Abstract
Multi-step cascade reactions have gained increasing attention in the biocatalysis field in recent years. In particular, multi-enzymatic cascades can achieve high molecular complexity without workup of reaction intermediates thanks to the enzymes’ intrinsic selectivity; and where enzymes fall short, organo- or metal catalysts can further expand the range of possible synthetic routes. Here, we present two enantiocomplementary (chemo)-enzymatic cascades composed of either a styrene monooxygenase (StyAB) or the Shi epoxidation catalyst for enantioselective alkene epoxidation in the first step, coupled with a halohydrin dehalogenase (HHDH)-catalysed regioselective epoxide ring opening in the second step for the synthesis of chiral aliphatic non-terminal azidoalcohols. Through the controlled formation of two new stereocenters, corresponding azidoalcohol products could be obtained with high regioselectivity and excellent enantioselectivity (99% ee) in the StyAB-HHDH cascade, while product enantiomeric excesses in the Shi-HHDH cascade ranged between 56 and 61%.
1. Introduction
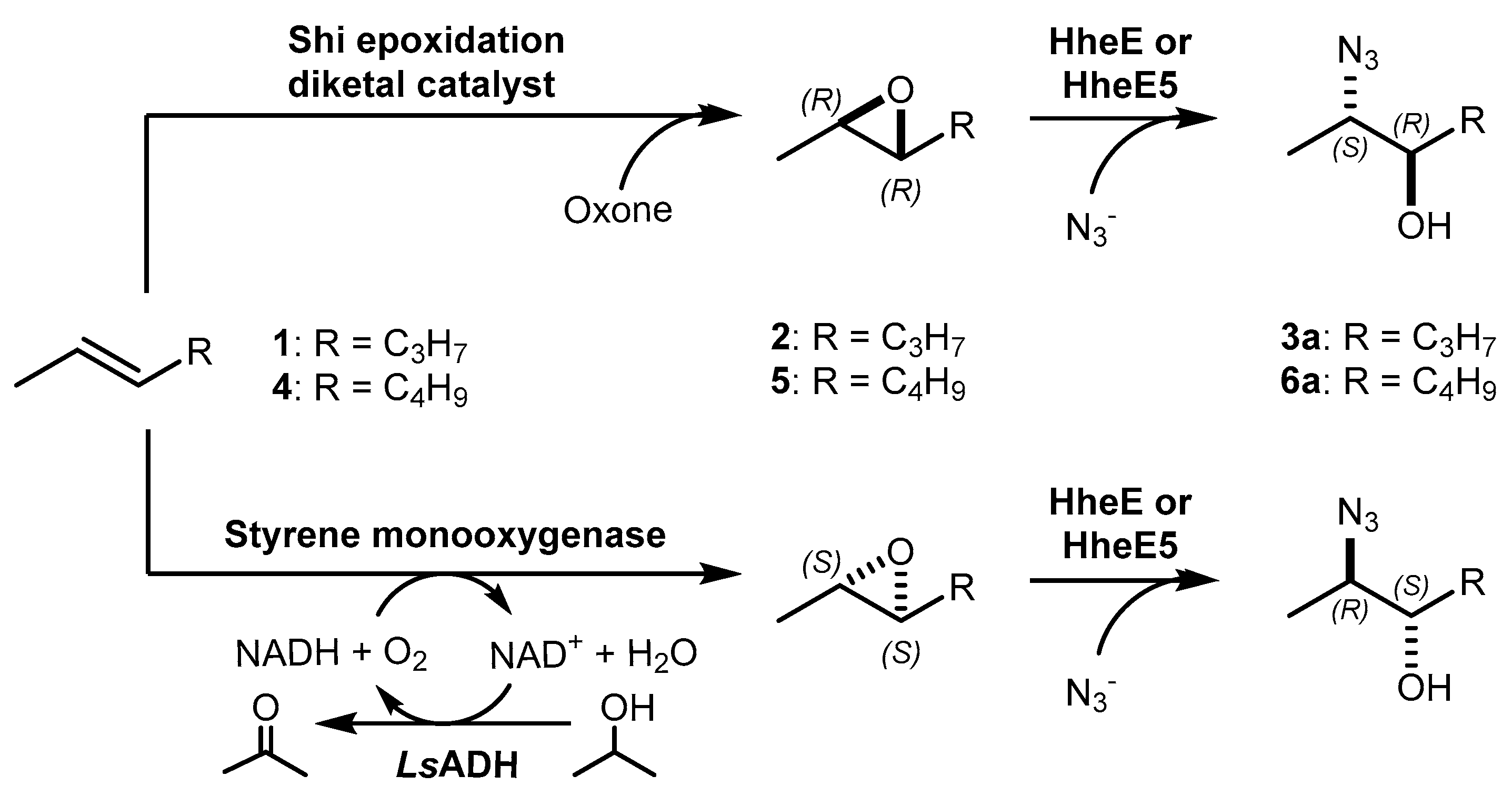
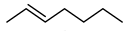
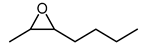
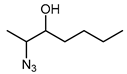
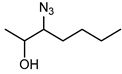
Biocatalytic and chemo-enzymatic cascade reactions have gained a lot of attention in recent years due to their environmental benefits and the ability to achieve high product yields and enantiomeric excesses over multi-step synthetic routes [1]. Their main advantages are (i) the avoidance of intermediate workup steps and (ii) the possibility to include unstable intermediates that are directly converted further in the next (bio)catalytic step [1,2,3]. However, these advantages are accompanied by new challenges arising from the combination of catalysts that require more or less different reaction conditions. Herein, we describe two (chemo)-enzymatic cascades for the synthesis of opposite enantiomers of 2,3-azidoalcohols starting from simple non-terminal alkenes (Scheme 1).
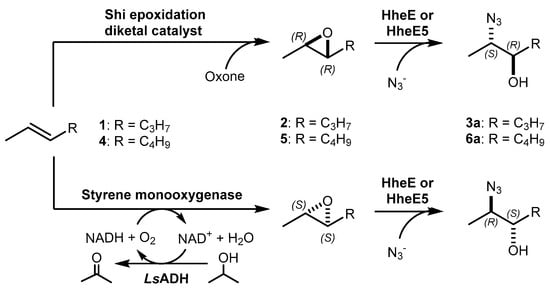
Scheme 1.
Reaction scheme for the herein established enantiocomplementary cascades towards chiral aliphatic azidoalcohols.
Compounds containing an azido group have a particular synthetic importance, and they caught the attention of organic chemists already in the previous century. They are key intermediates for the synthesis of aminoalcohols [4,5,6], lactams [7], amino sugars [8], oxazolines [9], and are important in the synthesis of carbohydrates and nucleosides [10,11].
So far, not many examples of chemo-enzymatic cascades to synthesise azidoalcohols are described. In a recent example, Martínez-Montero et al. reported a (chemo)-enzymatic cascade for the synthesis of aromatic α- and β-azidoalcohols. They coupled a styrene monooxygenase from Sphingopyxis fribergensis Kp5.2 either with chemical epoxide azidolysis or a set of HHDHs to synthesise different isomers of styrene-based azidoalcohols with high enantiopurity [12].
Here, we aimed to find a synthetic pathway towards enantiopure aliphatic azidoalcohols starting from more challenging linear alkenes as substrate. For this, we also coupled a stereoselective alkene epoxidation step with a regioselective epoxide ring-opening using azide to afford opposite enantiomers of model 2,3-azidoalcohols with the installation and control of two new stereocenters (Scheme 1).
Several possibilities to perform asymmetric epoxidations exist with Sharpless and Katsuki being pioneers in this field [13]. However, the so-called Sharpless epoxidation is very specific for allylic alcohols, as the substrate’s hydroxyl group is used as a pivot point [14,15]. A few years later, Jacobsen and Katsuki reported asymmetric epoxidations using salen complexes surrounded by a chiral environment [16]. With the Jacobsen–Katsuki epoxidation, high selectivity can be achieved mostly towards cis-di-substituted olefins [17], whereas selectivity towards trans-olefins was found to be low [18,19]. Dioxirane can perform epoxidations of olefines as well, and the enantioselectivity can be controlled by a chiral environment in the dioxirane’s surroundings. The most notable example is the Shi epoxidation [20], which makes use of a carbohydrate-derived rigid scaffold to enhance enantioselectivity [14]. This fructose-derived ketone (Shi epoxidation diketal catalyst), when activated by an oxidative agent such as Oxone© or H2O2, shows optimal performance with unfunctionalised trans-olefins in contrast to the Jacobsen catalysts (i.e., (salen)manganese complexes) [20,21]. Compared to those chemical approaches, nature has evolved different oxidative enzymes to perform asymmetric epoxidations of olefins using molecular oxygen. The most popular ones are monooxygenases, oxidases, and peroxygenases [22,23,24]. Among these, styrene monooxygenases have gained attention for their extremely high stereoselectivity in the epoxidation of C=C bonds. The substrate scope of these enzymes is generally limited to vinyl aromatic compounds with few exceptions [25,26]. However, a recently characterised styrene monooxygenase from Rhodococcus sp. ST-10, was found to convert also short-chain aliphatic alkenes with good performance [27]. Interestingly, most known enzymes preferentially produce (S)- or (S,S)-epoxide enantiomers [26,28,29,30], while (R)-selective styrene monooxygenases have only been reported very recently [31,32,33].
Epoxide ring-opening is a common reaction that has been used in organic synthesis for many years. Azidoalcohols are generally synthesised by azidolysis of epoxides in aqueous alcohol systems. However, this type of reaction requires high temperatures and long reaction times and no regio- or stereoselectivity is observed [11]. Due to the high value of some azidoalcohols, few methodologies were found to overcome such limitations. Among these, diethylaluminium azide is one of the most popular catalysts as it displays regioselectivity that is independent from the epoxide ring’s substitutents; however, this process is highly specific for 2,3-epoxyalcohols as substrates and often results in a considerable formation of 3-chloro-1,2-diols as side products [34]. Moreover, trimethylsilylazide in the presence of chiral (salen)Cr3+ complexes [35], similar to the Jacobsen catalysts for epoxidation, or β-cyclodextrin [36] could achieve high regio- and enantioselectivity in the azidolysis of terminal or cyclic epoxides. However, their molecular complexity and price constitute major limitations [35,37,38].
In contrast, a group of enzymes called halohydrin dehalogenases (HHDH) catalyse epoxide ring-opening reactions under mild conditions in the presence of different anionic nucleophiles such as N3−, CN−, and NO2−. Hence, HHDHs are particularly attractive in biocatalysis as they enable the formation of novel C-N, C-C, or C-O bonds without cofactor requirement [39,40]. Additionally, recent work on expanding the number of known HHDHs [41] in combination with the characterisation of new members showed that a wide range of epoxide substrates can be converted by HHDHs [42,43]. Depending on the individual enzyme and substrate to be converted, also high regio- and enantioselectivity can be achieved. In particular, members of subtype E were reported to display high regioselectivity in the epoxide ring-opening of aliphatic non-terminal epoxides, as desired in our cascade [42].
In order to obtain chiral non-terminal azidoalcohols from achiral alkenes, we herein aimed to control the formation of the two new stereocenters through the combination of an asymmetric, either chemical or enzymatic epoxidation and a regioselective, HHDH-catalysed epoxide ring-opening using azide.
2. Results and Discussion
2.1. Catalyst Selection for Asymmetric Epoxidation (First Cascade Step)
Initially, a set of chemical and enzyme catalysts was tested in the epoxidation of trans-2-heptene (4) as a model substrate. In particular, the styrene monooxygenase from Rhodococcus sp. ST-10, the unspecific peroxygenase from A. aegerita, the Shi epodixation diketal catalyst, and both (R,R)- and (S,S)-enantiomers of the Jacobsen catalyst were tested on an analytical scale (1 mL) for the conversion of 5 mM substrate using the respective optimal conditions described in the literature (see the Methods section for details).
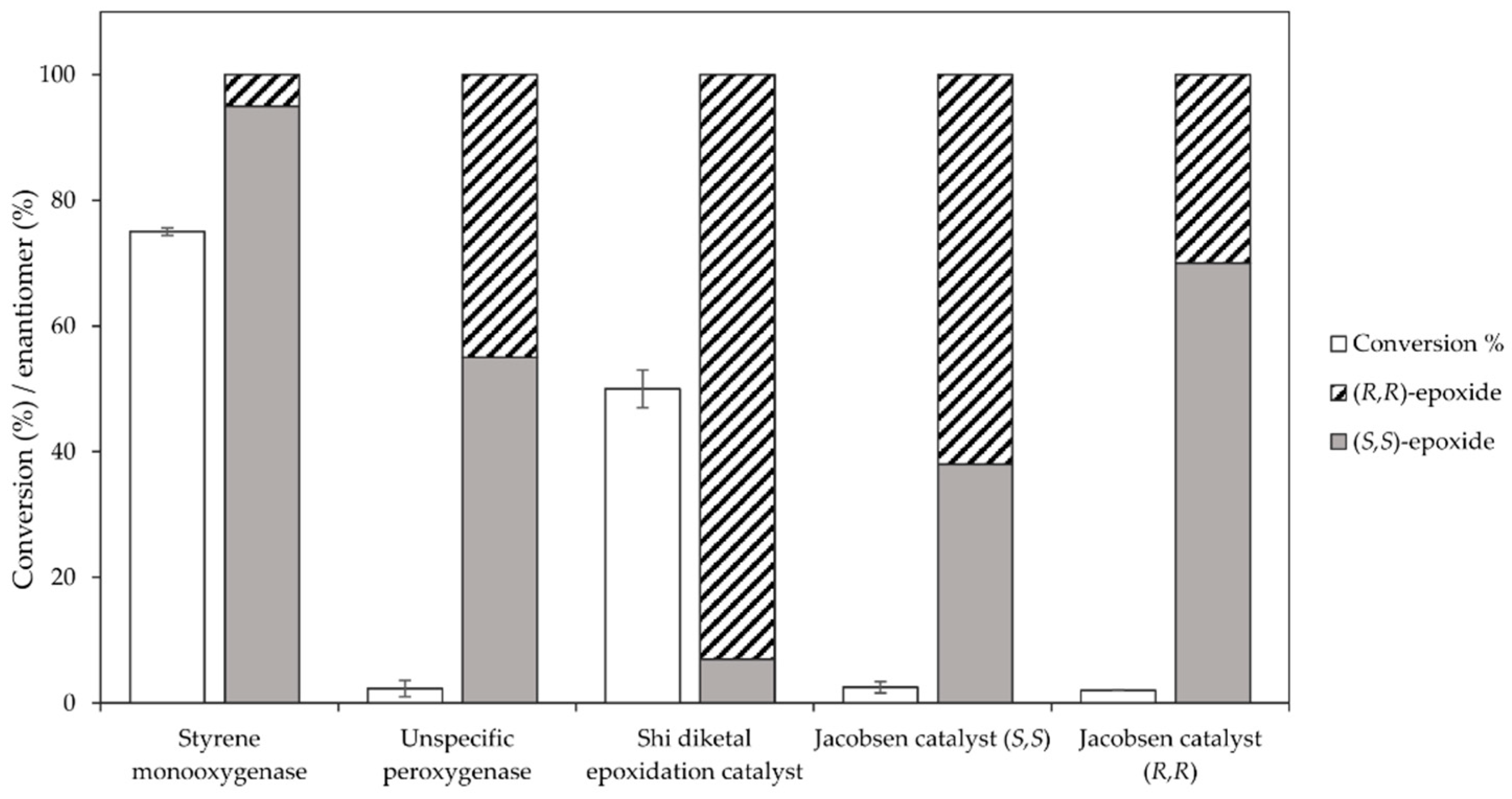
Among the tested catalysts, only two achieved a conversion ≥ 50% and a product enantiomeric excess (eeP) higher than 90% (Figure 1). Specifically, the styrene monooxygenase synthesised the (S,S)-epoxide with an eeP > 95%, whereas the Shi epoxidation diketal catalyst produced the opposite epoxide enantiomer with an eeP > 90%. The remaining catalysts displayed a conversion below 5% and insufficient selectivity. The identification of two epoxidation catalysts with opposite enantioselectivity opened up the possibility to synthesise opposite enantiomers of the final azidoalcohol products (Scheme 1).
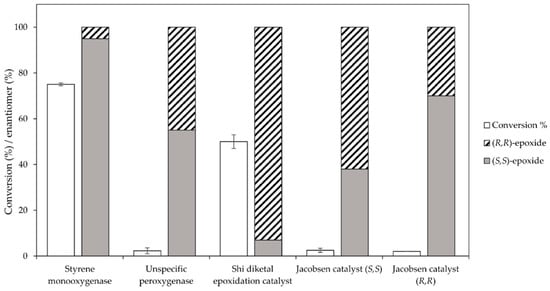
Figure 1.
Catalyst screening for the epoxidation of 5 mM trans-2-heptene (4) at 1 mL scale using the styrene monooxygenase from Rhodococcus sp. ST-10, the unspecific peroxygenase from A. aegerita, the Shi epodixation diketal catalyst and both (R,R)- and (S,S)-enantiomers of the Jacobsen catalyst.
2.2. Catalyst Selection for the Epoxide Ring-Opening Reaction (Second Cascade Step)
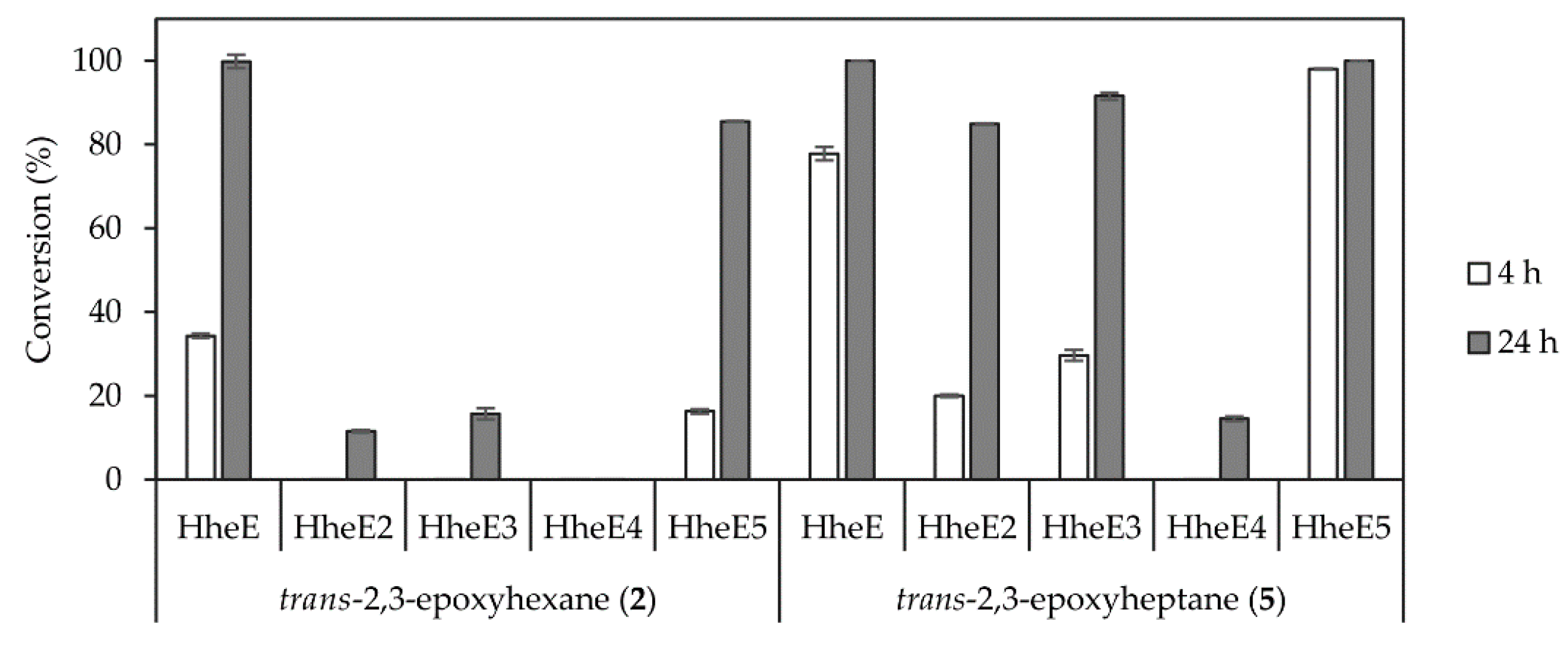
After enantioselective epoxidation in the first step, a highly regioselective catalyst was required for ring-opening of the epoxide intermediate to control the production of only one of the two possible azidoalcohol isomers (Scheme 1, compounds 3a and 6a). For this, we investigated different HHDHs from subtype E in the conversion of 10 mM epoxide substrate in the 1 mL scale. These HHDHs had previously been identified as regioselective towards non-terminal epoxides [42], but further experiments were required to select the most active one for the conversion of epoxides 2 and 5. The enzymes were applied as whole cell catalysts (60 g/L, wet cell weight), and HheE as well as HheE5 performed evidently better with both substrates compared to the other three HHDHs. HheE appeared to be the best enzyme in the epoxide ring-opening of trans-2,3-epoxyhexane (2) (Figure 2), whereas HheE5 seemed less active with this substrate and did not reach full conversion within 24 h. In contrast, HheE5 was faster in the conversion of trans-2,3-epoxyheptane (5) compared to HheE (Figure 2). Both enzymes preferentially formed 2-azido-3-hydroxyhexane (3a) and 2-azido-3-hydroxyheptane (6a) as products with a regioselectivity of ≥98%, which is in agreement with our previous data [42]. In both cases, only traces of the corresponding regioisomers 3-azido-2-hydroxyhexane (3b) and 3-azido-2-hydroxyheptane (6b) were obtained. Hence, HheE was selected for conversion of epoxide 2 in the final cascade, whereas HheE5 was selected for the azidolysis of epoxide 5.
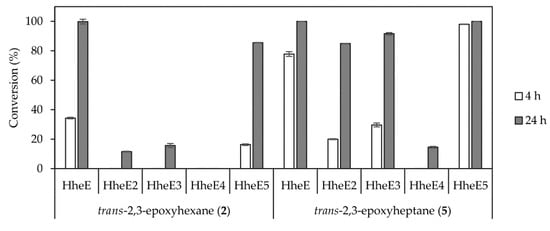
Figure 2.
Conversion of racemic epoxides 2 (left) and 5 (right) using the regioselective HHDHs from subtype E as whole cell catalysts (60 g/L, wet cell weight). Each reaction was carried out in duplicate using 10 mM substrate, 50 mM NaN3 in 1 mL 50 mM KPi buffer pH 7.5 at 25 °C.
2.3. Optimisation and Compatibility Tests of the Shi Epoxidation and the HHDH-Catalysed Epoxide Ring Opening (Cascade 1)
Compatibility of required reaction conditions of different cascade steps is one of the main challenges in setting up a cascade reaction. The required reaction conditions for asymmetric Shi epoxidation are constrained by the necessity to prevent catalyst autoxidation and consequently decomposition. Shi et al. determined that performing the reaction at low temperature (−2 °C) and at pH > 10 could help to reduce catalyst decomposition [44]. An additional strategy to decrease catalyst decomposition is to provide the oxidant (Oxone©) portion-wise during the course of the reaction. Thus, for reaction optimisation, the reaction volume was increased to 20 mL to make Oxone© feeding possible. To maintain the pH at 11, 1.1 equivalents (relative to Oxone©) of solid K2CO3 were added portion-wise during Oxone© feeding.
Shi et al. tested different acetonitrile and water mixtures and demonstrated that with an acetonitrile concentration of only 20% (v/v), the reaction would still occur with high enantioselectivity and conversion > 95% [44,45]. However, we anticipated that the HHDHs in the subsequent epoxide ring-opening reaction might still be sensitive to this amount of organic co-solvent. Hence, different acetonitrile concentrations were tested to determine their effect on epoxide yield and enantiopurity. As supposed, reactions with 20% (v/v) acetonitrile concentration resulted in the best conversion values for both alkene substrates. At the same time, the product enantiomeric excess decreased to 80–85% compared to small-scale reactions, where 30% (v/v) acetonitrile had been used (Table 1). Decreasing the amount of acetonitrile further resulted in significantly lower conversions for both substrates. However, in general, better conversions were obtained in epoxidation reactions of trans-2-hexene (1) compared to trans-2-heptene (4) (Table 1). Lowering the acetonitrile concentration to 10% (v/v) caused a decrease in conversion below 50%, while the product enantiomeric excess remained around 80–85% (Table 1). Moreover, using only 5% (v/v) of acetonitrile did not decrease conversion further, while the product enantiomeric excess dropped to approximately 50%. Possibly, Oxone© is more soluble at lower acetonitrile concentrations, causing higher auto-oxidation of the Shi diketal epoxidation catalyst. This auto-oxidation of the catalyst leads to the formation of acetone, which in turn will catalyse a non-selective alkene epoxidation together with Oxone© [20,44]. Moreover, acetonitrile has been proposed to participate in the reaction mechanism of the Shi epoxidation [45]. Hence, lower epoxide yields and enantiopurity at reduced acetonitrile concentration are in agreement with both hypotheses.

Table 1.
Conversion and product enantiomeric excess (eeP) obtained in the Shi epoxidation of alkenes 1 and 4 at different acetonitrile concentrations. The epoxidations were carried out at a 20 mL scale, and K2CO3 was used to maintain the pH at 11; 0.2 equivalents of the Shi epoxidation catalyst and 4 equivalents of Oxone© were used in the conversion of 50 mM substrate.
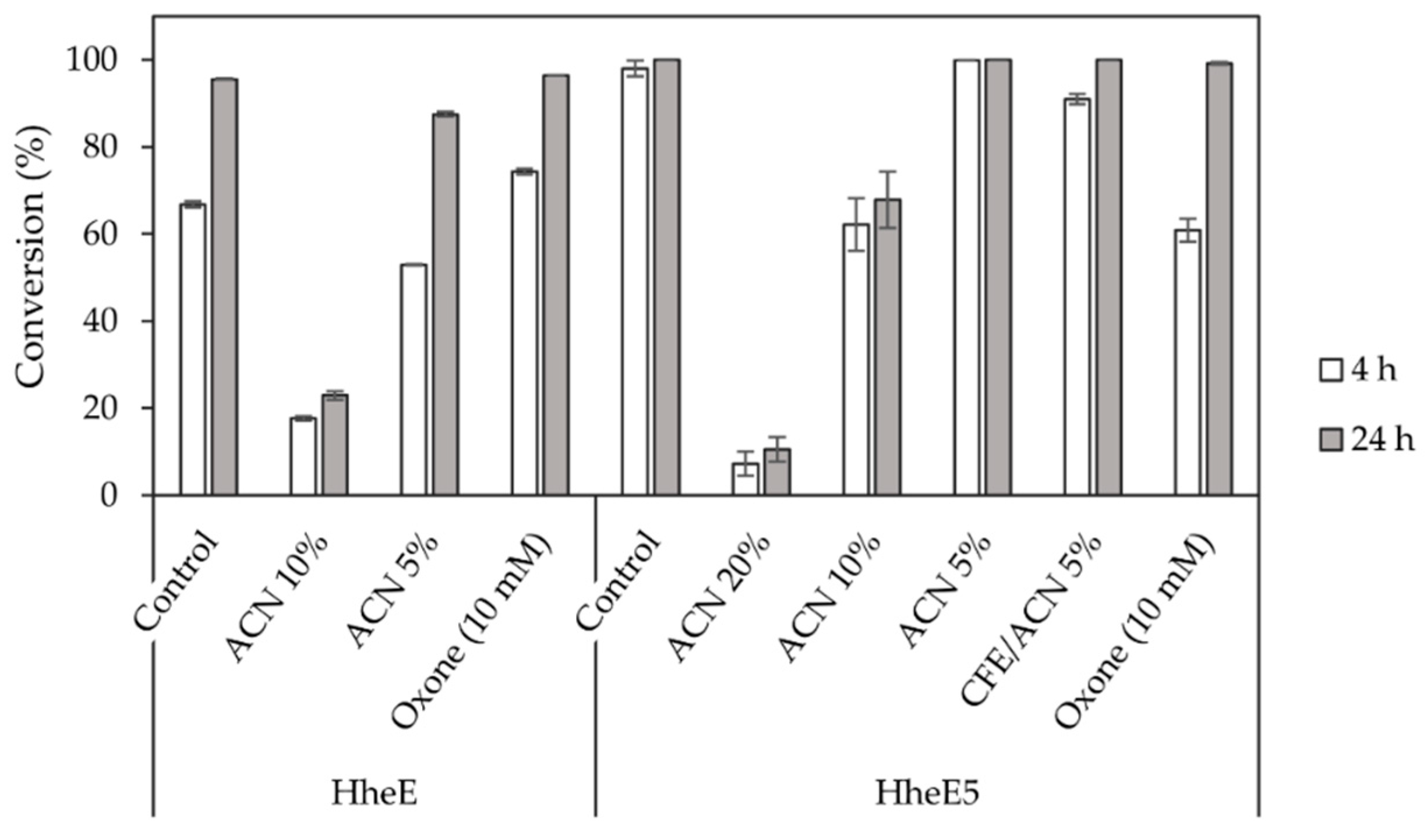
Given the extreme reaction conditions required in the Shi epoxidation (high pH and low temperature), a simultaneous combination of epoxidation and HHDH-catalysed epoxide ring opening was impossible. Therefore, we focused on a sequential combination of both steps, as temperature and pH could then be adjusted in between. However, in such a setup, the reaction components of the first step will still be present during the second cascade step. Hence, we investigated the sensitivity of halohydrin dehalogenases HheE and HheE5, applied as whole cells, towards acetonitrile and Oxone©, both reaction components of the Shi epoxidation (Figure 3). Both enzymes displayed reduced activity in the presence of 10% (v/v) acetonitrile. In contrast, 5% (v/v) acetonitrile had a significantly lower effect on HheE, while no difference in terms of conversion compared to the control reaction without acetonitrile was observed for HheE5. The latter enzyme even displayed very low residual activity at 20% (v/v) acetonitrile concentration. For comparison, we also investigated the effect of 5% (v/v) acetonitrile on the activity of HheE5 applied as cell-free extract. Here, the resulting conversion was slightly lower compared to the reaction with HheE5 applied as whole cells. This might indicate that the cellular envelope helps to protect the HHDH against the organic co-solvent. Therefore, we decided to use the HHDHs as whole-cell catalysts in the final cascade. Moreover, the use of 10% acetonitrile in the first step seemed to be an acceptable compromise between activity and selectivity for combination of both steps in a cascade.
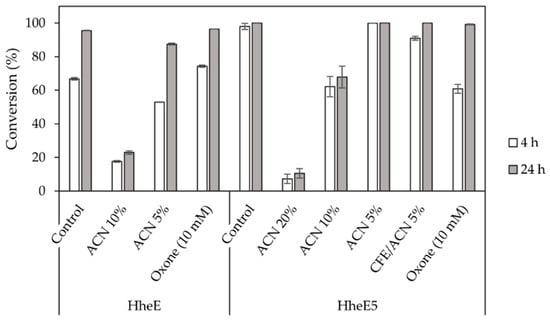
Figure 3.
Epoxide ring opening catalysed by HheE (using 10 mM racemic epoxide 2) and HheE5 (using 10 mM racemic epoxide 5) using whole cells (60 g/L wet cell weight) or cell-free extract (CFE, 9.2 g/L total protein content) and NaN3 (40 mM) in the presence of acetonitrile (ACN) and Oxone©, components of the Shi epoxidation reaction. All reactions were performed in duplicate.
We further expected that residual Oxone© from the Shi epoxidation would interfere with the HHDH step, as Oxone© was reported to oxidise azide, which is used as a nucleophile in the epoxide ring opening, to form N2 and N2O [46]. Thus, a concentration of 10 mM Oxone© was tested to determine its effect on the epoxide ring opening using 40 mM azide, assuming that most of the added Oxone© from the first step would either be consumed in the epoxidation or would spontaneously decompose during the course of the reaction. A 10 mM concentration of Oxone© reduced the reaction rate of the HheE5-catalysed reaction, but full conversion was still achieved within 24 h (Figure 3). In case of HheE, 10 mM Oxone© did not exhibit a negative effect on the reaction rate compared to the positive control reaction without Oxone©. If all Oxone© present would have reacted with azide, a theoretical amount of 30 mM azide (3 eq.) would still have been available for the epoxide ring-opening. The fact that this reduction in azide concentration affected only the reaction rate of HheE5 and not HheE might indicate a possible difference in the KM of both enzymes for azide as the nucleophile. When determining the enzyme kinetics of both HHDHs, indeed, an apparently higher affinity of HheE for azide was observed compared to HheE5 (Table S1). Overall, for the final cascade, it appears that Oxone© can negatively affect the subsequent cascade step, i.e., epoxide ring opening. However, most of the applied Oxone© will have reacted or spontaneously decomposed during epoxidation and the subsequent pH and temperature adjustment. Therefore, the main limiting factor for compatibility of Shi epoxidation and HHDH-catalysed epoxide ring opening appears to be the co-solvent acetonitrile, which seems to be essential for conversion and enantioselectivity in the Shi epoxidation, while it is detrimental for HHDH activity.
2.4. Optimisation of the Styrene Monooxygenase-Catalysed Epoxidation and Compatibility Tests with the HHDH-Catalysed Epoxide Ring Opening (Cascade 2)
The styrene monooxygenase (StyAB) from Rhodococcus sp. ST-10 is a two-component system that requires NADH to provide electrons necessary for oxygen activation at the bound flavin (FAD) cofactor. In general, monooxygenases are complex systems with multiple variables required for activity. Moreover, styrene monooxygenases usually display high activity at low substrate concentrations, while substrate and product inhibition can occur at higher substrate concentrations [27]. Hence, we considered a number of parameters to improve epoxide production such as different cofactor regeneration systems, different enzyme formulations, oxygen input, substrate and/or enzyme feeding, and the use of a two-phase system to enable the use of high substrate concentrations (50 mM) in the final cascade.
First, we tested two possible strategies for cofactor regeneration: (i) applying the biocatalyst as whole cells (WC) in combination with glucose for NADH regeneration within the cell, or (ii) applying the biocatalyst as a cell-free extract in combination with a commercial cofactor regeneration enzyme such as formate dehydrogenase (FDH) from Candida boidinii, which uses a cheap co-substrate to regenerate NADH. Cofactor regeneration is usually required to avoid the stoichiometric addition of expensive NADH. Generally, reactions using the whole-cell system resulted in lower conversions compared to cell-free extract (CFE) reactions with FDH-catalysed cofactor regeneration (Supplementary Materials, Figure S1). However, on a larger scale, the addition of a commercial cofactor regeneration enzyme gets more costly. Therefore, we tested coexpression together with StyAB of two alcohol dehydrogenases from Lactobacillus kefir (LkADH) and Leifsonia sp. S749 (LsADH) for NADH regeneration. LsADH was chosen as it had been applied together with this styrene monooxygenase before [47], whereas LkADH is a common alcohol dehydrogenase used for cofactor regeneration and was tested for comparison [48].
In general, the coexpression of LsADH resulted in higher substrate epoxidation by StyAB (applied as cell-free extract) compared to the coexpression of LkADH. In fact, only negligible epoxide amounts could be obtained in a reaction using cell-free extract of coexpressed StyAB and LkADH due to the strong overexpression of LkADH and thus diminished amounts of soluble StyAB during coexpression in E. coli (Supplementary Materials, Figure S2). In contrast, the coexpression of StyAB and LsADH resulted in more balanced amounts of both soluble enzymes. For comparison, an epoxidation reaction using StyAB-containing cell-free extract with LkADH added as separate cell-free extract did yield epoxide product (Supplementary Materials, Figure S3).
Secondly, active oxygen input (using compressed air) was tested to see if StyAB-catalysed epoxidation could be improved by providing more available oxygen for the reaction. In general, the availability of dissolved molecular oxygen is highly important for successful StyAB-catalysed epoxidation, as molecular oxygen is used as the oxygen donor in the reaction. The requirement for active oxygen input was particularly evident in reactions with the enzyme applied as a whole-cell catalyst in combination with glucose for NADH regeneration. Here, the cellular metabolism also consumed oxygen for efficient NADH regeneration and possibly cell growth, resulting in a fast drop in dissolved oxygen concentration (Supplementary Materials, Figure S4A). In contrast, in a reaction using cell-free extract with the styrene monooxygenase as the only oxygen consumer, the concentration of dissolved oxygen decreased less rapidly (Supplementary Materials, Figure S4A). Using compressed air with a constant flow rate of 3 L/h in a stirred 80 mL reaction enabled us to keep the dissolved oxygen concentration at a high level throughout the StyAB-catalysed epoxidation reaction (Supplementary Materials, Figure S4B).
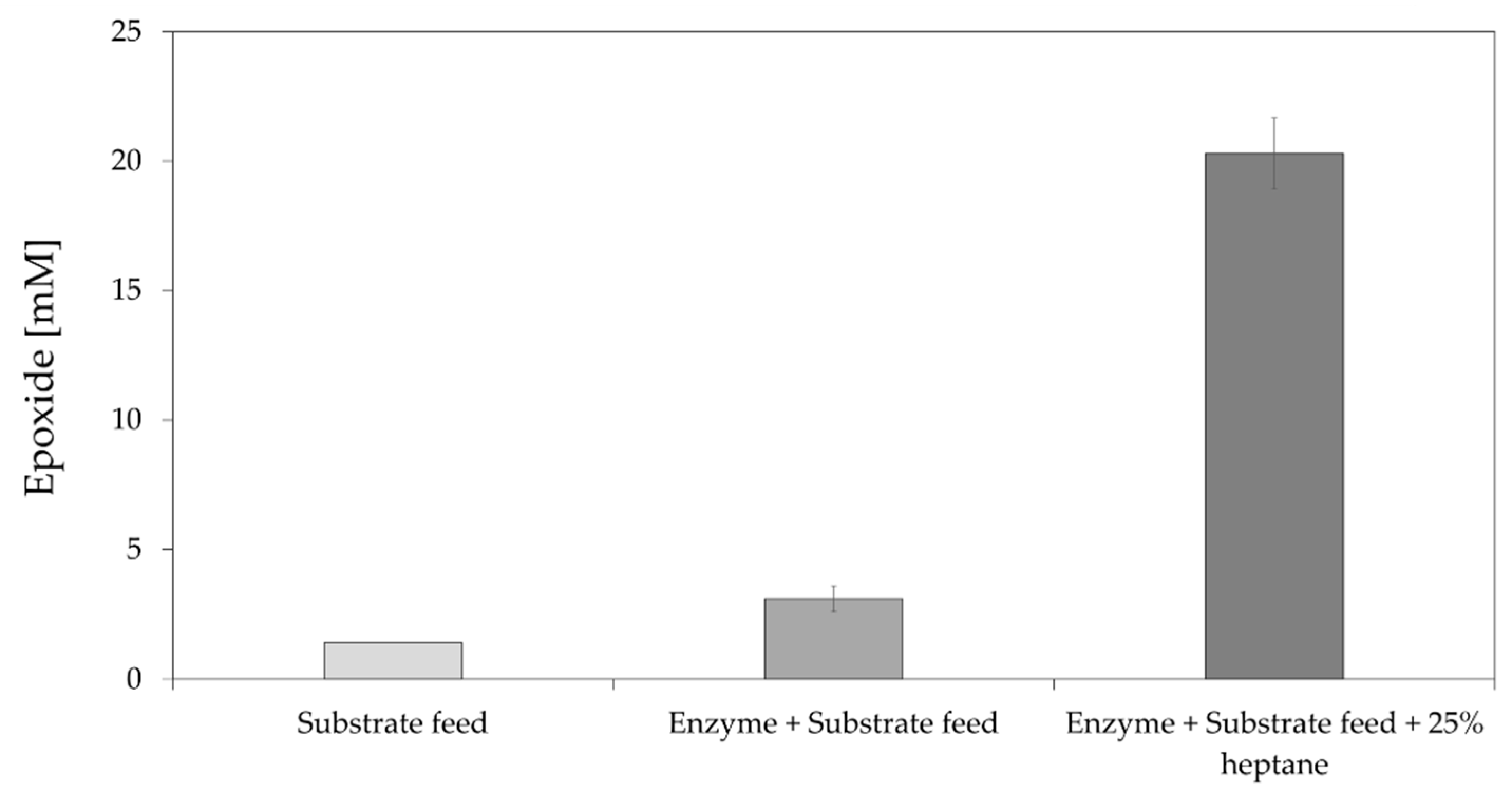
Thirdly, small-scale experiments indicated that the StyA component of StyAB was already inactivated within 6 h at 30 °C (Supplementary Materials, Figure S5), which seems to be due to the thermal instability of the enzyme. Moreover, StyAB was reported in previous studies to display substrate and product inhibition [27]. Substrate inhibition can be mitigated by using a substrate feeding strategy to keep the substrate concentration below a certain level. Respective portion-wise addition of alkene substrate to the reaction proved to be beneficial as higher epoxide production was achieved (Supplementary Materials, Figure S3). Hence, further optimisation tests were performed with reactions using cell-free extract of coexpressed StyAB and LsADH together with alkene feeding. Similarly, enzyme instability can be mitigated by feeding the enzyme over time to compensate for loss of activity instead of providing the full amount of StyAB- and LsADH-containing cell-free extract from the beginning of the reaction. This improved epoxide production further (Figure 4).
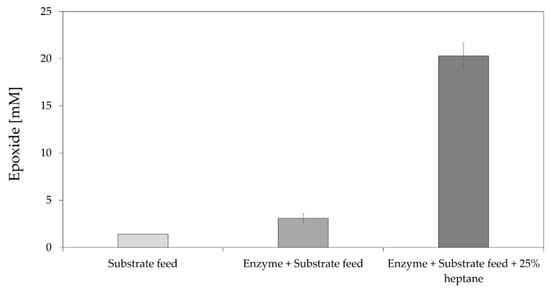
Figure 4.
Epoxidation of 50 mM trans-2-heptene (4) catalysed by StyAB at 80 mL scale. The reactions were performed at 30 °C in 50 mM potassium phosphate buffer pH 7.5 with or without 25% heptane using CFE containing StyAB and LsADH (9.2 g/L final total protein content). Compressed air was applied using an HPLC frit to finely disperse air bubbles, and overhead mechanical stirring was applied (300 rpm). Enzyme and substrate feeding were applied hourly for the first 8 h until the stated concentration was reached (50 mM substrate and 9.2 g/L total protein content).
Finally, we tested the addition of a second organic phase, acting as substrate and product sink, to improve product formation further by alleviating also product inhibition [25,49]. Several organic solvents of different polarity were tested and, in general, solvents with logP > 4, such as cyclohexane, heptane, and isooctane, improved epoxide production (Supplementary Materials, Figure S6), whereas the use of more polar organic solvents, such as ethyl acetate, pentyl acetate, or hexyl acetate [25], did not yield any epoxide (data not shown). Heptane was finally chosen as organic solvent for its lower boiling point compared to isooctane. Using heptane (25% v/v) as the second organic phase resulted in a four-fold increase in product concentration compared to the best StyAB-catalysed reaction without organic solvent (Figure 4). Overall, the use of an aqueous–organic two-phase system had the highest positive effect on product formation in the StyAB-catalysed epoxidation among all variables tested.
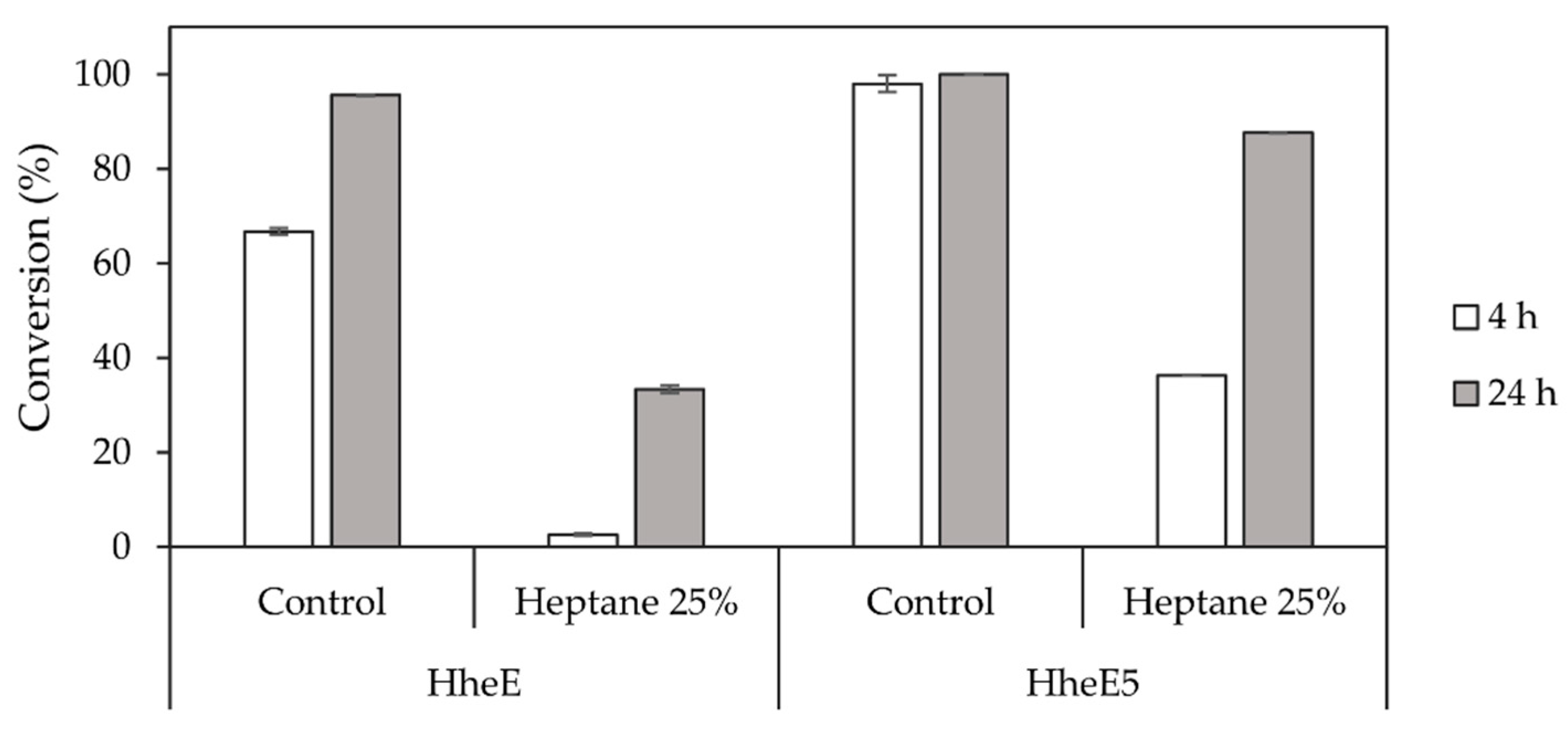
Since optimisation of the StyAB-catalysed epoxidation reaction highlighted the need for a second organic phase, the influence of heptane (25% v/v) on the HHDH-catalysed epoxide ring-opening reaction had to be investigated as well. The presence of organic solvent had a considerably negative effect on enzyme activity for both HHDHs applied as whole cells (Figure 5). Especially HheE achieved only 33% conversion within 24 h (Figure 5), whereas HheE5 still reached 87% conversion, however, after significantly longer reaction time compared to the control reaction without heptane (Figure 5). Even though the StyAB-catalysed epoxidation and the HHDH-catalysed epoxide ring-opening reactions could in theory be performed simultaneously, the observed reduction of HHDH activity in the presence of heptane as second organic phase speaks against a simultaneous cascade. Instead, running both steps in a sequential fashion would at least prevent contact between the HHDHs and heptane before a critical epoxide concentration is formed.
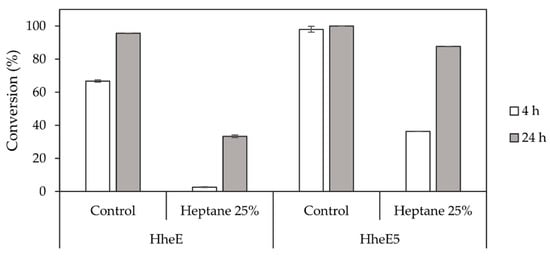
Figure 5.
Conversion of each 10 mM epoxide 2 and 5 by HheE and HheE5, respectively, applied as whole cells in the presence and absence of heptane (25% v/v) as organic phase. All experiments were performed in duplicate in 50 mM potassium phosphate buffer, pH 7.5 at 25 °C.
2.5. Cascade Runs
The results of the compatibility tests indicated that both cascades (cascade 1: Shi epoxidation diketal catalyst + HHDH; cascade 2: StyAB + HHDH) should be performed in a sequential two-step fashion. Moreover, we decided for cascade 1 to run the Shi epoxidation with only 10% v/v acetonitrile to limit the negative effect of the co-solvent on the subsequent HHDH-catalysed step. As a consequence, cascade 1 yielded the final azidoalcohols (2S,3R)-3a and (2S,3R)-6a with reduced product enantiomeric excess of around 60% (Table 2) due to insufficient enantioselectivity in the epoxidation reaction (Supplementary Materials, Table S2), and 30–36% conversion over the two steps. The epoxide ring-opening step even increased eeP a bit, obviously because of a slight enantioselectivity of both HHDHs [42]. In contrast, corresponding (2R,3S)-azidoalcohols were obtained with excellent enantiomeric excess (>99%) in the bi-enzymatic cascade 2 (Table 2), however, at reduced isolated yields compared to cascade 1. The latter is caused by incomplete conversions in both cascade steps of cascade 2 (Supplementary Materials, Table S2). Considering the high (50 mM) substrate concentration used in this cascade, 20% and 39% conversion in the StyAB-catalysed step still translates into 10 and almost 20 mM, respectively, of formed epoxide product. Moreover, regioisomers 3a and especially 6a were formed with high regioselectivity in the HHDH-catalysed step (Table 2). Thus, our StyAB + HHDH cascade for the synthesis of aliphatic 2,3-azidoalcohols compares favourably, in terms of regioselectivity, with the recently published cascade for the synthesis of aryl aliphatic azidoalcohols [12].
Table 2.
Summary of the cascade runs combining StyAB or the Shi epoxidation diketal catalyst with an HHDH (HheE or HheE5). Cascades starting from 50 mM trans-2-hexene (1) resulted in the formation of 2-azido-3-hydroxyhexane (3a), whereas cascades starting from 50 mM trans-2-heptene (4) produced 2-azido-3-hydroxyheptane (6a).
Overall, the poor organic solvent tolerance of the used HHDHs, HheE and HheE5, turned out to be a main limiting factor regarding product yield in both cascades, but also enantioselectivity in cascade 1. In the future, this could be addressed by enzyme engineering as previously shown for HheC, for which a mutant with high residual activity in 50% (v/v) acetonitrile could be generated [50]. Such a solvent-tolerant HHDH would enable to run the Shi epoxidation reaction at optimal acetonitrile concentration to improve epoxide eeP% and conversion. Furthermore, a solvent-tolerant HHDH would also facilitate a simultaneous combination of StyAB-catalysed epoxidation and HHDH-catalysed epoxide ring opening. This could improve the yield of cascade 2, as the formed epoxide product would quickly be further converted to the corresponding azidoalcohol, and hence, product inhibition of StyAB would be minimised. Alternatively, instead of protein engineering, also immobilisation of the HHDHs might be used to increase their solvent tolerance [51,52,53].
Moreover, besides the already mentioned substrate and product inhibition of StyAB, the enzyme’s low stability—especially also concerning the StyB subunit [26]—is another reason limiting the overall conversion of cascade 2. Even though we tried to overcome this by enzyme feeding, the problem could not be completely solved when starting from 50 mM alkene substrate. Enzyme engineering to further stabilise the monooxygenase, e.g., by fusion of subunits StyA and StyB as previously shown for the styrene monooxygenase from Pseudomonas sp. [54], or enzyme immobilisation [55], as well as more sophisticated reaction engineering could be used in the future to achieve high product formation rates even at these high substrate concentrations. The latter was already demonstrated by Panke et al., who could produce 388 g of styrene oxide in a 30 L fed-batch reactor using growing cells harbouring the styrene monooxygenase from Pseudomonas sp. strain VLB120 [56].
3. Materials and Methods
3.1. Chemicals
All chemicals were purchased from Sigma Aldrich (Steinheim, Germany), AppliChem (Darmstadt, Germany), abcr GmbH (Karlsruhe, Germany), Acros Organics (Geel, Belgium), and Carl Roth (Karlsruhe, Germany) in the highest purity available, unless otherwise stated. Trans-2-hexene (1) was purchased from Acros Organics (Geel, Belgium), and trans-2-heptene (4) was purchased from abcr GmbH (Karlsruhe, Germany). The Shi epoxidation diketal catalyst, (R,R)- and (S,S)-Jacobsen catalysts were purchased from Sigma Aldrich (Steinheim, Germany). Chemical standards that were not commercially available were synthesised according to the following protocols. An overview of all substrates, intermediates, and products used in this study is given in Table 3.
Table 3.
List of compounds used in this work.
3.2. Chemical Synthesis of Epoxides
The racemic epoxides trans-2,3-epoxyhexane (2) and trans-2,3-epoxyheptane (5) were synthesised from the corresponding alkenes using meta-chloroperbenzoic acid (m-CPBA) through a slightly modified protocol from Sharma et al. [57]. Epoxidations were carried out in CH2Cl2 (40 mL/g of alkene). m-CPBA (1.5 eq.) was added in small portions over 15 min at RT. After 1 h at room temperature, the mixture was stirred on ice for 5 min, and the white slurry was filtered.Then, 5% (w/v) aq. Na2SO3 (40 mL/g alkene) was added and stirred at RT for 15 min, after which the phases were separated. The aqueous layer was extracted two times with CH2Cl2. The combined organic layers were washed with sat. aq. NaHCO3 (1×) and brine (1×), dried over Na2SO4, and filtered before solvent removal by evaporation. The epoxides were purified by column chromatography using a mixture of cyclohexane/ethyl acetate, 95:5. The identity and purity of the epoxides (isolated yields of 40–60%) was confirmed by NMR, and the resulting NMR data were consistent with the literature data [58].
3.3. Chemical Synthesis of Azidoalcohol Standards
First, 1 mmol of either trans-2,3-epoxyhexane (2) (100 mg) or trans-2,3-epoxyheptane (5) (114 mg) was mixed with 202 mg of NaN3 (3.1 eq.) and 166 mg of NH4Cl (3.1 eq.) in 3.5 mL of methanol and refluxed until no more substrate was visible on TLC (5–6 h). Then, the reaction mixture was diluted in diethyl ether, washed with brine, and the water phase was extracted twice with diethyl ether. The organic layers were combined and dried over Na2SO4. The crude extracts were purified by column chromatography with cyclohexane/diethyl ether (90:10), yielding 52% azido-hydroxyhexane (3) and 48% azido-hydroxyheptane (6) [59]. The identity and purity of obtained azidoalcohols was confirmed by NMR, and the resulting NMR data were consistent with literature data [42].
3.4. Bacterial Strains and Plasmids
The Escherichia coli DH5α strain (Invitrogen, Carlsbad, CA, USA) was used for plasmid amplification and DNA manipulation, while E. coli BL21 (DE3) (Novagen, EMD Biosciences, San Diego, CA, USA) was used for all heterologous expressions.
The vector pMK-RQ containing the codon-optimised genes coding for StyA and StyB was obtained from Invitrogen (Thermo Fisher Scientific division, Wilmington, USA). Genes styA and styB were cloned into MCS1 and MCS2, respectively, of the expression vector pETDuet-1. In particular, styA was inserted between NcoI and BamHI restriction sites, whereas styB was inserted between XhoI and NdeI restriction sites.
The synthetic alcohol dehydrogenase gene from Leisfonia sp. S749 (LsADH) was ordered cloned in vector pCDF1Duet-1 (Genscript, NJ, USA), whereas LkADH was readily available in vector pET21a(+).
The HHDH genes were readily available in pET28a(+)-based vectors [41].
3.5. Biocatalyst Production
Heterologous protein production was performed in 400 mL of TB media (2 L shake flask) with addition of the respective antibiotic(s) (Table 4). A 5 mL overnight culture of the respective E. coli strain was used to inoculate a 50 mL pre-culture in TB media (250 mL flask). The cells were grown at 37 °C and 200 rpm until they reached an OD600 of approximately 2–4. Then, the main culture (400 mL) was inoculated with a required volume of the pre-culture to reach a starting OD600 of 0.1. The cells in the main culture were grown at 37 °C and 200 rpm until they reached OD600 of 0.8–1.2, when 0.2 mM isopropyl-β-D-thiogalactopyranoside (IPTG) was added as an inducer, and the expression temperature was lowered to 20 °C. Expression was performed overnight. Cells were harvested by centrifugation at 7000× g for 30 min at 4 °C, and cell pellets were washed and stored at −20 °C until further use. In experiments where cell-free extract was used, cell pellets were resuspended in 50 mM potassium phosphate buffer pH 7.5 to a concentration of 0.60 g/mL, and cell disruption was performed via sonication (6 cycles of 30 s at an amplitude of 50%, with 30 s pause on ice between cycles). Cell debris was removed by centrifugation at 33,000× g for 30 min at 4 °C. Glycerol (20% final concentration) was added to the resulting cell-free extract before storage at −20 °C.
Table 4.
List of vectors and corresponding antibiotics used in this work.
3.6. Epoxidation Catalysed by the Unspecific Peroxygenase from Agrocybe aegerita
Cell-free extract harbouring the unspecific peroxygenase from A. aegerita (42 μM) was applied to test for trans-2-heptene (4) epoxidation using a modified literature protocol [60]. The enzyme concentration was adjusted to 100 nM in 50 mM potassium phosphate buffer pH 7.5, 4 was added from a 500 mM stock in ethanol to a final concentration of 20 mM. Finally, H2O2 was added drop-wise over 8 h to a final concentration of 100 mM (5 eq.). After 24 h at 30 °C and at 900 rpm shaking, an aliquot was extracted according to the sample preparation protocol described previously.
3.7. StyAB-Catalysed Epoxidation—1 mL Scale
Cell-free extract of E. coli BL21(DE3) containing StyAB was diluted in 50 mM potassium phosphate buffer, pH 7.5 to reach a final total protein concentration of 9.2 g/L. Then, 5 mM substrate (1 or 4) was added, and the commercial formate dehydrogenase from C. boidinii (0.5 U/mL) was added together with 150 mM sodium formate for cofactor regeneration. After 24 h, samples were taken and analysed by GC.
3.8. StyAB-Catalysed Epoxidation—80 mL Scale
Epoxidations using CFE of E. coli BL21 (DE3) harbouring StyAB and LsADH at a final concentration of 9.2 g/L (total protein content) were carried out in 50 mM potassium phosphate buffer at pH 7.5 in a final volume of 80 mL. Trans-2-hexene (1) or trans-2-heptene (4) was added to a concentration of 50 mM from a 2 M stock in isopropanol (used as co-substrate for NADH regeneration). Compressed air was provided through an HPLC frit with a flow of 3 L/h. The reaction was mechanically stirred with overhead stirring for 24 h at 25 °C. At fixed time points, samples were taken and analysed by GC.
Epoxidations with substrate feeding: Substrate was manually fed 5 times every hour using an appropriate amount of the 2 M stock in isopropanol until the final 50 mM concentration was reached.
Epoxidations with enzyme feeding: Enzyme feeding was performed manually every hour for the first 5 h of reaction by addition of an appropriate amount of the stock CFE solution (75 g/L total protein content) until the final concentration of 9.2 g/L (total protein content) was reached.
If an organic phase was present, the total volume was kept to 80 mL split in 60 mL of 50 mM potassium phosphate buffer at pH 7.5 and 20 mL of organic phase. Substrate was fed on the organic phase, whereas cell-free extract was fed into the aqueous phase using a syringe and a needle. The reaction was monitored via GC analysis by taking samples directly from the organic phase by processing it as described below.
3.9. Jacobsen Epoxidation—1 mL Scale
The Jacobsen catalyst is generally designed for the epoxidation of terminal alkenes, but as it was readily available, it was tested with alkene 4 using the standard protocol [16]. To 10 mM of trans-2-heptene (4) in 1 mL saturated NaHCO3 pH 8.0 at room temperature, 0.2 equivalents of either (R,R)- or (S,S)-Jacobsen’s catalyst were added. Then, 2 equivalents of NaOCl were added in portions to support the epoxidation. After 24 h incubation at 900 rpm shaking, a sample of 400 μL was taken, extracted, and analysed by GC as described below.
3.10. Shi epoxidation
The typical Shi epoxidation was performed in either 1, 20, or 50 mL scale using different acetonitrile concentrations (5%, 10%, and 20% v/v). The test reaction was carried out in ddH2O in a final volume of 2 mL. Then, 2 mM of the Shi epoxidation catalyst were used to convert 10 mM of alkene 4 using 2 equivalents of Oxone©, which were added in portions over the first 2 h, at room temperature for 24 h at 900 rpm shaking. NaHCO3 was added to maintain the pH at 8.0. Optimisation reactions were carried out in 20 or 50 mL scale using K2CO3 to maintain the pH at 11. Different concentrations of alkene 1 or 4 were used in combination with 0.2 equivalents of the Shi epoxidation diketal catalyst and 4 equivalents of Oxone©, which were added in portions over the first 8 h together with K2CO3 to keep the pH constant. At fixed time points, samples were taken and analysed by GC.
3.11. HHDH-Catalysed Epoxide Ring-Opening
Whole cells (60 g/L wet cell weight) of E. coli BL21(DE3) harbouring the HHDHs from the E-type were used to convert 10 mM of epoxide 2 or 5, respectively. Then, 50 mM of NaN3 were added as nucleophile, and the reaction was carried out in 1 mL in 50 mM potassium phosphate buffer with 600 rpm shaking at 25 °C. Generally, after 4 h and 24 h, samples were taken and analysed by GC.
3.12. HHDH—Compatibility Tests
Compatibility test studies were performed as described above using either whole cells (60 g/L wet cell weight) or CFE (9.2 g/L total protein content) of E. coli BL21(DE3) harbouring HheE or HheE5 converting 10 mM of epoxide 2 or 5, respectively. The influence of heptane (25% v/v) on HheE and HheE5, applied as whole-cell biocatalyst with 60 g/L wet cell weight, was tested in 3 mL 50 mM potassium phosphate buffer at pH 7.5. The reaction volume was increased to have enough volume to sample directly from the organic phase. Then, 10 mM substrate (2 or 5) and 50 mM sodium azide (NaN3) were added, and the solution was magnetically stirred at 25 °C and 600 rpm. After 4 h and 24 h of reaction, samples were taken for GC analysis.
The influence of acetonitrile (5%, 10%, or 20% v/v) and Oxone© (10 mM) on the HHDHs was tested in 4 mL of 50 mM potassium phosphate buffer at pH 7.5. Then, 10 mM substrate (2 or 5 for HheE and HheE5, respectively) and 40 mM sodium azide (NaN3) were added and the solution was magnetically stirred at 25 °C and 600 rpm. After 4 h and 24 h of reaction, samples were taken for GC analysis.
3.13. Cascade 1—Shi Epoxidation Diketal Catalyst, HHDH—One-Pot Two-Step Mode
In the one-pot two-step cascade run, the Shi epoxidation was carried out at −2 °C in 45 mL ddH2O with 5 mL of acetonitrile (10% v/v). Then, 0.2 equivalents of Shi epoxidation diketal catalyst were used to convert 50 mM of alkene 1 or 4. Four equivalents of Oxone© were added in portions over the first 8 h together with solid K2CO3 to maintain a pH of 11. After incubation for 24 h at 300 rpm, the temperature was adjusted to 25 °C before lowering the pH to 7.5 using 95 % H2SO4. After this, whole cells (60 g/L wet cell weight) of E. coli harbouring HheE or HheE5 were added together with 50 mM of NaN3, and the reaction was continued for an additional 24 h. After 48 h of total reaction time (step 1 + step 2), NaCl was added until saturation. Afterwards, the reaction mixture was extracted 4 times with TBME. For better phase separation, a short centrifugation step (2 min at 2900× g) was performed. The combined organic phases were dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. The resulting crude (2S,3R)-2-azido-3-hydroxyhezane (3a) or (2S,3R)-2-azido-3-hydroxyheptane (6a) were purified as described in the section chemical synthesis of azidoalcohol standards.
3.14. Cascade 2—StyAB, HHDH—One-Pot Two-Step Mode
The epoxidation of 50 mM of either trans-2-hexene (2) or trans-2-heptene (4) was carried out in 60 mL of 50 mM potassium phosphate buffer, pH 7.5, and 20 mL of heptane. The reaction was mechanically stirred at 300 rpm in a water bath at 30 °C with compressed air input (3 L/h). CFE harbouring StyAB and LsADH as well as the substrate (as 2 M stock in isopropanol) were added hourly for the first 8 h to final concentrations of 9.2 g/L total protein content and 50 mM, respectively. After 24 h, whole cells (60 g/L wet cell weight) of E. coli harbouring HheE or HheE5 and 50 mM of sodium azide were added in one portion to start the epoxide ring-opening reaction. After another 24 h of reaction (48 h of total reaction time), NaCl was added until saturation. Afterwards, the reaction mixture was extracted 4 times with TBME. For better phase separation, a short centrifugation step (2 min at 2900× g) was performed. The combined organic phases were dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. The resulting crude (2R,3S)-2-azido-3-hydroxyheptane (3a) or (2R,3S)-2-azido-3-hydroxyheptane (6a) were purified as described in the section chemical synthesis of azidoalcohol standards.
3.15. Sample Preparation for GC Analysis
Samples taken from the aqueous reaction mixture were extracted with an equal volume of TBME or ethyl acetate containing each 0.1% (v/v) dodecane as internal standard. The organic phase was dried over anhydrous sodium sulphate and submitted to GC analysis. If a second organic phase was used (i.e., heptane) or the reaction was performed in an organic solvent, the sample was taken directly from the organic phase, dried over anhydrous sodium sulphate, and submitted to GC analysis.
3.16. GC Analysis
GC analyses have been performed on a Shimadzu GC2010 plus gas chromatograph with an FID detector using different chiral and achiral columns and hydrogen as carrier gas. The injector and detector temperature were set to 300 °C for achiral measurements, whereas they were set to 200 °C and 250 °C respectively for chiral measurements. A summary of the applied temperature programs and columns can be found in Table 5. Conversions of substrate into the corresponding products were determined by achiral GC based on relative peak areas or with a calibration curve. Enantiomeric excesses (ee) of azidoalcohol products 3a and 6a were determined using chiral GC according to a literature protocol [61].
Table 5.
Temperature programs and columns used for analysis of the different compounds on achiral or chiral GC as well as resulting retention times.
4. Conclusions
The aim of this work was to expand the range of accessible enantiopure azidoalcohols through a combination of alkene epoxidation and subsequent epoxide azidolysis, as recently reported for the generation of styrene-based azidoalcohols [12]. Here, we have demonstrated that this strategy can be extended to linear non-terminal alkene substrates, giving access to a broader range of possible azidoalcohol products. Due to the high regioselectivity of the applied halohydrin dehalogenases for epoxide ring opening and very low chemical background reaction, final azidoalcohol products were obtained with very high to excellent regioselectivity, resulting in high product purities. Moreover, the identification of stereocomplementary epoxidation catalysts even enabled us to synthesise opposite azidoalcohol enantiomers, albeit epoxide enantiopurity in the Shi epoxidation reaction requires further improvement. Alternatively, recently discovered (R)-selective styrene monooxygenases could be used in the future to obtain respective (2S,3R)-azidoalcohols with high enantiomeric excess [31,32,33].
With this, our approach nicely adds to existing synthetic routes and already accessible azidoalcohol products. With an additional simple hydrogenation step to reduce the azido group to an amino group, our cascade will further offer access to corresponding chiral aminoalcohols [62,63,64].
Our attempts of running both cascades at synthetically meaningful substrate concentrations (50 mM) also revealed current limitations that need to be addressed by enzyme and reaction engineering in the future to harness the full potential of our proposed cascades.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/catal11080982/s1, Additional methods, Table S1: HHDHs kinetic parameters, Table S2: Detailed description of cascade reactions run at preparative scale, Figure S1: StyAB-catalysed epoxidation whole cells vs. cell-free extract, Figure S2: SDS-PAGE showing coexpression of StyAB with either LkADH or LsADH, Figure S3: StyAB-catalysed epoxidation at 40 and 80 mL scale, Figure S4: Oxygen consumption during the StyAB-catalysed epoxidation using whole cells vs. cell-free extract, Figure S5: StyA residual activity, Figure S6: Comparison of different organic solvents for the StyAB-catalysed epoxidation.
Author Contributions
Conceptualisation, A.S. and R.W.; methodology, E.C., A.S. and P.S.; validation, E.C., A.S. and P.S.; investigation, E.C.; formal analysis, E.C.; resources, A.S., F.H., R.W.; writing—original draft preparation, E.C. and A.S.; writing—review and editing, P.S., F.H. and R.W.; visualisation, E.C.; supervision, A.S., F.H. and P.S.; project administration, A.S., R.W.; funding acquisition, A.S., F.H. and R.W. All authors have read and agreed to the published version of the manuscript.
Funding
This project was funded by the European Union’s Horizon 2020 MSCA ITN-EID program under grant agreement No 634200 (BIOCASCADES). This communication reflects only the beneficiary’s view and the European Commission is not responsible for any use that may be made of the information it contains.
Acknowledgments
We thank Melinda Fekete for valuable advice in the synthesis of authentic compounds and product purification. We acknowledge financial support from the German Research Foundation and the Open Access Publication Funds of the Technische Universität Braunschweig.
Conflicts of Interest
The company Enzymicals markets the respective halohydrin dehalogenases. Apart from that, the authors declare no conflict of interest.
References
- Ricklefs, E.; Girhard, M.; Koschorreck, K.; Smit, M.S.; Urlacher, V.B. Two-Step One-Pot Synthesis of Pinoresinol from Eugenol in an Enzymatic Cascade. Chem. Cat. Chem. 2015, 7, 1857–1864. [Google Scholar] [CrossRef]
- Bruggink, A.; Schoevaart, R.; Kieboom, T. Concepts of Nature in Organic Synthesis: Cascade Catalysis and Multistep Conversions in Concert. Org. Process. Res. Dev. 2003, 7, 622–640. [Google Scholar] [CrossRef]
- Schoevaart, R.; van Rantwijk, F.; Sheldon, R.A. A Four-Step Enzymatic Cascade for the One-Pot Synthesis of Non-Natural Carbohydrates from Glycerol. J. Org. Chem. 2000, 65, 6940–6943. [Google Scholar] [CrossRef] [PubMed]
- Schubert, J.; Schwesinger, R.; Prinzbach, H. Total Synthesis of a Fortimicin Aglycone. Angew. Chem. Int. Ed. 1984, 23, 167–169. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, P.T.; Nanda, S.; Rao, A.B. Stereoselective Synthesis of (R)-(−)-Denopamine, (R)-(−)-Tembamide and (R)-(−)-Aegeline via Asymmetric Reduction of Azidoketones by Daucus carota in Aqueous Medium. Tetrahedron Asymmetry 2002, 12, 3381–3385. [Google Scholar] [CrossRef]
- Gupta, P.; Mahajan, N. Biocatalytic Approaches towards the Stereoselective Synthesis of Vicinal Amino Alcohols. New J. Chem. 2018, 42, 12296–12327. [Google Scholar] [CrossRef]
- Smith, B.T.; Gracias, V.; Aubé, J. Regiochemical Studies of the Ring Expansion Reactions of Hydroxy Azides with Cyclic Ketones. J. Org. Chem. 2000, 65, 3771–3774. [Google Scholar] [CrossRef]
- Sonavane, S.U.; Chidambaram, M.; Khalil, S.; Almog, J.; Sasson, Y. Synthesis of Cyclic Disulfides Using Didecyldimethylammonium Bromide as Phase Transfer Catalyst. Tetrahedron Lett. 2008, 49, 520–522. [Google Scholar] [CrossRef]
- Badiang, J.G.; Aubé, J. One-Step Conversion of Aldehydes to Oxazolines and 5,6-Dihydro-4H-1,3-Oxazines Using 1,2- and 1,3-Azido Alcohols. J. Org. Chem. 1996, 61, 2484–2487. [Google Scholar] [CrossRef]
- Scriven, E.F.V.; Turnbull, K. Azides: Their Preparation and Synthetic Uses. Chem. Rev. 1988, 88, 297–368. [Google Scholar] [CrossRef]
- Fringuelli, F.; Piermatti, O.; Pizzo, F.; Vaccaro, L. Ring Opening of Epoxides with Sodium Azide in Water. A Regioselective PH-Controlled Reaction. J. Org. Chem. 1999, 64, 6094–6096. [Google Scholar] [CrossRef]
- Martínez-Montero, L.; Tischler, D.; Süss, P.; Schallmey, A.; Franssen, M.C.; Hollmann, F.; Paul, C.E. Asymmetric Azidohydroxylation of Styrene Derivatives Mediated by a Biomimetic Styrene Monooxygenase Enzymatic Cascade. Catal. Sci. Technol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Katsuki, T.; Sharpless, K.B. The First Practical Method for Asymmetric Epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar] [CrossRef]
- Xia, Q.-H.; Ge, H.-Q.; Ye, C.-P.; Liu, Z.-M.; Su, K.-X. Advances in Homogeneous and Heterogeneous Catalytic Asymmetric Epoxidation. Chem. Rev. 2005, 105, 1603–1662. [Google Scholar] [CrossRef]
- Wang, C.; Yamamoto, H. Asymmetric Epoxidation Using Hydrogen Peroxide as Oxidant. Chem. Asian J. 2015, 10, 2056–2068. [Google Scholar] [CrossRef]
- Zhang, W.; Loebach, J.L.; Wilson, S.R.; Jacobsen, E.N. Enantioselective Epoxidation of Unfunctionalized Olefins Catalyzed by Salen Manganese Complexes. J. Am. Chem. Soc. 1990, 112, 2801–2803. [Google Scholar] [CrossRef]
- Ito, Y.N.; Katsuki, T. Asymmetric Catalysis of New Generation Chiral Metallosalen Complexes. Bull. Chem. Soc. Jpn. 1999, 72, 603–619. [Google Scholar] [CrossRef]
- Miyazaki, T.; Katsuki, T. Nb(Salen)-Catalyzed Sulfoxidation. Synlett 2003, 1046–1048. [Google Scholar] [CrossRef]
- Brandes, B.D.; Jacobsen, E.N. Highly Enantioselective, Catalytic Epoxidation of Trisubstituted Olefins. J. Org. Chem. 1994, 59, 4378–4380. [Google Scholar] [CrossRef]
- Tu, Y.; Wang, Z.-X.; Shi, Y. An Efficient Asymmetric Epoxidation Method for Trans-Olefins Mediated by a Fructose-Derived Ketone. J. Am. Chem. Soc. 1996, 118, 9806–9807. [Google Scholar] [CrossRef]
- Shu, L.; Shi, Y. An Efficient Ketone-Catalyzed Epoxidation Using Hydrogen Peroxide as Oxidant. J. Org. Chem. 2000, 65, 8807–8810. [Google Scholar] [CrossRef]
- Liang, Y.; Wei, J.; Qiu, X.; Jiao, N. Homogeneous Oxygenase Catalysis. Chem. Rev. 2018, 118, 4912–4945. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, F.; Arends, I.W.C.; Buehler, K.; Schallmey, A.; Bühler, B. Enzyme -Mediated Oxidations for the Chemist. Green Chem. 2011, 13, 226–265. [Google Scholar] [CrossRef]
- Holtmann, D.; Fraaije, M.W.; Arends, I.W.C.; Opperman, D.J.; Hollmann, F. The Taming of Oxygen: Biocatalytic Oxyfunctionalisations. Chem. Comm. 2014, 50, 13180–13200. [Google Scholar] [CrossRef] [Green Version]
- Toda, H.; Imae, R.; Itoh, N. Bioproduction of Chiral Epoxyalkanes Using Styrene Monooxygenase from Rhodococcus sp. ST-10 (RhSMO). Adv. Synth. Catal. 2014, 356, 3443–3450. [Google Scholar] [CrossRef]
- Toda, H.; Imae, R.; Komio, T.; Itoh, N. Expression and Characterization of Styrene Monooxygenases of Rhodococcus sp. ST-5 and ST-10 for Synthesizing Enantiopure (S)-Epoxides. Appl. Microbiol. Biotechnol. 2012, 96, 407–418. [Google Scholar] [CrossRef]
- Toda, H.; Itoh, N. Isolation and Characterization of Styrene Metabolism Genes from Styrene-Assimilating Soil Bacteria Rhodococcus sp. ST-5 and ST-10. J. Biosci. Bioeng. 2012, 113, 12–19. [Google Scholar] [CrossRef]
- Tischler, D.; Schlömann, M.; van Berkel, W.J.H.; Gassner, G.T. FAD C(4a)-Hydroxide Stabilized in a Naturally Fused Styrene Monooxygenase. FEBS Lett. 2013, 587, 3848–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedel, A.; Heine, T.; Westphal, A.H.; Conrad, C.; Rathsack, P.; van Berkel, W.J.H.; Tischler, D. Catalytic and Hydrodynamic Properties of Styrene Monooxygenases from Rhodococcus opacus 1CP Are Modulated by Cofactor Binding. AMB Express 2015, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Oelschlägel, M.; Zimmerling, J.; Schlömann, M.; Tischler, D. Styrene Oxide Isomerase of Sphingopyxis sp. Kp5.2. Microbiology 2014, 160, 2481–2491. [Google Scholar] [CrossRef]
- Xiao, H.; Dong, S.; Liu, Y.; Pei, X.-Q.; Lin, H.; Wu, Z.-L. A New Clade of Styrene Monooxygenases for (R)-Selective Epoxidation. Catal. Sci. Technol. 2021, 11, 2195–2201. [Google Scholar] [CrossRef]
- Heine, T.; Scholtissek, A.; Hofmann, S.; Koch, R.; Tischler, D. Accessing Enantiopure Epoxides and Sulfoxides: Related Flavin-Dependent Monooxygenases Provide Reversed Enantioselectivity. ChemCatChem 2020, 12, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Guo, C.; Lin, H.; Ding, Z.-Y.; Liu, Y.; Wu, Z.-L. Functional Characterization of an (R)-Selective Styrene Monooxygenase from Streptomyces sp. NRRL S-31. Enzyme Microb. Technol. 2020, 132, 109391. [Google Scholar] [CrossRef]
- Benedetti, F.; Berti, F.; Norbedo, S. Regio- and Stereoselective Ring Opening of 2,3-Epoxyalcohols with Diethylaluminium Azide. Tetrahedron Lett. 1998, 39, 7971–7974. [Google Scholar] [CrossRef]
- Hansen, K.B.; Leighton, J.L.; Jacobsen, E.N. On the Mechanism of Asymmetric Nucleophilic Ring-Opening of Epoxides Catalyzed by (Salen)CrIII Complexes. J. Am. Chem. Soc. 1996, 118, 10924–10925. [Google Scholar] [CrossRef]
- Kamal, A.; Damayanthi, Y.; Reddy, B.S.N.; Lakminarayana, B.; Reddy, B.S.P. Novel Biocatalytic Reduction of Aryl Azides: Chemoenzymatic Synthesis of Pyrrolo [2,1–c][1,4] Benzodiazepine Antibiotics1. Chem. Comm. 1997, 11, 1015–1016. [Google Scholar] [CrossRef]
- Jacobsen, E.N. Asymmetric Catalysis of Epoxide Ring-Opening Reactions. Acc. Chem. Res. 2000, 33, 421–431. [Google Scholar] [CrossRef]
- White, D.E.; Tadross, P.M.; Lu, Z.; Jacobsen, E.N. A Broadly Applicable and Practical Oligomeric (Salen) Co Catalyst for Enantioselective Epoxide Ring-Opening Reactions. Tetrahedron 2014, 70, 4165–4180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schallmey, A.; Schallmey, M. Recent Advances on Halohydrin Dehalogenases—from Enzyme Identification to Novel Biocatalytic Applications. Appl. Microbiol. Biotechnol. 2016, 100, 7827–7839. [Google Scholar] [CrossRef] [Green Version]
- Blažević, Z.F.; Milčić, N.; Sudar, M.; Elenkov, M.M. Halohydrin Dehalogenases and Their Potential in Industrial Application—A Viewpoint of Enzyme Reaction Engineering. Adv. Syn. Catal. 2021, 363, 388–410. [Google Scholar] [CrossRef]
- Schallmey, M.; Koopmeiners, J.; Wells, E.; Wardenga, R.; Schallmey, A. Expanding the Halohydrin Dehalogenase Enzyme Family: Identification of Novel Enzymes by Database Mining. Appl. Environ. Microbiol. 2014, 80, 7303–7315. [Google Scholar] [CrossRef] [Green Version]
- Calderini, E.; Wessel, J.; Süss, P.; Schrepfer, P.; Wardenga, R.; Schallmey, A. Selective Ring-Opening of Di-Substituted Epoxides Catalysed by Halohydrin Dehalogenases. Chem. Cat. Chem. 2019, 11, 2099–2106. [Google Scholar] [CrossRef]
- Koopmeiners, J.; Diederich, C.; Solarczek, J.; Voß, H.; Mayer, J.; Blankenfeldt, W.; Schallmey, A. HheG, a Halohydrin Dehalogenase with Activity on Cyclic Epoxides. ACS Catal. 2017, 7, 6877–6886. [Google Scholar] [CrossRef]
- Shi, Y. Organocatalytic Asymmetric Epoxidation of Olefins by Chiral Ketones. Acc. Chem. Res. 2004, 37, 488–496. [Google Scholar] [CrossRef]
- Shu, L.; Shi, Y. An Efficient Ketone-Catalyzed Asymmetric Epoxidation Using Hydrogen Peroxide (H2O2) as Primary Oxidant. Tetrahedron 2001, 57, 5213–5218. [Google Scholar] [CrossRef]
- Thompson, R.C.; Wieland, P.; Appelman, E.H. Oxidation of Azide and Azidopentaamminechromium(III) by Peroxymonosulfate in Aqueous Solution. Inorg. Chem. 1979, 18, 1974–1977. [Google Scholar] [CrossRef]
- Toda, H.; Imae, R.; Itoh, N. Efficient Biocatalysis for the Production of Enantiopure (S)-Epoxides Using a Styrene Monooxygenase (SMO) and Leifsonia Alcohol Dehydrogenase (LSADH) System. Tetrahedron Asymmetry 2012, 23, 1542–1549. [Google Scholar] [CrossRef]
- Bradshaw, C.W.; Hummel, W.; Wong, C.H. Lactobacillus Kefir Alcohol Dehydrogenase: A Useful Catalyst for Synthesis. J. Org. Chem. 1992, 57, 1532–1536. [Google Scholar] [CrossRef]
- Panke, S.; Wubbolts, M.G.; Schmid, A.; Witholt, B. Production of Enantiopure Styrene Oxide by Recombinant Escherichia coli Synthesizing a Two-Component Styrene Monooxygenase. Biotechnol. Bioeng. 2000, 69, 91–100. [Google Scholar] [CrossRef]
- Janssen, D.B.; Majerić-Elenkov, M.; Hasnaoui, G.; Hauer, B.; Spelberg, J.H.L. Enantioselective Formation and Ring-Opening of Epoxides Catalysed by Halohydrin Dehalogenases. Biochem. Soc. Trans. 2006, 34, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Bernal, C.; Rodríguez, K.; Martínez, R. Integrating Enzyme Immobilization and Protein Engineering: An Alternative Path for the Development of Novel and Improved Industrial Biocatalysts. Biotechnol. Adv. 2018, 36, 1470–1480. [Google Scholar] [CrossRef]
- Hermanová, S.; Zarevúcká, M.; Bouša, D.; Pumera, M.; Sofer, Z. Graphene Oxide Immobilized Enzymes Show High Thermal and Solvent Stability. Nanoscale 2015, 7, 5852–5858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Li, B.; Sasaki, T.; Miyazaki, C.; Kajino, T.; Inagaki, S. Immobilized Enzymes in Ordered Mesoporous Silica Materials and Improvement of Their Stability and Catalytic Activity in an Organic Solvent. Microporous Mesoporous Mater. 2001, 44–45, 755–762. [Google Scholar] [CrossRef]
- Corrado, M.L.; Knaus, T.; Mutti, F.G. A Chimeric Styrene Monooxygenase with Increased Efficiency in Asymmetric Biocatalytic Epoxidation. Chembiochem 2018, 19, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Ruinatscha, R.; Karande, R.; Buehler, K.; Schmid, A. Integrated One-Pot Enrichment and Immobilization of Styrene Monooxygenase (StyA) Using SEPABEAD EC-EA and EC-Q1A Anion-Exchange Carriers. Molecules 2011, 16, 5975–5988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panke, S.; Held, M.; Wubbolts, M.G.; Witholt, B.; Schmid, A. Pilot-Scale Production of (S)-Styrene Oxide from Styrene by Recombinant Escherichia coli Synthesizing Styrene Monooxygenase. Biotechnol. Bioeng. 2002, 80, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Bulger, P.G.; McNevin, M.; Dormer, P.G.; Ball, R.G.; Streckfuss, E.; Cuff, J.F.; Yin, J.; Chen, C. A Cascade Approach to Cyclic Aminonitrones: Reaction Discovery, Mechanism, and Scope. Org. Lett. 2009, 11, 3194–3197. [Google Scholar] [CrossRef]
- Kroutil, W.; Mischitz, M.; Faber, K. Deracemization of (±)-2,3-Disubstituted Oxiranes via Biocatalytic Hydrolysis Using Bacterial Epoxide Hydrolases: Kinetics of an Enantioconvergent Process. J. Chem. Soc. Perkin Trans. 1997, 3629–3636. [Google Scholar] [CrossRef]
- Righi, G.; Pietrantonio, S.; Bonini, C. Stereocontrolled ‘One Pot’ Organometallic Addition–Ring Opening Reaction of α,β-Aziridine Aldehydes. A New Entry to Syn 1,2-Amino Alcohols. Tetrahedron 2001, 57, 10039–10046. [Google Scholar] [CrossRef]
- Wang, Y.; Lan, D.; Durrani, R.; Hollmann, F. Peroxygenases En Route to Becoming Dream Catalysts. What Are the Opportunities and Challenges? Curr. Opin. Chem. Biol. 2017, 37, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Fujimoto, Y.; Girdaukas, G.; Sih, C.J. Quantitative Analyses of Biochemical Kinetic Resolutions of Enantiomers. J. Am. Chem. Soc. 1982, 104, 7294–7299. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, Y.; Wang, T.; Too, H.-P.; Wang, D.I.C.; Li, Z. Highly Regio- and Enantioselective Multiple Oxy- and Amino-Functionalizations of Alkenes by Modular Cascade Biocatalysis. Nat. Commun. 2016, 7, 11917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrittwieser, J.H.; Coccia, F.; Kara, S.; Grischek, B.; Kroutil, W.; d’Alessandro, N.; Hollmann, F. One-Pot Combination of Enzyme and Pd Nanoparticle Catalysis for the Synthesis of Enantiomerically Pure 1,2-Amino Alcohols. Green Chem. 2013, 15, 3318–3331. [Google Scholar] [CrossRef] [Green Version]
- Ingram, C.U.; Bommer, M.; Smith, M.E.B.; Dalby, P.A.; Ward, J.M.; Hailes, H.C.; Lye, G.J. One-Pot Synthesis of Amino-Alcohols Using a de-Novo Transketolase and β-Alanine: Pyruvate Transaminase Pathway in Escherichia coli. Biotechnol. Bioeng. 2007, 96, 559–569. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).