
The Advancement of Supported Bimetallic Catalysts for the Elimination of Chlorinated Volatile Organic Compounds



 , ,
, , 
Abstract

1. Introduction
2. Synthesis of Bimetallic Nanoparticles
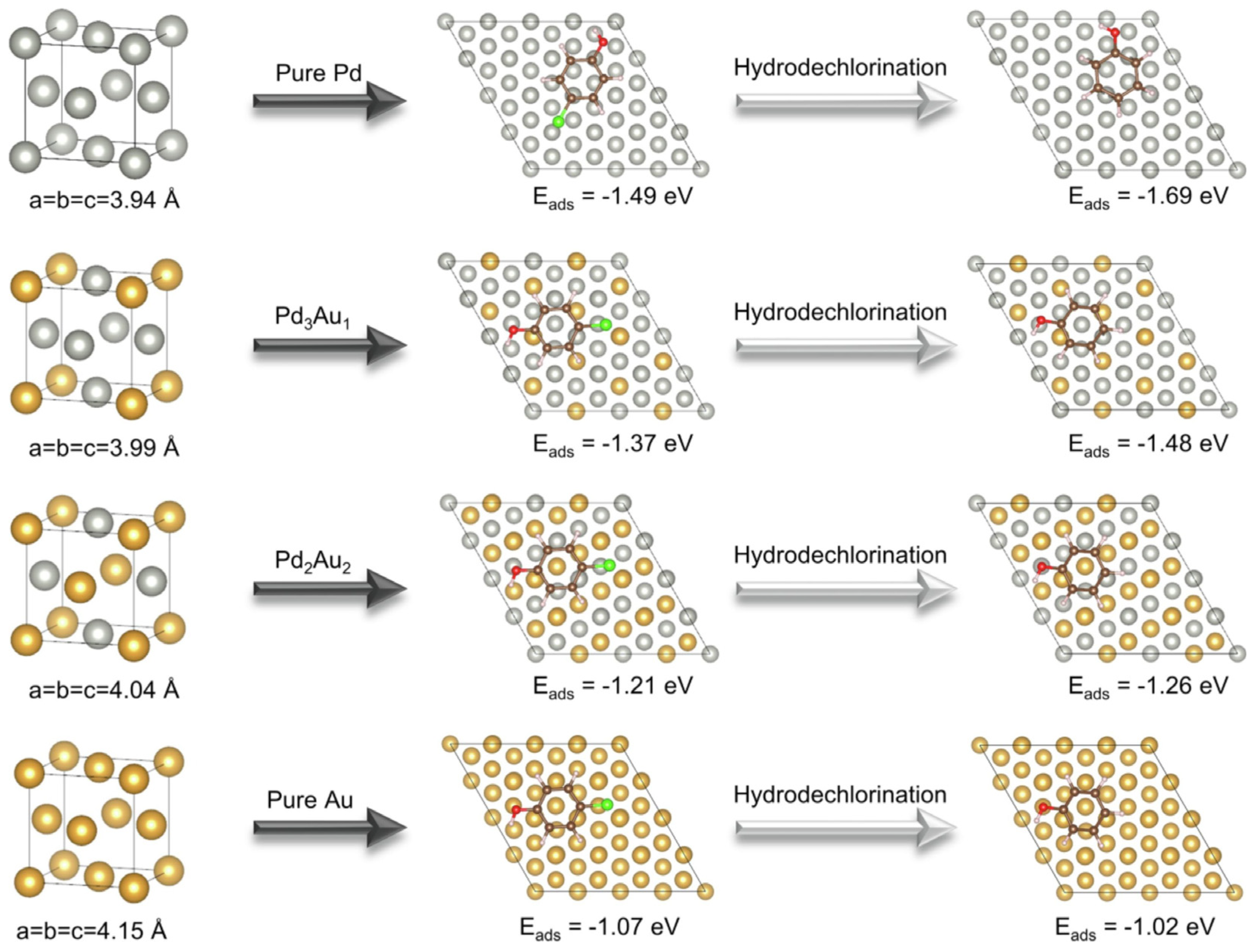
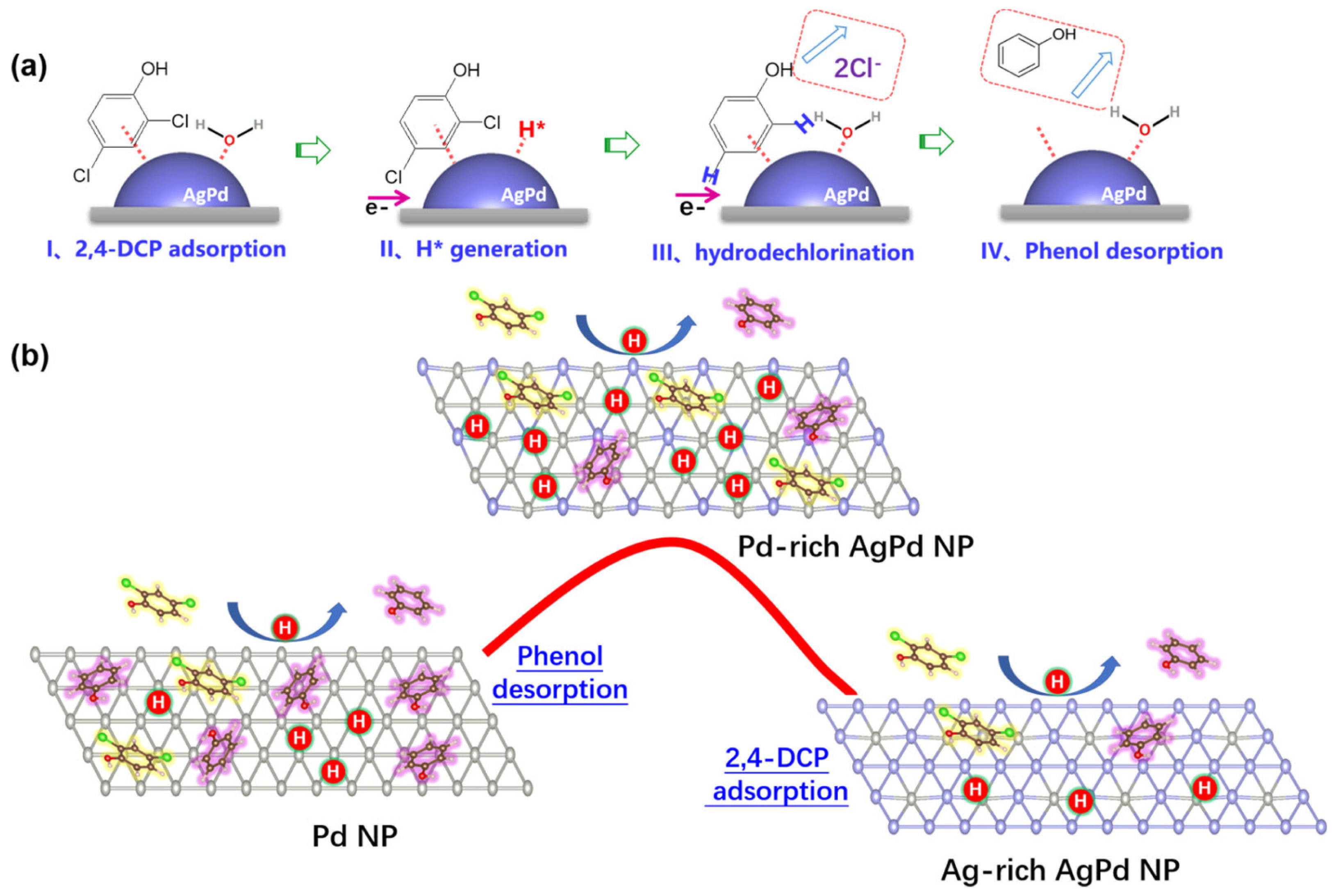
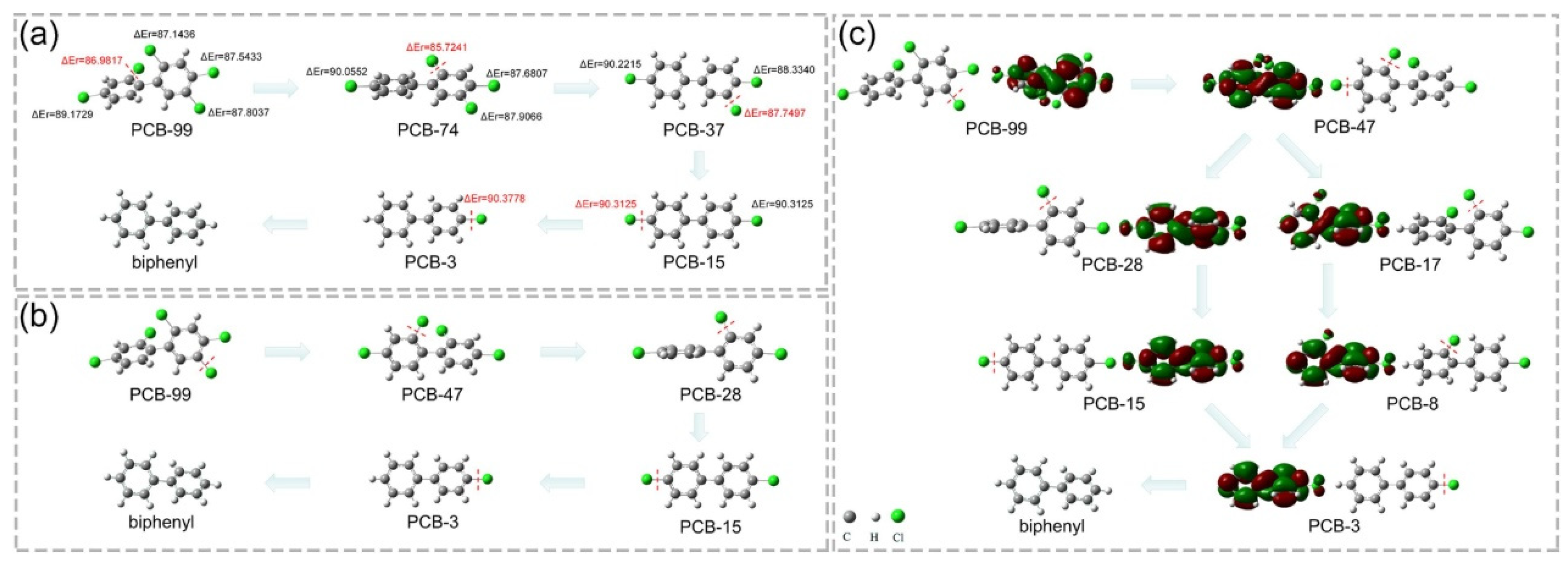
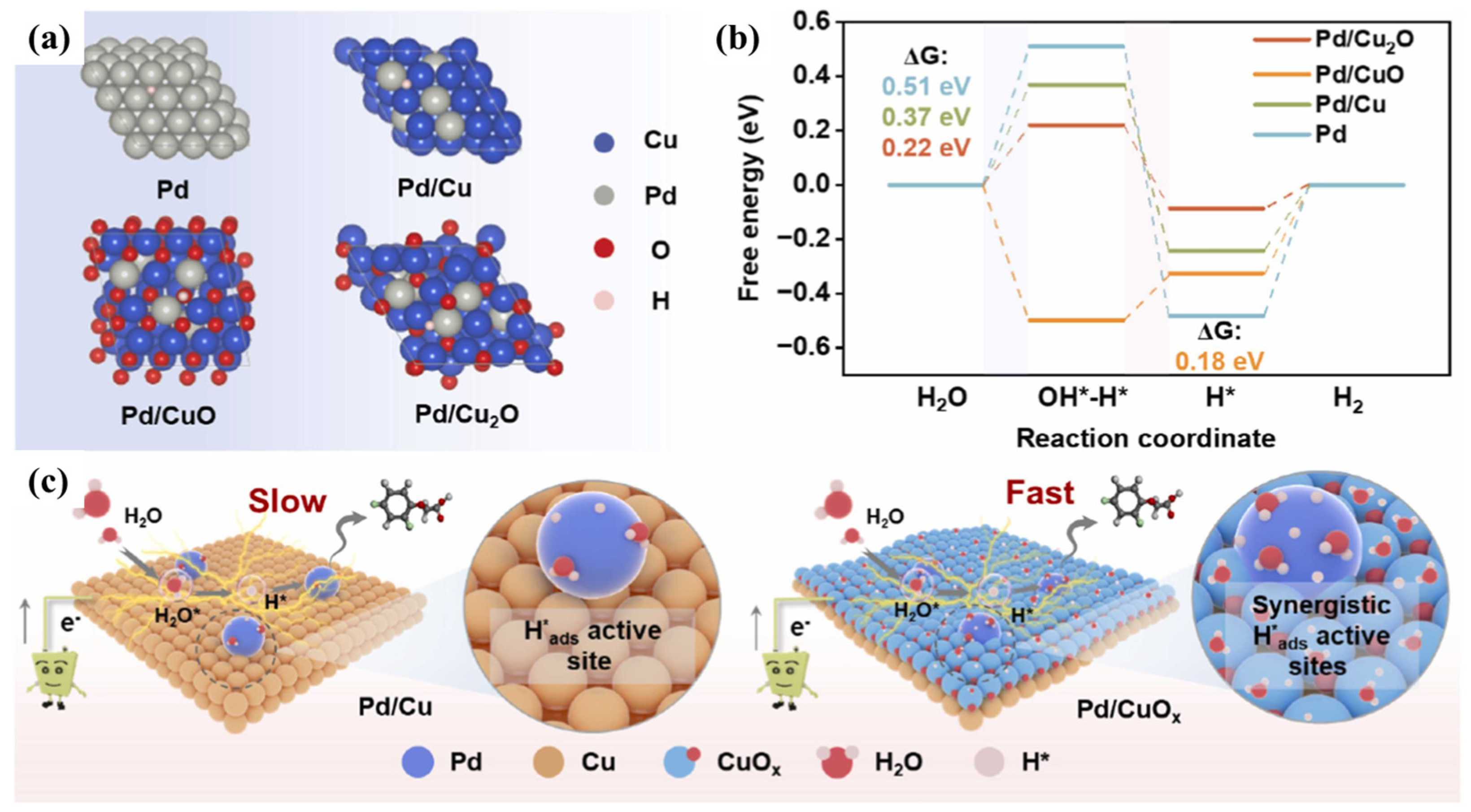
3. Catalytic Applications for CVOC Elimination

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Method | Particle Size (nm) | Reaction Condition | Pollutant | Elimination Efficiency | Ref. |
|---|---|---|---|---|---|---|
| PdPt | Co-impregnation method | – | 267 ppm, SV = 10,000 h−1 | CH2Cl2 | T50 = 360 °C, T90 = 500 °C | [43] |
| PdPt | Co-impregnation method | – | 188 ppm, SV = 10,000 h−1 | CHCl3 | T50 = 315 °C, T90 = 409 °C | [43] |
| PdPt | Co-impregnation method | – | 145 ppm, SV = 10,000 h−1 | CCl4 | T50 = 275 °C, T90 = 410 °C | [43] |
| CeNb | Impregnation method | 4.6–8.7 | 1000 ppm, SV = 15,000 h−1 | 1,2-Dichlorobenzene | T50 = 297 °C, T90 = 335 °C | [44] |
| CeMn | Impregnation method | 19.9–29.8 | 500 ppm, SV = 15,000 h−1 | 1,2-Dichlorobenzene | T50 = 300 °C, T90 = 360 °C | [44] |
| NiV | Citrate sol–gel method | 132–148 | 1000 ppm, SV = 15,000 h−1 | Dichloromethane | T50 = 168 °C, T90 = 203 °C | [46] |
| PtRu | Co-impregnation method | 8.7–10.1 | 1000 ppm, SV = 40,000 mL/(g h) | Chlorobenzene | T50 = 234 °C, T90 = 270 °C | [48] |
| PtW | Co-impregnation method | 3.6–4.0 | 1000 ppm, SV = 60,000 h−1 | Chlorobenzene | T50 = 263 °C, T90 = 327 °C | [52] |
| PtW | Co-impregnation method | 3.6–4.0 | 1000 ppm, SV = 60,000 h−1 | 1,2-Dichlorobenzene | T50 = 293 °C, T90 = 363 °C | [52] |
| PtW | Co-impregnation method | 3.6–4.0 | 1000 ppm, SV = 60,000 h−1 | 1,2-Dichloroethane | T50 = 235 °C, T90 = 302 °C | [52] |
| PtW | Co-impregnation method | 3.6–4.0 | 1000 ppm, SV = 60,000 h−1 | Dichloromethane | T50 = 235 °C, T90 = 335 °C | [52] |
| PtW | Co-impregnation method | 3.6–4.0 | 1000 ppm, SV = 60,000 h−1 | Trichloroethylene | T50 = 282 °C, T90 = 400 °C | [52] |
| RuCe | Equal-volume impregnation method | 20.2 | 3000 mg/m3, SV = 30,000 h−1 | Chlorobenzene | T50 = 165 °C, T90 = 180 °C | [53] |
| AuPt | Atomic layer deposition method | 5.3 | H2 pressure, 0.3 MPa; T = 65 °C; p-CNb, 8 mmol | p-Chloronitrobenzene | p-CAN selectivity of 99% with a specific activity of 1.75 × 104 molp-CNB/(molPt h) | [48] |
| AgPt | Photo-deposition method | 2.2–3.5 | 7300 ppm 1,2-dichloroethane, 36,800 ppm H2, He (balance) | Hydrodechlorination of 1,2-dichloroethane to ethylene | Pt1.03Ag0.93/TiO2 exhibited 68.1% ethylene selectivity | [54] |
| AgPd | Galvanic replacement method | 4.3 | 7300 ppm 1,2-dichloroethane, 36,800 ppm H2, He (balance) | Hydrodechlorination of 1,2-dichloroethane to ethylene | Pt0.13Ag0.84/Al2O3 exhibited 94.6% ethylene selectivity | [55] |
| AgPd | Co-impregnation method | – | 7300 ppm 1,2-dichloroethane, 36,800 ppm H2, He (balance) | Hydrodechlorination of 1,2-dichloroethane to ethylene | Ag1.99Pd0.099/ZrO2 exhibited 100% ethylene selectivity | [56] |
| AuPd | Aqueous phase co-reduction method | 8.1 | Initial concentration = 50 mg/L | 4-Chlorophenol hydrodechlorination | 98.35% 4-chlorophenol hydrodechlorination efficiency in 4 h of reaction and mass activity = 0.47 h−1 mgPd−1 | [57] |
| CeZr | Solvothermal method | 100 | 0.1 μL of HCB solution, SV = 12,000 mL/(g h) | Hexachlorobenzene | The degradation efficiency of HCB over Ce0.2Zr0.8/UiO-66 was the highest (86.5%) at 100 °C | [51] |
| AgPd | Solvothermal method | – | Initial concentration = 50 mg/L | 2,4-Dichlorophenol | Mass activity of Ag32Pd68 reached 2.58 min−1 gPd−1 | [58] |
| FeNi | Aqueous phase co-reduction method | 6.5 | 50 mL of pentachlorobiphenyl (100 μg/L) | 2,2′,4,4′,5-Pentachlorobiphenyl | The dechlorination efficiency at 25 °C reached 75.9% within 2 h of reaction | [59] |
| NiFe | Co-precipitation method | 600 | pH = 7.5, initial concentration = 42 μmol/L | Trichloroethylene | The removal efficiency reached 80.9% after 110 min of reaction | [25] |
| CoFe | Solvothermal method | – | pH = 6.0, initial concentration = 10 mg/L | 2,4-Dichlorophenol | 95.46% dechlorination activity | [61] |
| NiFe | Aqueous phase co-reduction method | 50–100 | Initial concentration = 7.5 mg/L | Pentachlorophenol | The dechlorination efficiency was close to 100% in 5 min of reaction | [60] |
| PdCu | Atomic reconstruction method | – | Initial concentration = 40 μmol/L | 2,4-Dichlorophenoxyacetic acid | Degradation efficiency = 94% in 60 min of reaction | [62] |
| PdCu | Atomic reconstruction method | – | Initial concentration = 40 μmol/L | 4-Chlorophenol | Degradation efficiency = 85% in 60 min of reaction | [62] |
| PdCu | Atomic reconstruction method | – | Initial concentration = 40 μmol/L | 4-Chloroaniline | Degradation efficiency = 99% in 60 min of reaction | [62] |
| PdCu | Chemical reduction method | 6.16 ± 0.31 | Initial concentration = 5 mg/L | Diclofenac | Degradation efficiency = 93.3 ± 0.1% in 10 h of reaction | [27] |
| NiFe | Thermal decomposition method | – | pH = 5, initial concentration = 20 mg/L | Monochlorobenzene | Degradation efficiency = 98.9% in 30 min of reaction | [63] |
| NiFe | Thermal decomposition method | – | pH = 5, initial concentration = 20 mg/L | Dichlorobenzene | Degradation efficiency = 90.4% in 30 min of reaction | [63] |
| NiFe | Thermal decomposition method | – | pH = 5, initial concentration = 20 mg/L | Trichlorobenzene | degradation efficiency of 85.7% in 30 min of reaction | [63] |
4. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xiong, Y.C.; Yu, F.; Ma, J. Research progress in chlorine ion removal electrodes for desalination by capacitive deionization. Acta Phys. Chim. Sin. 2022, 38, 2006037. [Google Scholar] [CrossRef]
- Kifle, G.A.; Huang, Y.; Xiang, M.H.; Wang, W.B.; Wang, C.; Li, C.Y.; Li, H. Heterogeneous activation of peroxygens by iron-based bimetallic nanostructures for the efficient remediation of contaminated water. A review. Chem. Eng. J. 2022, 442, 136187. [Google Scholar] [CrossRef]
- Ferrando, R.; Jellinek, J.; Johnston, R.L. Nanoalloys: From theory to applications of alloy clusters and nanoparticles. Chem. Rev. 2008, 108, 845–910. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Gao, Z.R.; Sun, B.W.; Mu, S.Y.; Dang, F.X.; Guo, X.W.; Ma, D.; Shi, C. Engineering metal-support interaction to construct catalytic interfaces and redisperse metal nanoparticles. Chem. Catal. 2023, 3, 100768. [Google Scholar] [CrossRef]
- Zheng, F.B.; Lin, T.; Wang, K.; Wang, Y.L.; Li, G.D. Recent advances in bimetallic metal-organic frameworks and their derivatives for thermal catalysis. Nano Res. 2023, 16, 12919–12935. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, X.Y.; Dai, Q.G.; Li, D. Effect of Ce and La on the structure and activity of MnOx catalyst in catalytic combustion of chlorobenzene. Appl. Catal. B 2012, 111–112, 141–149. [Google Scholar] [CrossRef]
- Sun, P.F.; Wang, W.L.; Dai, X.X.; Weng, X.L.; Wu, Z.B. Mechanism study on catalytic oxidation of chlorobenzene over MnxCe1-xO2/H-ZSM5 catalysts under dry and humid conditions. Appl. Catal. B 2016, 198, 389–397. [Google Scholar] [CrossRef]
- Huang, C.C.; Lo, S.L.; Tsai, S.M.; Lien, H.L. Catalytic hydrodechlorination of 1,2-dichloroethane using copper nanoparticles under reduction conditions of sodium borohydride. J. Environ. Monit. 2011, 13, 2406–2412. [Google Scholar] [CrossRef]
- Zell, T.; Feierabend, M.; Halfter, B.; Radius, U. Stoichiometric and catalytic CeCl activation of Aryl Chlorides using an NHC-stabilized nickel(0) complex. J. Organomet. Chem. 2011, 696, 1380–1387. [Google Scholar] [CrossRef]
- Urbano, F.J.; Marinas, J.M. Hydrogenolysis of organohalogen compounds over palladium supported catalysts. J. Mol. Catal. A 2001, 173, 329–345. [Google Scholar] [CrossRef]
- Huang, H.; Dai, Q.G.; Wang, X.Y. Morphology effect of Ru/CeO2 catalysts for the catalytic combustion of chlorobenzene. Appl. Catal. B 2014, 158–159, 96–105. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, Z.; Guo, Y.; Wang, L.; Guo, Y.L.; Zhang, J.; Zhan, W.C. Total oxidation of propane over a Ru/CeO2 catalyst at low temperature. Environ. Sci. Technol. 2018, 52, 9531–9541. [Google Scholar] [CrossRef]
- Lin, F.W.; Zhang, Z.M.; Li, N.; Yan, B.B.; He, C.; Hao, Z.P.; Chen, G.Y. How to achieve complete elimination of Cl-VOCs: A critical review on byproducts formation and inhibition strategies during catalytic oxidation. Chem. Eng. J. 2021, 404, 126534. [Google Scholar] [CrossRef]
- Gil, S.; Garcia-Vargas, J.M.; Liotta, L.F.; Pantaleo, G.; Ousmane, M.; Retailleau, L.; Giroir-Fendler, A. Catalytic oxidation of propene over Pd catalysts supported on CeO2, TiO2, Al2O3 and M/Al2O3 oxides (M = Ce, Ti, Fe, Mn). Catalysts 2015, 5, 671–689. [Google Scholar] [CrossRef]
- Wang, Q.L.; Huang, Q.X.; Wu, H.F.; Lu, S.Y.; Wu, H.L.; Li, X.D.; Yan, J.H. Catalytic decomposition of gaseous 1,2-dichlorobenzene over CuOx/TiO2 and CuOx/TiO2-CNTs catalysts: Mechanism and PCDD/Fs formation. Chemosphere 2016, 144, 2343–2350. [Google Scholar] [CrossRef] [PubMed]
- Weng, X.L.; Long, Y.; Wang, W.L.; Shao, M.; Wu, Z.B. Structural effect and reaction mechanism of MnO2 catalysts in the catalytic oxidation of chlorinated aromatics. Chin. J. Catal. 2019, 40, 638–646. [Google Scholar] [CrossRef]
- Yang, P.; Shi, Z.N.; Yang, S.S.; Zhou, R.X. High catalytic performances of CeO2-CrOx catalysts for chlorinated VOCs elimination. Chem. Eng. Sci. 2015, 126, 361–369. [Google Scholar] [CrossRef]
- Tajima, M.; Niwa, M.; Fujii, Y.; Koinuma, Y.; Aizawa, R.; Kushiyama, S.; Kobayashi, S.; Mizuno, K.; Ohuchi, H. Decomposition of chlorofluorocarbons in the presence of water over zeolite catalyst. Appl. Catal. B 1996, 9, 167–177. [Google Scholar] [CrossRef]
- Aranzabal, A.; González-Marcos, J.A.; Romero-Sáez, M.; González-Velasco, J.R.; Guillemot, M.; Magnoux, P. Stability of protonic zeolites in the catalytic oxidation of chlorinated VOCs (1,2-dichloroethane). Appl. Catal. B 2009, 88, 533–541. [Google Scholar] [CrossRef]
- Aranzabal, A.; Romero-Sáez, M.; Elizundia, U.; González-Velasco, J.R.; González-Marcos, J.A. Deactivation of H-zeolites during catalytic oxidation of trichloroethylene. J. Catal. 2012, 296, 165–174. [Google Scholar] [CrossRef]
- Lu, Y.J.; Dai, Q.G.; Wang, X.Y. Catalytic combustion of chlorobenzene on modified LaMnO3 catalysts. Catal. Commun. 2014, 54, 114–117. [Google Scholar] [CrossRef]
- Zhang, C.H.; Hua, W.C.; Wang, C.; Guo, Y.L.; Guo, Y.; Lu, G.Z.; Baylet, A.; Giroir-Fendler, A. The effect of A-site substitution by Sr, Mg and Ce on the catalytic performance of LaMnO3 catalysts for the oxidation of vinyl chloride emission. Appl. Catal. B 2013, 134–135, 310–315. [Google Scholar] [CrossRef]
- Gao, R.Y.; Tian, X.R.; Ding, X.L.; Hou, Z.Q.; Li, Z.Y.; Yu, X.H.; Wang, J.; Wu, L.K.; Jing, L.; Deng, J.G.; et al. Regulating catalytic stability of PtSnM/CeO2 (M = Mn, W, Nb) catalysts via the closely coupled multi-active sites to promote multicomponent VOCs oxidation. Chem. Eng. J. 2023, 471, 144456. [Google Scholar] [CrossRef]
- Li, J.J.; Wang, Y.; Zhao, B.; Ding, J.; Zhang, J.; Yin, M.H.; Zhang, Z.H.; Ma, S.M.; Liu, Y.Q.; Tan, Z.L.; et al. Unraveling kinetics and mechanism of electrocatalytic hydrodechlorination of chlorinated PPCPs by nickel-cobalt metal organic framework supported palladium composite electrode. Appl. Catal. B 2023, 332, 122754. [Google Scholar] [CrossRef]
- Deng, J.; Gao, E.L.; Wu, F.; You, Z.X.; Li, X.Z.; Gao, S.X.; Huang, L.Z. Generation of atomic hydrogen by Ni-Fe hydroxides: Mechanism and activity for hydrodechlorination of trichloroethylene. Water Res. 2021, 207, 117802. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Chen, J.T.; Hu, Y.; He, K.; Bylaska, E.J.; Tratnyek, P.G.; He, F. Degradation of chloroform by zerovalent iron: Effects of mechanochemical sulfidation and nitridation on the kinetics and mechanism. Environ. Sci. Technol. 2023, 57, 9811–9821. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Y.; Cui, M.H.; Ambuchi, J.J.; Niu, S.M.; Li, X.H.; Wang, W.L.; Liu, H.; Liu, G.S.; Wang, A.J. H*ads dynamics engineering via bimetallic Pd-Cu@MXene catalyst for enhanced electrocatalytic hydrodechlorination. Environ. Res. 2024, 252, 118859. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.F.; Qin, W.H.; Sun, X.L.; Zhu, Y.Q.; Niu, J.F. Synergistic effects on d-band center via coordination sites of M-N3P1 (M = Co and Ni) in dual single atoms that enhances photocatalytic dechlorination from tetrachlorobispheonl A. J. Hazard. Mater. 2022, 430, 128419. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Zhang, W.X. Iron nanoparticles: The core-shell structure and unique properties for Ni (II) sequestration. Langmuir 2006, 22, 4638–4642. [Google Scholar] [CrossRef]
- Rai, R.K.; Tyagi, D.; Gupta, K.; Singh, S.K. Activated nanostructured bimetallic catalysts for C–C coupling reactions: Recent progress. Catal. Sci. Technol. 2016, 6, 3341–3361. [Google Scholar] [CrossRef]
- Kim, P.; Kim, H.; Joo, J.B.; Kim, W.; Song, I.K.; Yi, J. Effect of nickel precursor on the catalytic performance of Ni/Al2O3 catalysts in the hydrodechlorination of 1,1,2-trichloroethane. J. Mol. Catal. A 2006, 256, 178–183. [Google Scholar] [CrossRef]
- Zanella, R.; Giorgio, S.; Shin, C.H.; Henry, C.R.; Louis, C. Characterization and reactivity in CO oxidation of gold nanoparticles supported on TiO2 prepared by deposition-precipitation with NaOH and urea. J. Catal. 2004, 222, 357–367. [Google Scholar] [CrossRef]
- Lu, X.M.; Tuan, H.Y.; Chen, J.Y.; Li, Z.Y.; Korgel, B.A.; Xia, Y.N. Mechanistic studies on the galvanic replacement reaction between multiply twinned particles of Ag and HAuCl4 in an organic medium. J. Am. Chem. Soc. 2007, 129, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.G.; Xia, Y.N. Alloying and dealloying processes involved in the preparation of metal nanoshells through a galvanic replacement reaction. Nano Lett. 2003, 3, 1569–1572. [Google Scholar] [CrossRef]
- Chen, J.Y.; Wiley, B.; McLellan, J.; Xiong, Y.J.; Li, Z.Y.; Xia, Y.N. Optical properties of Pd-Ag and Pt-Ag nanoboxes synthesized via galvanic replacement reactions. Nano Lett. 2005, 5, 2058–2062. [Google Scholar] [CrossRef]
- Ismail, A.A.; Al-Sayari, S.A.; Bahnemann, D.W. Photodeposition of precious metals onto mesoporous TiO2 nanocrystals with enhanced their photocatalytic activity for methanol oxidation. Catal. Today 2013, 209, 2–7. [Google Scholar] [CrossRef]
- Hidalgo, M.C.; Murcia, J.J.; Navío, J.A.; Colón, G. Photodeposition of gold on titanium dioxide for photocatalytic phenol oxidation. Appl. Catal. A 2011, 397, 112–120. [Google Scholar] [CrossRef]
- Kraeutler, B.; Bard, A.J. Heterogeneous photocatalytic preparation of supported catalysts. Photodeposition of platinum on titanium dioxide powder and other substrates. J. Am. Chem. Soc. 1978, 100, 4317–4318. [Google Scholar] [CrossRef]
- Epron, F.; Gauthard, F.; Pinéda, C.; Barbier, J. Catalytic reduction of nitrate and nitrite on Pt-Cu/Al2O3 catalysts in aqueous solution: Role of the interaction between copper and platinum in the reaction. J. Catal. 2001, 198, 309–318. [Google Scholar] [CrossRef]
- Gauthard, F.; Epron, F.; Barbier, J. Palladium and platinum-based catalysts in the catalytic reduction of nitrate in water: Effect of copper, silver, or gold addition. J. Catal. 2003, 220, 182–191. [Google Scholar] [CrossRef]
- Wang, D.S.; Peng, Q.; Li, Y.D. Nanocrystalline intermetallics and alloys. Nano Res. 2010, 3, 574–580. [Google Scholar] [CrossRef]
- Wang, D.S.; Li, Y.D. One-pot protocol for Au-based hybrid magnetic nanostructures via a noble-metal-induced reduction process. J. Am. Chem. Soc. 2010, 132, 6280–6281. [Google Scholar] [CrossRef] [PubMed]
- Koyer-Gołkowska, A.; Musialik-Piotrowska, A.; Rutkowski, J.D. Oxidation of chlorinated hydrocarbons over Pt-Pd-based catalyst: Part 1. Chlorinated methanes. Catal. Today 2004, 90, 133–138. [Google Scholar] [CrossRef]
- Zhao, H.J.; Han, W.L.; Dong, F.; Tang, Z.C. Enhanced catalytic performance of Nb doping Ce supported on ordered mesoporous TiO2-SiO2 catalysts for catalytic elimination of 1,2-dichlorobenzene. Mol. Catal. 2019, 479, 110638. [Google Scholar] [CrossRef]
- Zhao, H.J.; Han, W.L.; Dong, F.; Tang, Z.C. Highly-efficient catalytic combustion performance of 1,2-dichlorobenzene over mesoporous TiO2-SiO2 supported CeMn oxides: The effect of acid sites and redox sites. J. Ind. Eng. Chem. 2018, 64, 194–205. [Google Scholar] [CrossRef]
- Zhou, B.; Zhang, X.X.; Wang, Y.; Xie, J.; Xi, K.; Zhou, Y.; Lu, H.F. Effect of Ni-V loading on the performance of hollow anatase TiO2 in the catalytic combustion of dichloromethane. J. Environ. Sci. 2019, 84, 59–68. [Google Scholar] [CrossRef]
- Wang, H.W.; Gu, X.K.; Zheng, X.S.; Pan, H.B.; Zhu, J.F.; Chen, S.; Cao, L.N.; Li, W.X.; Lu, J.L. Disentangling the size-dependent geometric and electronic effects of palladium nanocatalysts beyond selectivity. Sci. Adv. 2019, 5, eaat6413. [Google Scholar] [CrossRef]
- Guan, Q.Q.; Zhu, C.W.; Lin, Y.; Vovk, E.I.; Zhou, X.H.; Yang, Y.; Yu, H.C.; Cao, L.N.; Wang, H.W.; Zhang, X.H.; et al. Bimetallic monolayer catalyst breaks the activity-selectivity trade-off on metal particle size for efficient chemoselective hydrogenations. Nat. Catal. 2021, 4, 840–849. [Google Scholar] [CrossRef]
- Wu, M.; Wang, X.Y.; Dai, Q.G.; Gu, Y.X.; Li, D. Low temperature catalytic combustion of chlorobenzene over Mn-Ce-O/γ-Al2O3 mixed oxides catalyst. Catal. Today 2010, 158, 336–342. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Zhang, L.; Wang, G.; Deng, W.; Guo, L.M. Total catalytic oxidation of chlorinated aromatics over bimetallic Pt-Ru supported on hierarchical HZSM-5 zeolite. Microporous Mesoporous Mater. 2020, 308, 110538. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, H.J.; Ren, M.H.; Zhang, Y.C.; Li, Y.; Wang, L.X.; Chen, J.P. Low-temperature catalytic degradation of chlorinated aromatic hydrocarbons over bimetallic Ce-Zr/UiO-66 catalysts. Chem. Eng. J. 2021, 414, 128782. [Google Scholar] [CrossRef]
- Gu, Y.F.; Shao, S.J.; Sun, W.; Xia, H.Q.; Gao, X.H.; Dai, Q.G.; Zhan, W.C.; Wang, X.Y. The oxidation of chlorinated organic compounds over W-modified Pt/CeO2 catalysts. J. Catal. 2019, 380, 375–386. [Google Scholar] [CrossRef]
- Liang, W.J.; Zhu, Y.X.; Ren, S.D.; Li, Q.L.; Song, L.Y.; Shi, X.J. Catalytic combustion of chlorobenzene at low temperature over Ru-Ce/TiO2: High activity and high selectivity. Appl. Catal. A 2021, 623, 118257. [Google Scholar] [CrossRef]
- Han, Y.X.; Zhou, J.; Wang, W.J.; Wan, H.Q.; Xu, Z.Y.; Zheng, S.R.; Zhu, D.Q. Enhanced selective hydrodechlorination of 1,2-dichloroethane to ethylene on Pt-Ag/TiO2 catalysts prepared by sequential photodeposition. Appl. Catal. B 2012, 125, 172–179. [Google Scholar] [CrossRef]
- Sun, J.Y.; Han, Y.X.; Fu, H.Y.; Wan, H.Q.; Xu, Z.Y.; Zheng, S.R. Selective hydrodechlorination of 1,2-dichloroethane catalyzed by trace Pd decorated Ag/Al2O3 catalysts prepared by galvanic replacement. Appl. Surf. Sci. 2018, 428, 703–709. [Google Scholar] [CrossRef]
- Han, Y.X.; Sun, J.Y.; Fu, H.Y.; Qu, X.L.; Wan, H.Q.; Xu, Z.Y.; Zheng, S.R. Highly selective hydrodechlorination of 1,2-dichloroethane to ethylene over Ag-Pd/ZrO2 catalysts with trace Pd. Appl. Catal. A 2016, 519, 1–6. [Google Scholar] [CrossRef]
- Chen, Y.J.; Feng, C.; Wang, W.H.; Liu, Z.; Li, J.X.; Liu, C.G.; Pan, Y.; Liu, Y.Q. Electronic structure engineering of bimetallic Pd-Au alloy nanocatalysts for improving electrocatalytic hydrodechlorination performance. Sep. Purif. Technol. 2022, 289, 120731. [Google Scholar] [CrossRef]
- Peng, Y.Y.; Cui, M.Y.; Zhang, Z.Y.; Shu, S.; Shi, X.L.; Brosnahan, J.T.; Liu, C.; Zhang, Y.L.; Godbold, P.; Zhang, X.M.; et al. Bimetallic composition-promoted electrocatalytic hydrodechlorination reaction on silver-palladium alloy nanoparticles. ACS Catal. 2019, 9, 10803–10811. [Google Scholar] [CrossRef]
- Wu, Y.X.; Zhou, J.Y.; Wu, Z.H.; Ye, Q.Y.; Wu, W.C.; Liu, X.W.; He, D.C.; Lv, G.F.; Zhang, J. Electron transfer process in dechlorination of polychlorinated biphenyls (PCBs) by nickel/zero-valent iron: Effects of temperature and selectivity pattern. Chem. Eng. J. 2023, 470, 144053. [Google Scholar] [CrossRef]
- Lin, F.Y.; Lien, H.L.; Kuo, D.T.F.; Wang, K.; Shih, Y.H. Degradation of pentachlorophenol by CTAB-modified Ni/Fe bimetallic nanoparticles in the soil solution. ACS EST Engg. 2024, 4, 318–329. [Google Scholar] [CrossRef]
- Kong, Z.Y.; Li, D.H.; Cai, R.S.; Li, T.; Diao, L.P.; Chen, X.K.; Wang, X.X.; Zheng, H.J.; Jia, Y.; Yang, D.J. Electron-rich palladium regulated by cationic vacancies in CoFe layered double hydroxide boosts electrocatalytic hydrodechlorination. J. Hazard. Mater. 2024, 463, 132964. [Google Scholar] [CrossRef]
- Guo, Y.; Li, Y.; Shi, W.; Yuan, J.; Wang, L.; Wang, Z.W. In situ redox growth of Pd/CuOx for enhanced electrocatalytic degradation of organic chlorides via indirect atomic H* reduction. Appl. Catal. B 2024, 356, 124252. [Google Scholar] [CrossRef]
- Tang, H.Y.; Ma, B.; Bian, Z.Y.; Wang, H. Selective dechlorination degradation of chlorobenzenes by dual single-atomic Fe/Ni catalyst with M-N/M-O active sites synergistic. J. Hazard. Mater. 2023, 443, 130315. [Google Scholar] [CrossRef]
- Lin, F.W.; Xiang, L.; Zhang, Z.M.; Li, N.; Yan, B.B.; He, C.; Hao, Z.P.; Chen, G.Y. Comprehensive review on catalytic degradation of Cl-VOCs under the practical application conditions. Crit. Rev. Environ. Sci. Technol. 2022, 52, 311–355. [Google Scholar] [CrossRef]
- Dai, X.X.; Wang, X.W.; Long, Y.P.; Pattisson, S.; Lu, Y.H.; Morgan, D.J.; Taylor, S.H.; Carter, J.H.; Hutchings, G.J.; Wu, Z.B.; et al. Efficient elimination of chlorinated organics on a phosphoric acid modified CeO2 catalyst: A hydrolytic destruction route. Environ. Sci. Technol. 2019, 53, 12697–12705. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.G.; Wu, J.Y.; Deng, W.; Hu, J.S.; Wu, Q.Q.; Guo, L.M.; Sun, W.; Zhan, W.C.; Wang, X.Y. Comparative studies of P/CeO2 and Ru/CeO2 catalysts for catalytic combustion of dichloromethane: From effects of H2O to distribution of chlorinated by-products. Appl. Catal. B 2019, 249, 9–18. [Google Scholar] [CrossRef]
- Dai, Q.G.; Bai, S.X.; Wang, Z.Y.; Wang, X.Y.; Lu, G.Z. Catalytic combustion of chlorobenzene over Ru-doped ceria catalysts. Appl. Catal. B 2012, 126, 64–75. [Google Scholar] [CrossRef]
- de Jong, V.; Cieplik, M.K.; Reints, W.A.; Fernandez-Reino, F.; Louw, R. A mechanistic study on the catalytic combustion of benzene and chlorobenzene. J. Catal. 2002, 211, 355–365. [Google Scholar] [CrossRef]
- Bertinchamps, F.; Treinen, M.; Eloy, P.; Dos Santos, A.-M.; Mestdagh, M.M.; Gaigneaux, E.M. Understanding the activation mechanism induced by NOx on the performances of VOx/TiO2 based catalysts in the total oxidation of chlorinated VOCs. Appl. Catal. B 2007, 70, 360–369. [Google Scholar] [CrossRef]
- Feng, X.B.; Tian, M.J.; He, C.; Li, L.; Shi, J.-W.; Yu, Y.K.; Cheng, J. Yolk-shell-like mesoporous CoCrOx with superior activity and chlorine resistance in dichloromethane destruction. Appl. Catal. B 2020, 264, 118493. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.; Liu, Y.; Deng, J.; Jing, L.; Wang, Z.; Wei, L.; Wei, Z.; Hou, Z.; Tao, J.; Dai, H. The Advancement of Supported Bimetallic Catalysts for the Elimination of Chlorinated Volatile Organic Compounds. Catalysts 2024, 14, 531. https://doi.org/10.3390/catal14080531
Lin H, Liu Y, Deng J, Jing L, Wang Z, Wei L, Wei Z, Hou Z, Tao J, Dai H. The Advancement of Supported Bimetallic Catalysts for the Elimination of Chlorinated Volatile Organic Compounds. Catalysts. 2024; 14(8):531. https://doi.org/10.3390/catal14080531
Chicago/Turabian StyleLin, Hongxia, Yuxi Liu, Jiguang Deng, Lin Jing, Zhiwei Wang, Lu Wei, Zhen Wei, Zhiquan Hou, Jinxiong Tao, and Hongxing Dai. 2024. "The Advancement of Supported Bimetallic Catalysts for the Elimination of Chlorinated Volatile Organic Compounds" Catalysts 14, no. 8: 531. https://doi.org/10.3390/catal14080531
APA StyleLin, H., Liu, Y., Deng, J., Jing, L., Wang, Z., Wei, L., Wei, Z., Hou, Z., Tao, J., & Dai, H. (2024). The Advancement of Supported Bimetallic Catalysts for the Elimination of Chlorinated Volatile Organic Compounds. Catalysts, 14(8), 531. https://doi.org/10.3390/catal14080531