
Hydroxyl-Decorated Diiron Complex as a [FeFe]-Hydrogenase Active Site Model Complex: Light-Driven Photocatalytic Activity and Heterogenization on Ethylene-Bridged Periodic Mesoporous Organosilica

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
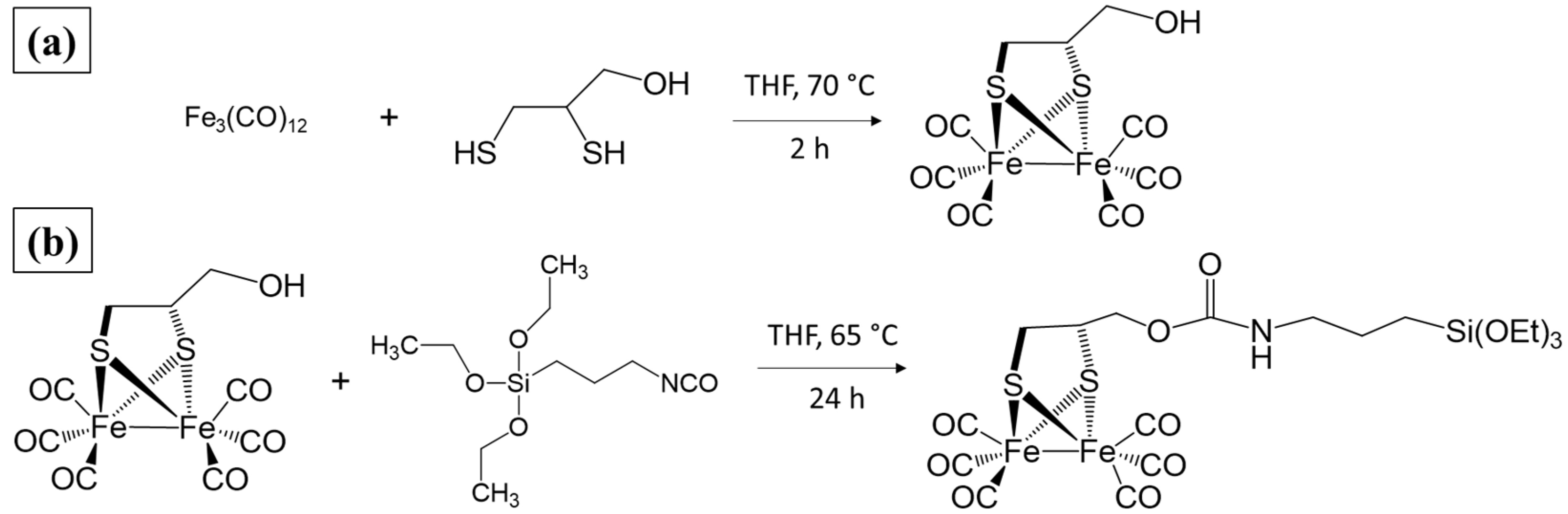
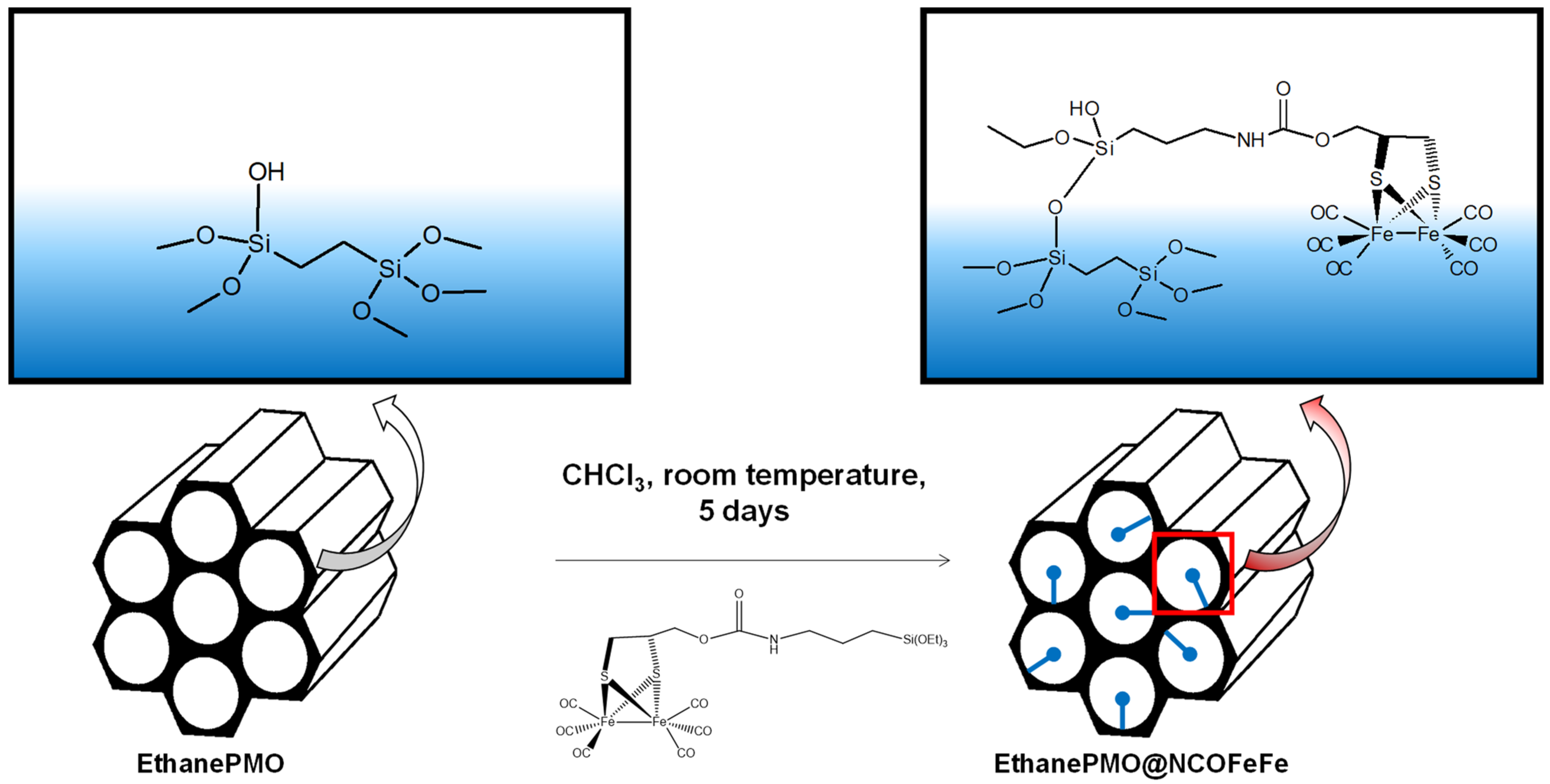
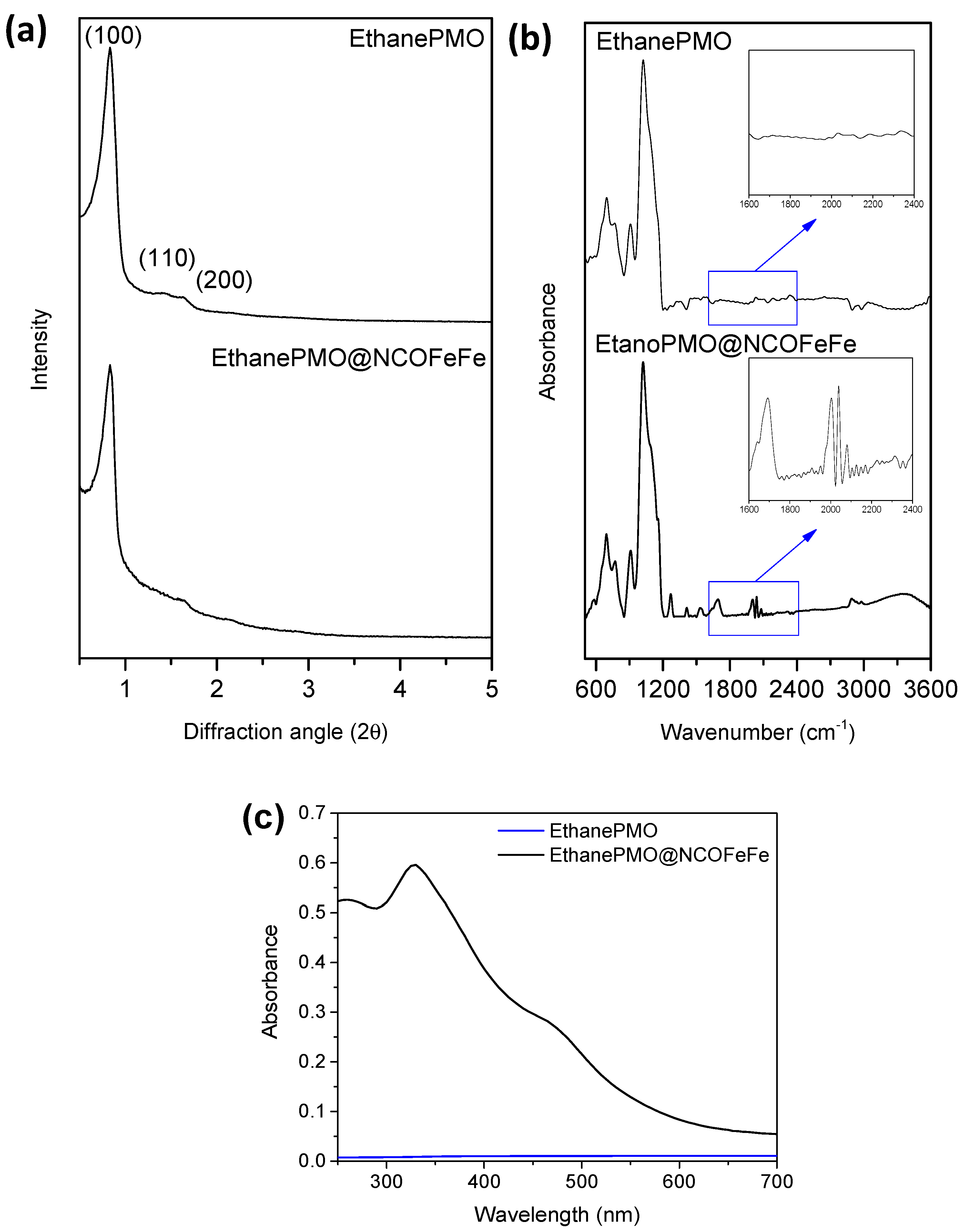
2.1. Synthesis and Characterization of Diiron Catalysts
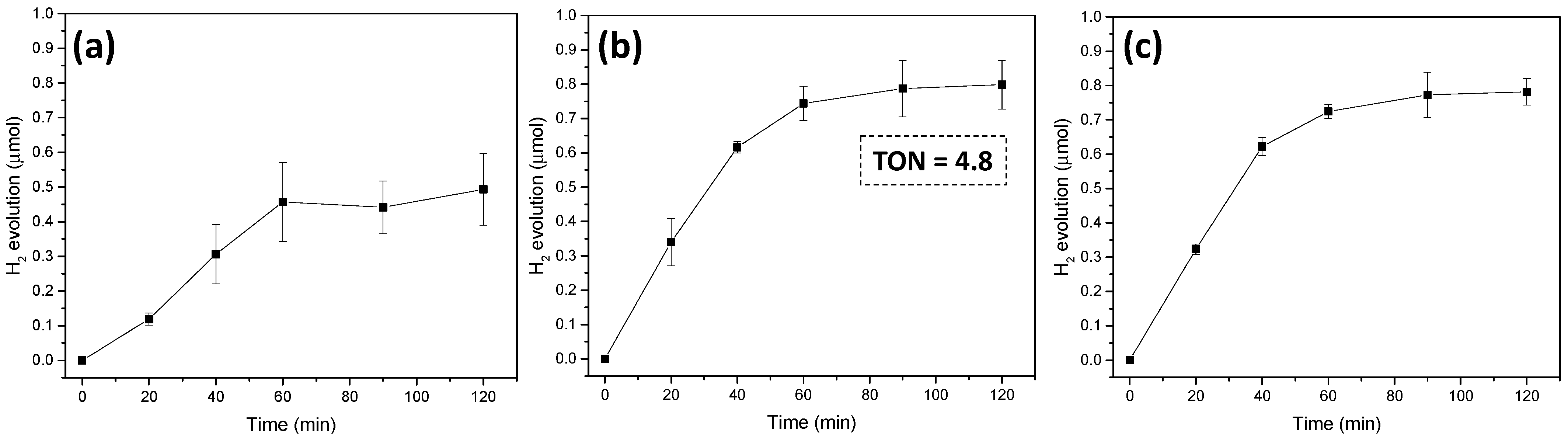
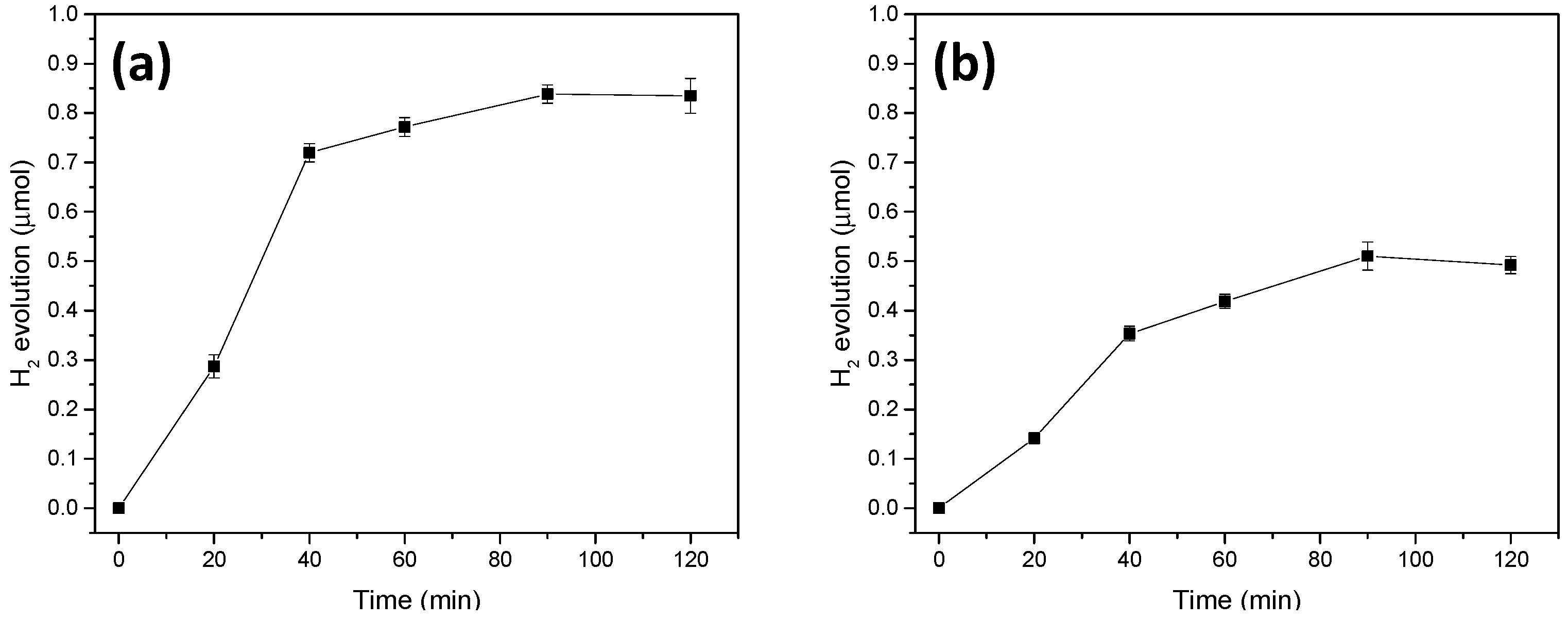
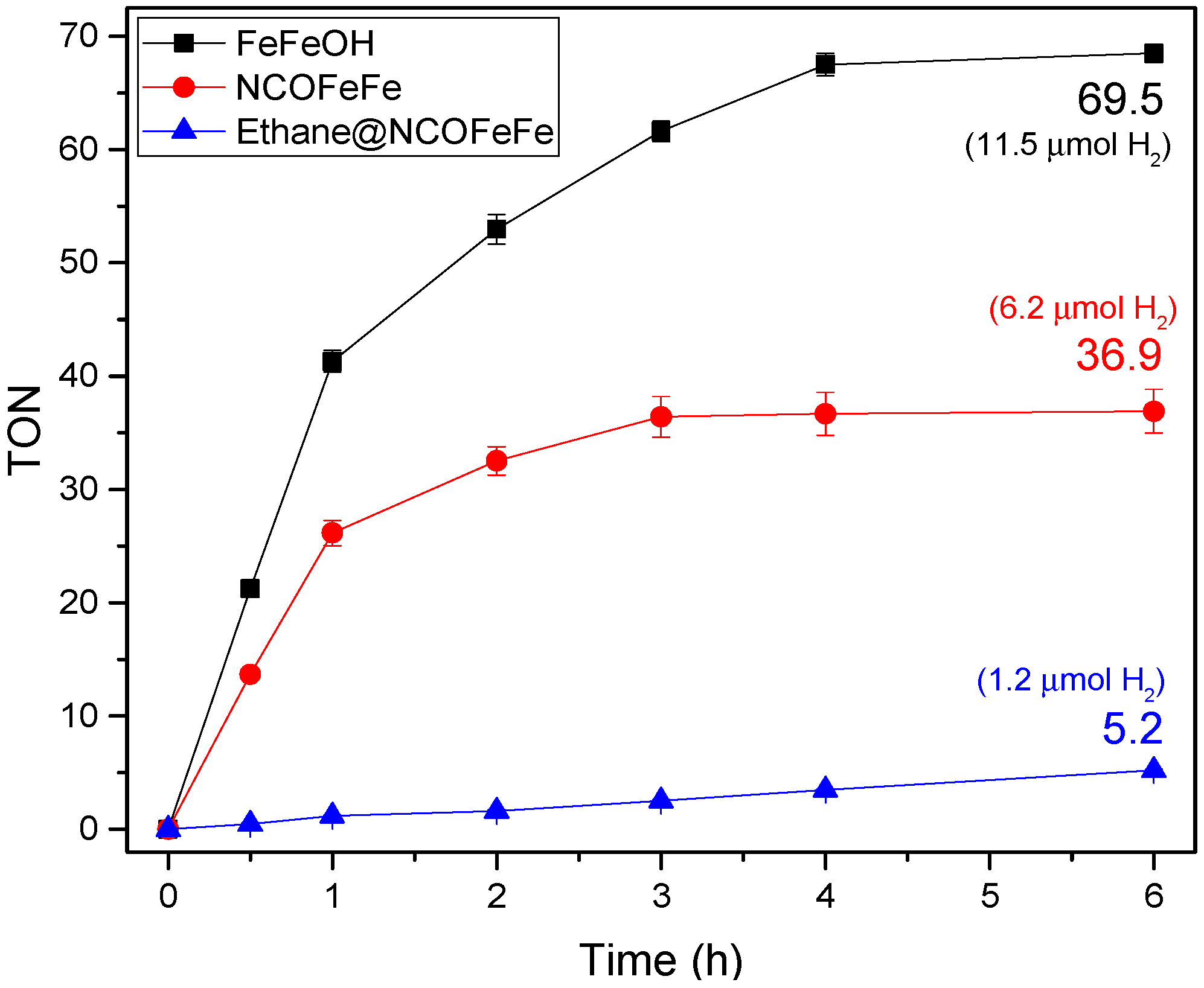
2.2. Light-Driven Hydrogen Production
3. Materials and Methods
3.1. Reagents and Materials
3.2. Synthesis of the Catalysts
3.3. Characterization of the Catalysts
3.4. Photocatalytic Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dresselhaus, M.S.; Thomas, I.L. Alternative energy technologies. Nature 2001, 414, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Panwar, N.L.; Kaushik, S.C.; Kothari, S. Role of renewable energy sources in environmental protection: A review. Renew. Sustain. Energy Rev. 2011, 15, 1513–1524. [Google Scholar] [CrossRef]
- Gielen, D.; Boshell, F.; Saygin, D.; Bazilian, M.D.; Wagner, N.; Gorini, R. The role of renewable energy in the global energy transformation. Energy Strateg. Rev. 2019, 24, 38–50. [Google Scholar] [CrossRef]
- Ismail, A.A.; Bahnemann, D.W. Photochemical splitting of water for hydrogen production by photocatalysis: A review. Sol. Energy Mater. Sol. Cells 2014, 128, 85–101. [Google Scholar] [CrossRef]
- Acar, C.; Dincer, I.; Naterer, G.F. Review of photocatalytic water-splitting methods for sustainable hydrogen production. Int. J. Energy Res. 2016, 40, 1449–1473. [Google Scholar] [CrossRef]
- Chen, S.; Takata, T.; Domen, K. Particulate photocatalysts for overall water splitting. Nat. Rev. Mater. 2017, 2, 17050. [Google Scholar] [CrossRef]
- Kabir, E.; Kumar, P.; Kumar, S.; Adelodun, A.A.; Kim, K.-H. Solar energy: Potential and future prospects. Renew. Sustain. Energy Rev. 2018, 82, 894–900. [Google Scholar] [CrossRef]
- Corredor, J.; Rivero, M.J.; Rangel, C.M.; Gloaguen, F.; Ortiz, I. Comprehensive review and future perspectives on the photocatalytic hydrogen production. J. Chem. Technol. Biotechnol. 2019, 94, 3049–3063. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, S.E.; Wahid, M.A. Hydrogen production from renewable and sustainable energy resources: Promising green energy carrier for clean development. Renew. Sustain. Energy Rev. 2016, 57, 850–866. [Google Scholar] [CrossRef]
- Abe, J.O.; Popoola, A.P.I.; Ajenifuja, E.; Popoola, O.M. Hydrogen energy, economy and storage: Review and recommendation. Int. J. Hydrogen Energy 2019, 44, 15072–15086. [Google Scholar] [CrossRef]
- Frey, M. Hydrogenases: Hydrogen-Activating Enzymes. ChemBioChem 2002, 3, 153–160. [Google Scholar] [CrossRef]
- Vignais, P.M.; Billoud, B. Occurrence, Classification, and Biological Function of Hydrogenases: An Overview. Chem. Rev. 2007, 107, 4206–4272. [Google Scholar] [CrossRef]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Akermark, B.; Ott, S. Iron hydrogenase active site mimics in supramolecular systems aiming for light-driven hydrogen production. Coord. Chem. Rev. 2005, 249, 1653–1663. [Google Scholar] [CrossRef]
- Simmons, T.R.; Berggren, G.; Bacchi, M.; Fontecave, M.; Artero, V. Mimicking hydrogenases: From biomimetics to artificial enzymes. Coord. Chem. Rev. 2014, 270–271, 127–150. [Google Scholar] [CrossRef]
- Lomoth, R.; Ott, S. Introducing a dark reaction to photochemistry: Photocatalytic hydrogen from [FeFe] hydrogenase active site model complexes. Dalton Trans. 2009, 9952–9959. [Google Scholar] [CrossRef]
- Junge, H.; Rockstroh, N.; Fischer, S.; Brückner, A.; Ludwig, R.; Lochbrunner, S.; Kühn, O.; Beller, M. Light to Hydrogen: Photocatalytic Hydrogen Generation from Water with Molecularly-Defined Iron Complexes. Inorganics 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Na, Y.; Wang, M.; Pan, J.; Zhang, P.; Åkermark, B.; Sun, L. Visible Light-Driven Electron Transfer and Hydrogen Generation Catalyzed by Bioinspired [2Fe2S] Complexes. Inorg. Chem. 2008, 47, 2805–2810. [Google Scholar] [CrossRef]
- Streich, D.; Astuti, Y.; Orlandi, M.; Schwartz, L.; Lomoth, R.; Hammarström, L.; Ott, S. High-Turnover Photochemical Hydrogen Production Catalyzed by a Model Complex of the [FeFe]-Hydrogenase Active Site. Chem. Eur. J. 2010, 16, 60–63. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, M.; Na, Y.; Li, X.; Jiang, Y.; Sun, L. Homogeneous photocatalytic production of hydrogen from water by a bioinspired [Fe2S2] catalyst with high turnover numbers. Dalton Trans. 2010, 39, 1204–1206. [Google Scholar] [CrossRef]
- Wang, F.; Wang, W.-G.; Wang, X.-J.; Wang, H.-Y.; Tung, C.-H.; Wu, L.-Z. A Highly Efficient Photocatalytic System for Hydrogen Production by a Robust Hydrogenase Mimic in an Aqueous Solution. Angew. Chem. Int. Ed. 2011, 50, 3193–3197. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.-N.; Wang, F.; Wang, H.-Y.; Chen, B.; Feng, K.; Tung, C.-H.; Wu, L.-Z. Photocatalytic hydrogen production from a simple water-soluble [FeFe]-hydrogenase model system. Chem. Commun. 2012, 48, 8081. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, M.; Chen, L.; Wang, X.; Dong, J.; Sun, L. Photocatalytic Water Reduction and Study of the Formation of FeIFe0 Species in Diiron Catalyst Sytems. ChemSusChem 2012, 5, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Pullen, S.; Fei, H.; Orthaber, A.; Cohen, S.M.; Ott, S. Enhanced Photochemical Hydrogen Production by a Molecular Diiron Catalyst Incorporated into a Metal–Organic Framework. J. Am. Chem. Soc. 2013, 135, 16997–17003. [Google Scholar] [CrossRef] [PubMed]
- Jian, J.-X.; Ye, C.; Wang, X.-Z.; Wen, M.; Li, Z.-J.; Li, X.-B.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Comparison of H2 photogeneration by [FeFe]-hydrogenase mimics with CdSe QDs and Ru(bpy)3Cl2 in aqueous solution. Energy Environ. Sci. 2016, 9, 2083–2089. [Google Scholar] [CrossRef]
- Wang, X.-B.; Zheng, H.-Q.; Rao, H.; Yao, H.-C.; Fan, Y.-T.; Hou, H.-W. Synthesis of a new iron-sulfur cluster compound and its photocatalytic H2 evolution activity through visible light irradiation. Appl. Organomet. Chem. 2016, 30, 638–644. [Google Scholar] [CrossRef]
- Himiyama, T.; Waki, M.; Esquivel, D.; Onoda, A.; Hayashi, T.; Van Der Voort, P.; Inagaki, S. A Heterogeneous Hydrogen-Evolution Catalyst Based on a Mesoporous Organosilica with a Diiron Catalytic Center Modelling [FeFe]-Hydrogenase. ChemCatChem 2018, 10, 4894–4899. [Google Scholar] [CrossRef]
- Rakowski DuBois, M.; DuBois, D.L. The roles of the first and second coordination spheres in the design of molecular catalysts for H2 production and oxidation. Chem. Soc. Rev. 2009, 38, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Felton, G.A.N.; Mebi, C.A.; Petro, B.J.; Vannucci, A.K.; Evans, D.H.; Glass, R.S.; Lichtenberger, D.L. Review of electrochemical studies of complexes containing the Fe2S2 core characteristic of [FeFe]-hydrogenases including catalysis by these complexes of the reduction of acids to form dihydrogen. J. Organomet. Chem. 2009, 694, 2681–2699. [Google Scholar] [CrossRef]
- Wang, M.; Chen, L.; Li, X.; Sun, L. Approaches to efficient molecular catalyst systems for photochemical H2 production using [FeFe]-hydrogenase active site mimics. Dalton Trans. 2011, 40, 12793. [Google Scholar] [CrossRef]
- Rauchfuss, T.B. Diiron Azadithiolates as Models for the [FeFe]-Hydrogenase Active Site and Paradigm for the Role of the Second Coordination Sphere. Acc. Chem. Res. 2015, 48, 2107–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darmon, J.M.; Kumar, N.; Hulley, E.B.; Weiss, C.J.; Raugei, S.; Bullock, R.M.; Helm, M.L. Increasing the rate of hydrogen oxidation without increasing the overpotential: A bio-inspired iron molecular electrocatalyst with an outer coordination sphere proton relay. Chem. Sci. 2015, 6, 2737–2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, M.; Li, X.-B.; Jian, J.-X.; Wang, X.-Z.; Wu, H.-L.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Secondary coordination sphere accelerates hole transfer for enhanced hydrogen photogeneration from [FeFe]-hydrogenase mimic and CdSe QDs in water. Sci. Rep. 2016, 6, 29851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brezinski, W.P.; Karayilan, M.; Clary, K.E.; McCleary-Petersen, K.C.; Fu, L.; Matyjaszewski, K.; Evans, D.H.; Lichtenberger, D.L.; Glass, R.S.; Pyun, J. Macromolecular Engineering of the Outer Coordination Sphere of [2Fe-2S] Metallopolymers to Enhance Catalytic Activity for H2 Production. ACS Macro Lett. 2018, 7, 1383–1387. [Google Scholar] [CrossRef]
- Amaro-Gahete, J.; Pavliuk, M.V.; Tian, H.; Esquivel, D.; Romero-Salguero, F.J.; Ott, S. Catalytic systems mimicking the [FeFe]-hydrogenase active site for visible-light-driven hydrogen production. Coord. Chem. Rev. 2021, 448, 214172. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, H.; Feng, Y.; Gu, E.; Liu, X. Electrochemically probing the correlation between photo-induced CO-releasing behaviours and their LUMO energies of three diiron carbonyl complexes. Inorganica Chim. Acta 2017, 464, 125–131. [Google Scholar] [CrossRef]
- Katz, S.; Noth, J.; Horch, M.; Shafaat, H.S.; Happe, T.; Hildebrandt, P.; Zebger, I. Vibrational spectroscopy reveals the initial steps of biological hydrogen evolution. Chem. Sci. 2016, 7, 6746–6752. [Google Scholar] [CrossRef] [Green Version]
- Attaei, M.; Loureiro, M.; do Vale, M.; Condeço, J.; Pinho, I.; Bordado, J.; Marques, A. Isophorone Diisocyanate (IPDI) Microencapsulation for Mono-Component Adhesives: Effect of the Active H and NCO Sources. Polymers 2018, 10, 825. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Hu, M.; Wen, H.; Chai, G.; Ma, C.; Chen, H.; Chen, C. Efficient [FeFe] hydrogenase mimic dyads covalently linking to iridium photosensitizer for photocatalytic hydrogen evolution. Dalton Trans. 2012, 41, 13899. [Google Scholar] [CrossRef]
- Caplins, B.W.; Lomont, J.P.; Nguyen, S.C.; Harris, C.B. Vibrational cooling dynamics of a [FeFe]-hydrogenase mimic probed by time-resolved infrared spectroscopy. J. Phys. Chem. A 2014, 118, 11529–11540. [Google Scholar] [CrossRef]
- Haley, A.L.; Broadbent, L.N.; McDaniel, L.S.; Heckman, S.T.; Hinkle, C.H.; Gerasimchuk, N.N.; Hershberger, J.C.; Mebi, C.A. [Fe–Fe] hydrogenase models: Iron(I)-carbonyl clusters coupled to alpha- and para-toluenethiolate ligands. Polyhedron 2016, 114, 218–224. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, T.; Li, B.; Zhang, G.; Hai, L.; Ma, X.; Wu, W. Direct synthesis of phenol by novel [FeFe]-hydrogenase model complexes as catalysts of benzene hydroxylation with H2O2. RSC Adv. 2017, 7, 2934–2942. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Liu, L.; Li, Y. Synthesis and Benzene Hydroxylation Properties of Amino Substituted [FeFe]-Hydrogenase Model Compounds. Catal. Lett. 2020, 150, 2879–2885. [Google Scholar] [CrossRef]
- Van Der Voort, P.; Esquivel, D.; De Canck, E.; Goethals, F.; Van Driessche, I.; Romero-Salguero, F.J. Periodic Mesoporous Organosilicas: From simple to complex bridges; a comprehensive overview of functions, morphologies and applications. Chem. Soc. Rev. 2013, 42, 3913–3955. [Google Scholar] [CrossRef] [PubMed]
- Esquivel, D.; Amaro-Gahete, J.; Caballero-Casero, N.; Jiménez-Sanchidrián, C.; Ruiz, J.R.; Rubio, S.; Van Der Voort, P.; Romero-Salguero, F.J. Tailoring Bifunctional Periodic Mesoporous Organosilicas for Cooperative Catalysis. ACS Appl. Nano Mater. 2020, 3, 2373–2382. [Google Scholar] [CrossRef]
- López, M.I.; Esquivel, D.; Jiménez-Sanchidrián, C.; Romero-Salguero, F.J.; Van Der Voort, P. A “one-step” sulfonic acid PMO as a recyclable acid catalyst. J. Catal. 2015, 326, 139–148. [Google Scholar] [CrossRef]
- Kaczmarek, A.M.; Abednatanzi, S.; Esquivel, D.; Krishnaraj, C.; Jena, H.S.; Wang, G.; Leus, K.; Van Deun, R.; Romero–Salguero, F.J.; Van Der Voort, P. Amine-containing (nano-) Periodic Mesoporous Organosilica and its application in catalysis, sorption and luminescence. Microporous Mesoporous Mater. 2020, 291, 109687. [Google Scholar] [CrossRef]
- Cornelius, M.; Hoffmann, F.; Fröba, M. Periodic Mesoporous Organosilicas with a Bifunctional Conjugated Organic Unit and Crystal-like Pore Walls. Chem. Mater. 2005, 17, 6674–6678. [Google Scholar] [CrossRef]
- López, M.I.; Esquivel, D.; Jiménez-Sanchidrián, C.; Van Der Voort, P.; Romero-Salguero, F.J. Thiol-Functionalized Ethylene Periodic Mesoporous Organosilica as an Efficient Scavenger for Palladium: Confirming the Homogeneous Character of the Suzuki Reaction. Materials 2020, 13, 623. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarek, A.M.; Van Der Voort, P. Light-Emitting Lanthanide Periodic Mesoporous Organosilica (PMO) Hybrid Materials. Materials 2020, 13, 566. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, F.; Jin, J. Self-quenching in the electrochemiluminescence of Ru(bpy)32+ using ascorbic acid as co-reactant. Luminescence 2008, 23, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, B.; Baine, T.; Ma, X.A.N.; Zhao, X.; Schmehl, R.H. Mechanistic Details for Cobalt Catalyzed Photochemical Hydrogen Production in Aqueous Solution: Efficiencies of the Photochemical and Non-Photochemical Steps. Inorg. Chem. 2013, 52, 4853–4859. [Google Scholar] [CrossRef]
- Zhu, D.; Xiao, Z.; Liu, X. Introducing polyethyleneimine (PEI) into the electrospun fibrous membranes containing diiron mimics of [FeFe]-hydrogenase: Membrane electrodes and their electrocatalysis on proton reduction in aqueous media. Int. J. Hydrogen Energy 2015, 40, 5081–5091. [Google Scholar] [CrossRef]
- Wang, W.; Yu, T.; Zeng, Y.; Chen, J.; Yang, G.; Li, Y. Enhanced photocatalytic hydrogen production from an MCM-41-immobilized photosensitizer–[Fe–Fe] hydrogenase mimic dyad. Photochem. Photobiol. Sci. 2014, 13, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Shylin, S.I.; Pavliuk, M.V.; D’Amario, L.; Mamedov, F.; Sá, J.; Berggren, G.; Fritsky, I.O. Efficient visible light-driven water oxidation catalysed by an iron(IV) clathrochelate complex. Chem. Commun. 2019, 55, 3335–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayanuma, M.; Stoll, T.; Daniel, C.; Odobel, F.; Fortage, J.; Deronzier, A.; Collomb, M.-N. A computational mechanistic investigation of hydrogen production in water using the [Rh III (dmbpy)2Cl2]+/[Ru II (bpy)3]2+/ascorbic acid photocatalytic system. Phys. Chem. Chem. Phys. 2015, 17, 10497–10509. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, J.; Yang, J.; Zhang, L.; Feng, Z.; Zhang, J.; Li, C. Acid catalyzed synthesis of ordered bifunctionalized mesoporous organosilicas with large pore. Microporous Mesoporous Mater. 2005, 77, 257–264. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, J.; Yang, J.; Kapoor, M.P.; Inagaki, S.; Li, C. Synthesis, characterization, and catalytic activity of sulfonic acid-functionalized periodic mesoporous organosilicas. J. Catal. 2004, 228, 265–272. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Brunauer–Emmett–Teller (BET) Surface Area (m2/g) | Dp (nm) a | Vp (cm3/g) a | External Surface Area (m2/g) b | Micropore Area (m2/g) b | Micropore Volume (cm3/g) b |
|---|---|---|---|---|---|---|
| EthanePMO | 789 | 5.2 | 0.71 | 363 | 426 | 0.18 |
| EthanePMO@NCOFeFe | 481 | 4.9 | 0.55 | 375 | 106 | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amaro-Gahete, J.; Esquivel, D.; Pavliuk, M.V.; Jiménez-Sanchidrián, C.; Tian, H.; Ott, S.; Romero-Salguero, F.J. Hydroxyl-Decorated Diiron Complex as a [FeFe]-Hydrogenase Active Site Model Complex: Light-Driven Photocatalytic Activity and Heterogenization on Ethylene-Bridged Periodic Mesoporous Organosilica. Catalysts 2022, 12, 254. https://doi.org/10.3390/catal12030254
Amaro-Gahete J, Esquivel D, Pavliuk MV, Jiménez-Sanchidrián C, Tian H, Ott S, Romero-Salguero FJ. Hydroxyl-Decorated Diiron Complex as a [FeFe]-Hydrogenase Active Site Model Complex: Light-Driven Photocatalytic Activity and Heterogenization on Ethylene-Bridged Periodic Mesoporous Organosilica. Catalysts. 2022; 12(3):254. https://doi.org/10.3390/catal12030254
Chicago/Turabian StyleAmaro-Gahete, Juan, Dolores Esquivel, Mariia V. Pavliuk, César Jiménez-Sanchidrián, Haining Tian, Sascha Ott, and Francisco J. Romero-Salguero. 2022. "Hydroxyl-Decorated Diiron Complex as a [FeFe]-Hydrogenase Active Site Model Complex: Light-Driven Photocatalytic Activity and Heterogenization on Ethylene-Bridged Periodic Mesoporous Organosilica" Catalysts 12, no. 3: 254. https://doi.org/10.3390/catal12030254