% under deuterium. A similar inhibition in the production of cyclohexane was observed with a delay of 20 min when using deuterium whereas when using hydrogen, cyclohexane formation occurred immediately.
The reaction of catechol under deuterium (
Figure 12) had a significantly higher rate than under hydrogen, resulting in a marked inverse kinetic isotope effect. A similar inhibition to that found with the other dihydroxybenzenes isomers regarding the production of cyclohexane was observed with a delay of 20 min when using deuterium.
We could find no literature directly related to the use of deuterium to hydrogenate dihydroxybenzenes; however, hydrogen-deuterium studies of phenol, anisole and 4-methoxyphenol have been reported [
14]. Both phenol and anisole gave standard kinetic isotope effects but 4-methoxyphenol gave an inverse kinetic isotope effect (KIE), which is in contrast to hydroquinone (4-hydroxyphenol) in this study which gave the only positive KIE. Clearly the change from –OH to –OCH
3 resulted in a change of mechanism. The deuteration of alkyl-substituted benzenes has been reported [
12]. A comparison of hydrogenation and deuteration of toluene, ethylbenzene and propyl benzene revealed that all three displayed an inverse kinetic isotope effect, which was concluded to be due to the change in hybridisation of the carbon atom from sp
2 to sp
3 which takes place during the hydrogenation of the aromatic ring—an explanation which may explain the inverse KIE values calculated for catechol and resorcinol. A study on xylenes hydrogenation [
12] also found a disparity amongst isomers, however in this instance the
ortho-isomer exhibited a positive KIE and while the
meta- and
para-isomers displayed an inverse KIE. It was concluded the
ortho-xylene must have a different rate-determining step (RDS) from the
meta- and
para-xylene. In our study, something similar may apply with both catechol and resorcinol having a different RDS from hydroquinone. This was a surprise as resorcinol and hydroquinone exhibited similar hydrogenation/HDO behaviour.
One common feature to all three isomers is the delay in cyclohexane formation. Under our reaction conditions no benzene or phenol is detected and cyclohexanol is stable, therefore cyclohexane is formed directly from dihydroxybenzene. As cyclohexane is formed directly it is likely that the inhibition is due to changes on the rhodium surface. It has been proposed for anisole HDO [
25] that the hydrodeoxygenation reaction is favoured by small rhodium crystallites with reaction on the low coordination number sites. At the outset of the reaction the rhodium surface will be covered in deuterium, however as the reaction progresses the surface will contain a mixture of H and D due to exchange processes. The Rh-D bond is stronger than the Rh–H bond [
26] and in deuterium exchange reactions over rhodium a delay in reaction initiating has also been observed before the reaction moved to a steady state [
27] Therefore we propose that the adsorbed deuterium on the low coordination number sites inhibits the reaction until there is sufficient exchange of D for H.




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}