Copper-Catalyzed C–H Arylation of Fused-Pyrimidinone Derivatives Using Diaryliodonium Salts

 , and
, and
Abstract
:
1. Introduction
2. Results
3. Materials and Methods
3.1. General Information
3.2. Chemistry
General Procedure for the Synthesis of 2-Aryl-N8-substitued-thiazolo[5,4-f]quinazolin-9(8H)-Ones (3a–s) and Fused Pyrimidines (5aa–5ha).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wencel-Delord, J.; Glorius, F. C-H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem. 2013, 5, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Cernak, T.; Dykstra, K.D.; Tyagarajan, S.; Vachal, P.; Krska, S.W. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.; Sharman, G.; Martínez-Olid, F.; Cañellas, S.; Gomez, J.E. Unlocking the potential of late-stage functionalisation: An accurate and fully automated method for the rapid characterisation of multiple regioisomeric products. React. Chem. Eng. 2020, 5, 779–792. [Google Scholar] [CrossRef]
- Fruit, C. Three catalysts for activating carbon-hydrogen bonds. Science 2016, 352, 1277–1278. [Google Scholar] [CrossRef] [PubMed]
- Théveau, L.; Schneider, C.; Fruit, C.; Hoarau, C. Orthogonal Palladium-Catalyzed Direct C-H Bond Arylation of Heteroaromatics with Aryl Halides. ChemCatChem 2016, 8, 3183–3194. [Google Scholar] [CrossRef]
- Ping, L.; Chung, D.S.; Bouffard, J.; Lee, S. Transition metal-catalyzed site- and regio-divergent C–H bond functionalization. Chem. Soc. Rev. 2017, 46, 4299–4328. [Google Scholar] [CrossRef]
- Tiwari, V.K.; Kapur, M. Catalyst-controlled positional-selectivity in C–H functionalizations. Org. Biomol. Chem. 2019, 17, 1007–1026. [Google Scholar] [CrossRef]
- Struble, T.J.; Coley, C.W.; Jensen, K.F. Multitask prediction of site selectivity in aromatic C–H functionalization reactions. React. Chem. Eng. 2020, 5, 896–902. [Google Scholar] [CrossRef] [Green Version]
- Laclef, S.; Harari, M.; Godeau, J.; Schmitz-Afonso, I.; Bischoff, L.; Hoarau, C.; Levacher, V.; Fruit, C.; Besson, T. Ligand-Free Pd-Catalyzed and Copper-Assisted C−H Arylation of Quinazolin-4-ones with Aryl Iodides under Microwave Heating. Org. Lett. 2015, 17, 1700–1703. [Google Scholar] [CrossRef]
- Godeau, J.; Harari, M.; Laclef, S.; Deau, E.; Fruit, C.; Besson, T. Cu/Pd-Catalyzed C2-H Arylation of Quinazolin-4(3H)-ones with (Hetero)Aryl Halides. Eur. J. Org. Chem. 2015, 7705–7717. [Google Scholar] [CrossRef]
- Harari, M.; Couly, F.; Fruit, C.; Besson, T. Pd-Catalyzed and Copper Assisted Regioselective Sequential C2 and C7 Arylation of Thiazolo[5,4-f]quinazolin-9(8H)-one with Aryl Halides. Org. Lett. 2016, 18, 3282–3285. [Google Scholar] [CrossRef]
- Couly, F.; Dubouilh-Benard, C.; Besson, T.; Fruit, C. Late-Stage C–H Arylation of Thiazolo[5,4-f]quinazolin-9(8H)-one Backbone: Synthesis of an Array of Potential Kinase Inhibitors. Synthesis 2017, 49, 4615–4622. [Google Scholar] [CrossRef]
- Couly, F.; Harari, M.; Dubouilh-Benard, C.; Bailly, L.; Petit, E.; Diharce, J.; Bonnet, P.; Meijer, L.; Fruit, C.; Besson, T. Development of Kinase Inhibitors via MetalCatalyzed C–H Arylation of 8-Alkyl-thiazolo[5,4-f]quinazolin-9-ones Designed by Fragment-Growing Studies. Molecules 2018, 23, 2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Huo, X.; Zhang, W. For a recent review focused on Pd/Cu Catalysis, Synergistic Pd/Cu Catalysis in Organic Synthesis. Chem. A Eur. J. 2020, 26, 4895–4916. [Google Scholar] [CrossRef] [PubMed]
- Abbassi, R.; Johns, T.G.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.; Fruit, C.; Hérault, Y.; Meijer, L.; Besson, T. Dual-specificity tyrosine phosphorylation-regulated kinase 1a (dyrk1a) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017, 11, 1183–1199. [Google Scholar] [CrossRef]
- Jarhad, D.B.; Mashelkar, K.K.; Kim, H.-R.; Noh, M.; Jeong, L.S. Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A (DYRK1A) Inhibitors as Potential Therapeutics. J. Med. Chem. 2018, 61, 9791–9810. [Google Scholar] [CrossRef]
- Branca, C.; Shaw, D.M.; Belfiore, R.; Gokhale, V.; Shaw, A.Y.; Foley, C.; Smith, B.; Hulme, C.; Dunckley, T.; Meechoovet, B.; et al. Dyrk1 inhibition improves Alzheimer’s disease-like pathology. Aging Cell. 2017, 1–9. [Google Scholar] [CrossRef]
- Feki, A.; Hibaoui, Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci. 2018, 8, 187. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Srivastava, P.; Seth, A.; Tripathi, P.N.; Banerjee, A.G.; Shrivastava, S.K. Comprehensive review of mechanisms of pathogenes is involved in Alzheimer’s disease and potential therapeutic strategies. Prog. Neurobiol. 2019, 174, 53–89. [Google Scholar] [CrossRef]
- Beringer, F.M.; Brierley, A.; Drexler, M.; Gindler, E.M.; Lumpkin, C.C. Diaryliodonium Salts. II. The Phenylation of Organic and Inorganic Bases. J. Am. Chem. Soc. 1953, 75, 2708–2712. [Google Scholar] [CrossRef]
- Merritt, E.A.; Olofsson, B. Diaryliodonium Salts: A Journey from Obscurity to Fame. Angew. Chem. Int. Ed. 2009, 48, 9052–9070. [Google Scholar] [CrossRef] [PubMed]
- Malmgren, J.; Santoro, S.; Jalalian, N.; Himo, F.; Olofsson, B. Arylation with Unsymmetrical Diaryliodonium Salts: A Chemoselectivity Study. Chem. A Eur. J. 2013, 19, 10334–10342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roopan, S.M.; Palaniraja, J. Synthetic journey towards transition metal-free arylations. Res. Chem. Intermed. 2015, 41, 8111–8146. [Google Scholar] [CrossRef]
- Olofsson, B. Arylation with Diaryliodonium Salts. Top. Curr. Chem. 2016, 373, 135–166. [Google Scholar] [CrossRef]
- Aradi, K.; Tóth, B.L.; Tolnai, G.L.; Novák, Z. Diaryliodonium Salts in Organic Syntheses: A Useful Compound Class for Novel Arylation Strategies. Synlett 2016, 27, 1456–1485. [Google Scholar] [CrossRef]
- Wang, M.; Chen, S.; Jiang, X. Atom-Economical Applications of Diaryliodonium Salts. Chem. Asian J. 2018, 13, 2195–2207. [Google Scholar] [CrossRef]
- Hyatt, I.D.; Dave, L.; David, N.; Kaur, K.; Medard, M.; Mowdawallla, C. Hypervalent iodine reactions utilized in carbon–carbon bond formations. Org. Biomol. Chem. 2019, 17, 7822–7848. [Google Scholar] [CrossRef]
- Pacheco-Benichou, A.; Besson, T.; Fruit, C. Diaryliodoniums Salts as Coupling Partners for Transition-Metal Catalyzed C- and N-Arylation of Heteroarenes. Catalysts 2020, 10, 483. [Google Scholar] [CrossRef]
- Bielawski, M.; Zhu, M.; Olofsson, B. Efficient and General One-Pot Synthesis of Diaryliodonium Triflates: Optimization, Scope and Limitations. Adv. Synth. Catal. 2007, 349, 2610–2618. [Google Scholar] [CrossRef]
- Bielawski, M.; Olofsson, B. High-yielding one-pot synthesis of diaryliodonium triflates from arenes and iodine or aryl iodides. Chem. Commun. 2007, 2521–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohi, T.; Ito, M.; Morimoto, K.; Minamitsuji, Y.; Takenaga, N.; Kita, Y. Versatile direct dehydrative approach for diaryliodonium(iii) salts in fluoroalcohol media. Chem. Commun. 2007, 4152–4154. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, M.; Aili, D.; Olofsson, B. Regiospecific One-Pot Synthesis of Diaryliodonium Tetrafluoroborates from Arylboronic Acids and Aryl Iodides. J. Org. Chem. 2008, 73, 4602–4607. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.-H.; Pike, V.W. Regiospecific Syntheses of Functionalized Diaryliodonium Tosylates via [Hydroxy(tosyloxy)iodo]arenes Generated in Situ from (Diacetoxyiodo)arenes. J. Org. Chem. 2012, 77, 1931–1938. [Google Scholar] [CrossRef] [Green Version]
- Seidl, T.L.; Sundalam, S.K.; McCullough, B.; Stuart, D.R. Unsymmetrical Aryl(2,4,6-trimethoxyphenyl)iodonium Salts: One-Pot Synthesis, Scope, Stability, and Synthetic Studies. J. Org. Chem. 2016, 81, 1998–2009. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef]
- Lindstedt, E.; Reitti, M.; Olofsson, B. One-Pot Synthesis of Unsymmetric Diaryliodonium Salts from Iodine and Arenes. J. Org. Chem. 2017, 82, 11909–11914. [Google Scholar] [CrossRef]
- Soldatova, N.; Postnikov, P.; Kukurina, O.; Zhdankin, V.V.; Yoshimura, A.; Wirth, T.; Yusubov, M.S. One-pot synthesis of diaryliodonium salts from arenes and aryl iodides with Oxone–sulfuric acid. Beilstein J. Org. Chem. 2018, 14, 849–855. [Google Scholar] [CrossRef]
- Dohi, T.; Hayashi, T.; Ueda, S.; Shoji, T.; Komiyama, K.; Takeuchi, H.; Kita, Y. Recyclable synthesis of mesityl iodonium(III) salts. Tetrahedron 2019, 75, 3617–3627. [Google Scholar] [CrossRef]
- Gallagher, R.T.; Seidl, T.L.; Bader, J.; Orella, C.; Vickery, T.; Stuart, D.R. Anion Metathesis of Diaryliodonium Tosylate Salts with a Solid Phase Column Constructed from Readily Available Laboratory Consumables. Org. Process Res. Dev. 2019, 23, 1269–1274. [Google Scholar] [CrossRef]
- Budhwan, R.; Yadav, S.; Murarka, S. Late stage functionalization of heterocycles using hypervalent iodine(III) reagents. Org. Biomol. Chem. 2019, 17, 6326–6341. [Google Scholar] [CrossRef]
- Deprez, N.R.; Kalyani, D.; Sanford, M.S. Room Temperature Palladium-Catalyzed 2-Arylation of Indoles. J. Am. Chem. Soc. 2006, 128, 4972–4973. [Google Scholar] [CrossRef] [PubMed]
- Phipps, R.J.; Grimster, N.P.; Gaunt, M.J. Cu(II)-Catalyzed Direct and Site-Selective Arylation of Indoles Under Mild Conditions. J. Am. Chem. Soc. 2008, 130, 8172–8174. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Chang, J.; Wu, Q.; Zhang, B. A simple phenylation of heteroaromatic compounds using diphenyliodonium triflate. Res. Chem. Intermed. 2012, 38, 1335–1340. [Google Scholar] [CrossRef]
- Kumar, D.; Pilania, M.; Arun, V.; Pooniya, S. C–H arylation of azaheterocycles: A direct ligand-free and Cu-catalyzed approach using diaryliodonium salts. Org. Biomol. Chem. 2014, 12, 6340–6344. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Wang, R.; Wu, H.; Du, Z.; Fu, Y. Pd-Catalyzed C-H Arylation of Benzothiazoles with Diaryliodonium Salt: One-Pot Synthesis of 2-Arylbenzothiazoles. Asian, J. Org. Chem. 2017, 6, 184–188. [Google Scholar] [CrossRef]
- Fañanás-Mastral, M. Copper-Catalyzed Arylation with Diaryliodonium Salts. Synthesis 2017, 49, 1905–1930. [Google Scholar] [CrossRef]
- Gandeepan, P.; Kaplaneris, N.; Santoro, S.; Vaccaro, L.; Ackermann, L. Biomass-Derived Solvents for Sustainable Transition Metal-Catalyzed C-H Activation. ACS Sustain. Chem. Eng. 2019, 7, 8023–8040. [Google Scholar] [CrossRef]
- Anastasiou, I.; Van Velthoven, N.; Tomarelli, E.; Lombi, A.; Lanari, D.; Liu, P.; Bals, S.; De Vos, D.E.; Vaccaro, L. For a recent example of C-H arylation using diaryliodonium salts as coupling partners in a biomass-derived solvent, C2–H Arylation of indoles catalyzed by Palladium containing Metal-Organic-Framework (Pd-MOF) in bioderived γ-valerolactone (GVL). ChemSusChem. 2020, 13, 2786–2791. [Google Scholar] [CrossRef]
- Kuriyama, M.; Hamaguchi, N.; Onomura, O. Copper(II)-Catalyzed Monoarylation of Vicinal Diols with Diaryliodonium Salts. Chem. A Eur. J. 2012, 18, 1591–1594. [Google Scholar] [CrossRef]
- Liu, F.; Yang, H.; Hu, X.; Jiang, G. Metal-Free Synthesis of ortho-CHO Diaryl Ethers by a Three-Component Sequential Coupling. Org. Lett. 2014, 16, 6408–6411. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Chang, D.; Leng, X.; Gu, Y.; Shen, Q. Well-Defined, Shelf-Stable (NHC)Ag (CF2H) Complexes for Difluoromethylation. Organometallics 2015, 34, 3065–3071. [Google Scholar] [CrossRef]
- Hickman, A.J.; Sanford, M.S. High-valent organometallic copper and palladium in catalysis. Nature 2012, 484, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Casitasa, A.; Ribas, X. The role of organometallic copper(iii) complexes in homogeneous catalysis. Chem. Sci. 2013, 4, 2301–2318. [Google Scholar] [CrossRef]


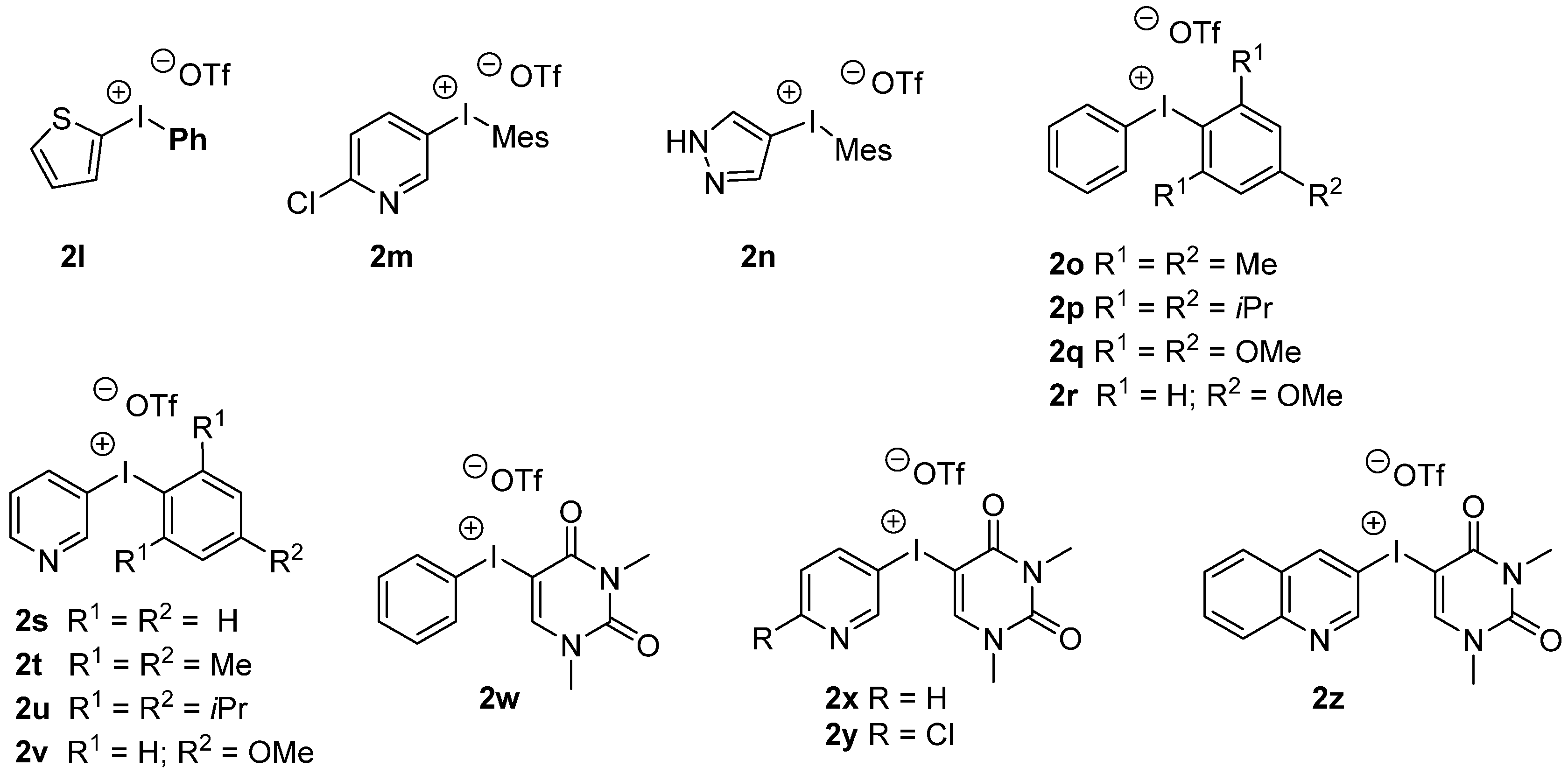

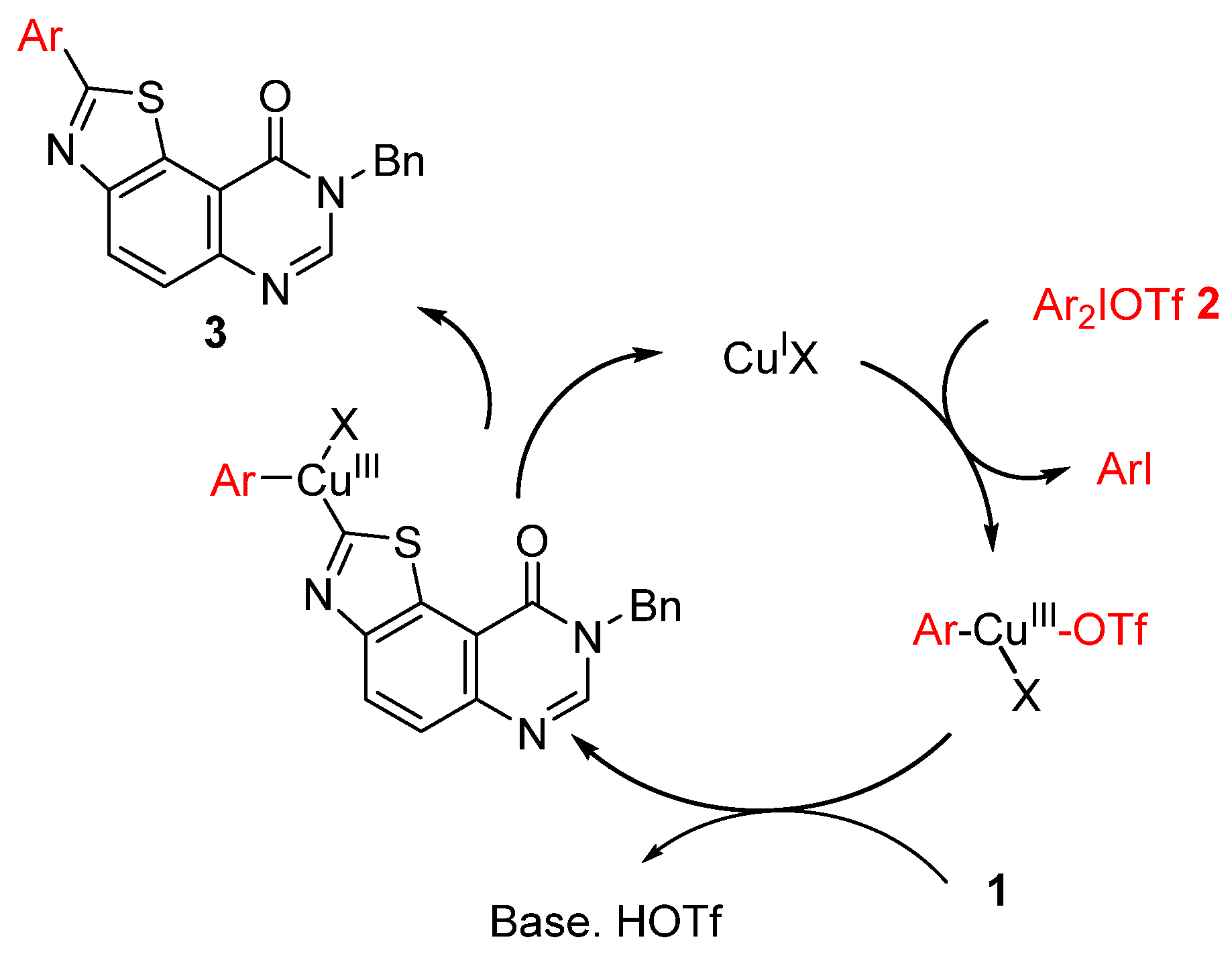

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
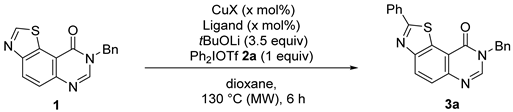
| Entries 1 | Copper Source | mol% | Ligand | NMR Yield 2 of 3a (%) |
|---|---|---|---|---|
| 1 | CuI | 30 | - | 64 (21) 3 |
| 2 | CuBr | 30 | - | 36 (23) |
| 3 | CuCl | 30 | - | 48 (28) |
| 4 | CuOAc | 30 | - | 59 (31) |
| 5 | CuSCN | 30 | - | 61 (24) |
| 6 | CuTC | 30 | - | 42 (33) |
| 7 | CuCl2 | 30 | - | 27 (49) |
| 8 | Cu(OAc)2 | 30 | - | 50 (38) |
| 9 | Cu(OTf)2 | 30 | - | 41 (35) |
| 10 | Cu2O | 30 | - | 7 (49) |
| 11 | Cu2O | 30 | - | NR 4 |
| 12 | CuI | 15 | - | 36 (45) |
| 13 | CuI | 50 | - | 63 (23) |
| 14 | CuI | 100 | - | 66 (10) |
| 15 | CuI | 30 | 1,10-Phen | 54 (40) |
| 16 | CuI | 30 | XantPhos | 55 (43) |
| 17 | CuI | 30 | Bipy | 36 (35) |
| 18 | CuI | 30 | 1,10-Phen | 60 (18) 5 |
| 19 | CuI | 30 | Bipy | 49 (48) 5 |
| 20 | CuI | 100 | 1,10-Phen | 59 (33) 5 |
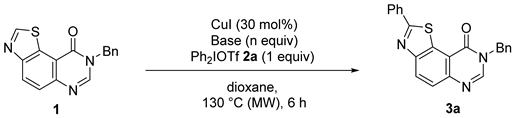
| Entries 1 | Base | N Equiv | NMR Yield 2 of 3a (%) |
|---|---|---|---|
| 1 | tBuOLi | 3.5 | 64 (21) 3 |
| 2 | tBuOK | 3.5 | NR |
| 3 | Cs2CO3 | 3.5 | 7 (85) |
| 4 | Li2CO3 | 3.5 | 0 (65) 4 |
| 5 | K3PO4 | 3.5 | NR |
| 6 | NaOAc | 3.5 | NR |
| 7 | DBU | 3.5 | 2 (62) |
| 8 | DIPEA | 3.5 | 6 (89) |
| 9 | DABCO | 3.5 | 4 (92) |
| 10 | dtbpy | 3.5 | NR |
| 11 | tBuOLi | 1 | 7 (87) |
| 12 | tBuOLi | 2 | 4 (92) |
| 13 | tBuOLi | 5 | 68 (15) |
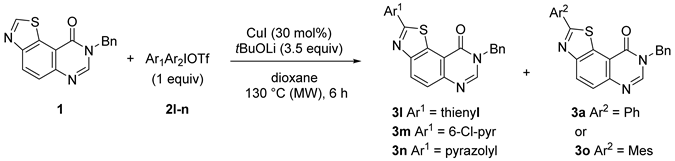
| Entries | Ar1Ar2IOTf | Products | NMR Yield (%) 2 |
|---|---|---|---|
| 1 | 2l | 3l/3a | 22/23 |
| 2 | 2m | 3m/3o | 6/53 |
| 3 | 2n | 3o | 69 |

| Entries | Ar1Ar2IOTf | Products | Isolated Yields (%) |
|---|---|---|---|
| 1 | 2o | 3a/3o | 24/36 |
| 2 | 2p | 3a/3p | 21/31 |
| 3 | 2q | 3a/3q | 18/35 |
| 4 | 2r | 3a/3q | 16/23 |
| 5 | 2w | 3a | 16 |
| 6 | 2s | 3a/3s | 13/10 |
| 7 | 2t | 3o/3s | 56/8 |
| 8 | 2u | 3p/3s | 50/6 |
| 9 | 2v | 3q/3s | 12/10 |
| 10 | 2x | 3s | 22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacheco-Benichou, A.; Ivendengani, E.; Kostakis, I.K.; Besson, T.; Fruit, C. Copper-Catalyzed C–H Arylation of Fused-Pyrimidinone Derivatives Using Diaryliodonium Salts. Catalysts 2021, 11, 28. https://doi.org/10.3390/catal11010028
Pacheco-Benichou A, Ivendengani E, Kostakis IK, Besson T, Fruit C. Copper-Catalyzed C–H Arylation of Fused-Pyrimidinone Derivatives Using Diaryliodonium Salts. Catalysts. 2021; 11(1):28. https://doi.org/10.3390/catal11010028
Chicago/Turabian StylePacheco-Benichou, Alexandra, Eugénie Ivendengani, Ioannis K. Kostakis, Thierry Besson, and Corinne Fruit. 2021. "Copper-Catalyzed C–H Arylation of Fused-Pyrimidinone Derivatives Using Diaryliodonium Salts" Catalysts 11, no. 1: 28. https://doi.org/10.3390/catal11010028