
Recent Advances in Enantioselective Transition Metal Catalysis Mediated by Ligand–Substrate Noncovalent Interactions


Abstract
:
1. Introduction
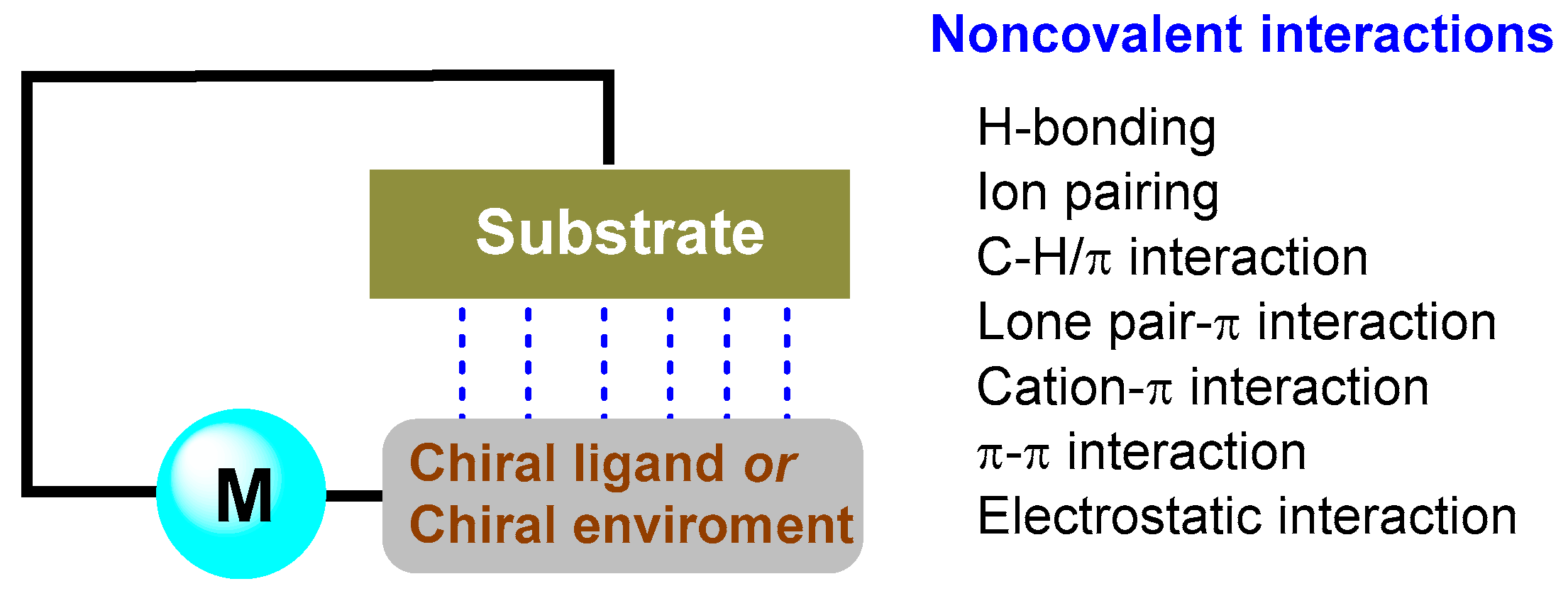
2. Ligand–Substrate Noncovalent Interactions
2.1. Representative Examples Involve NCIs Before 2020
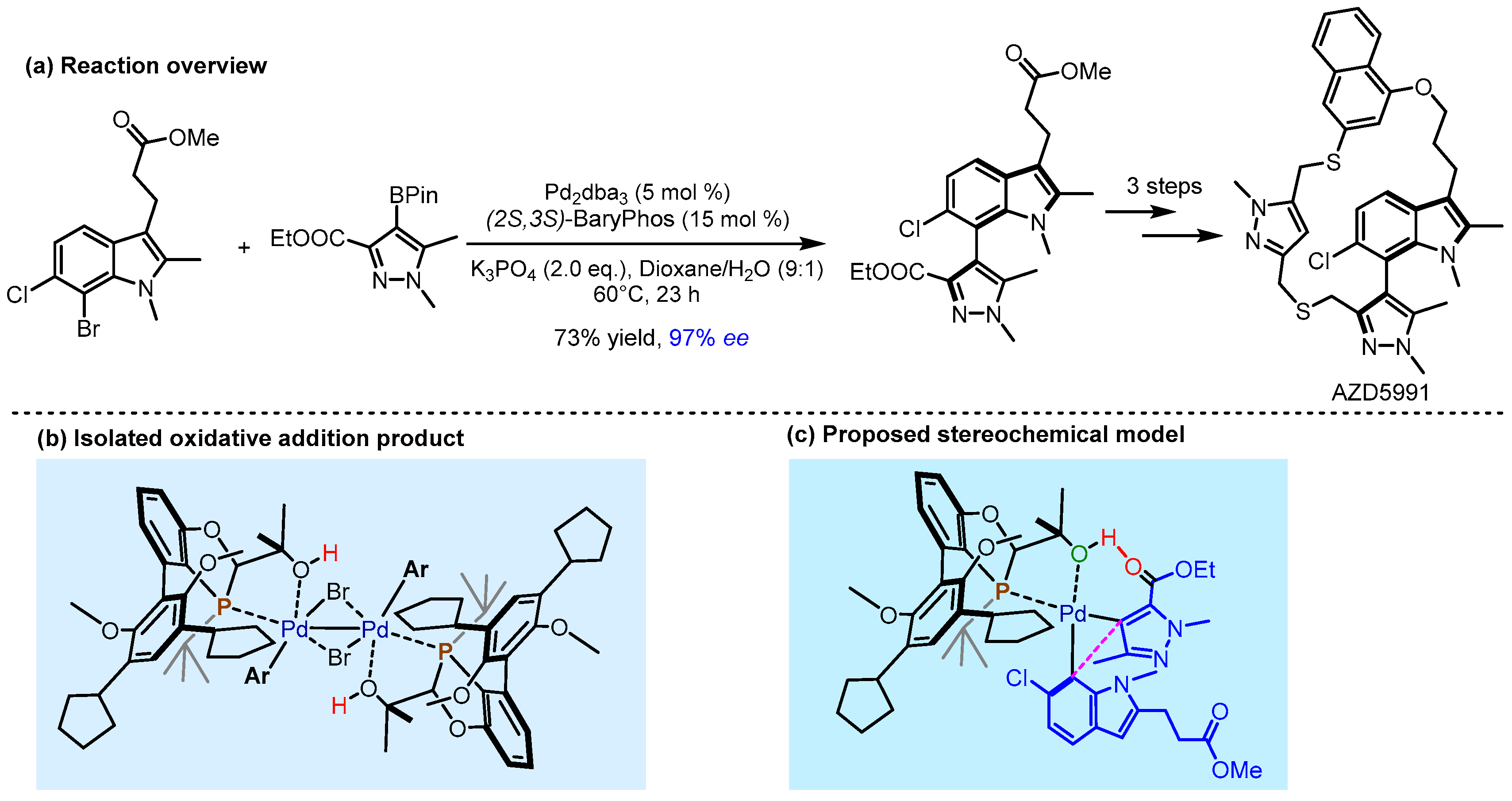
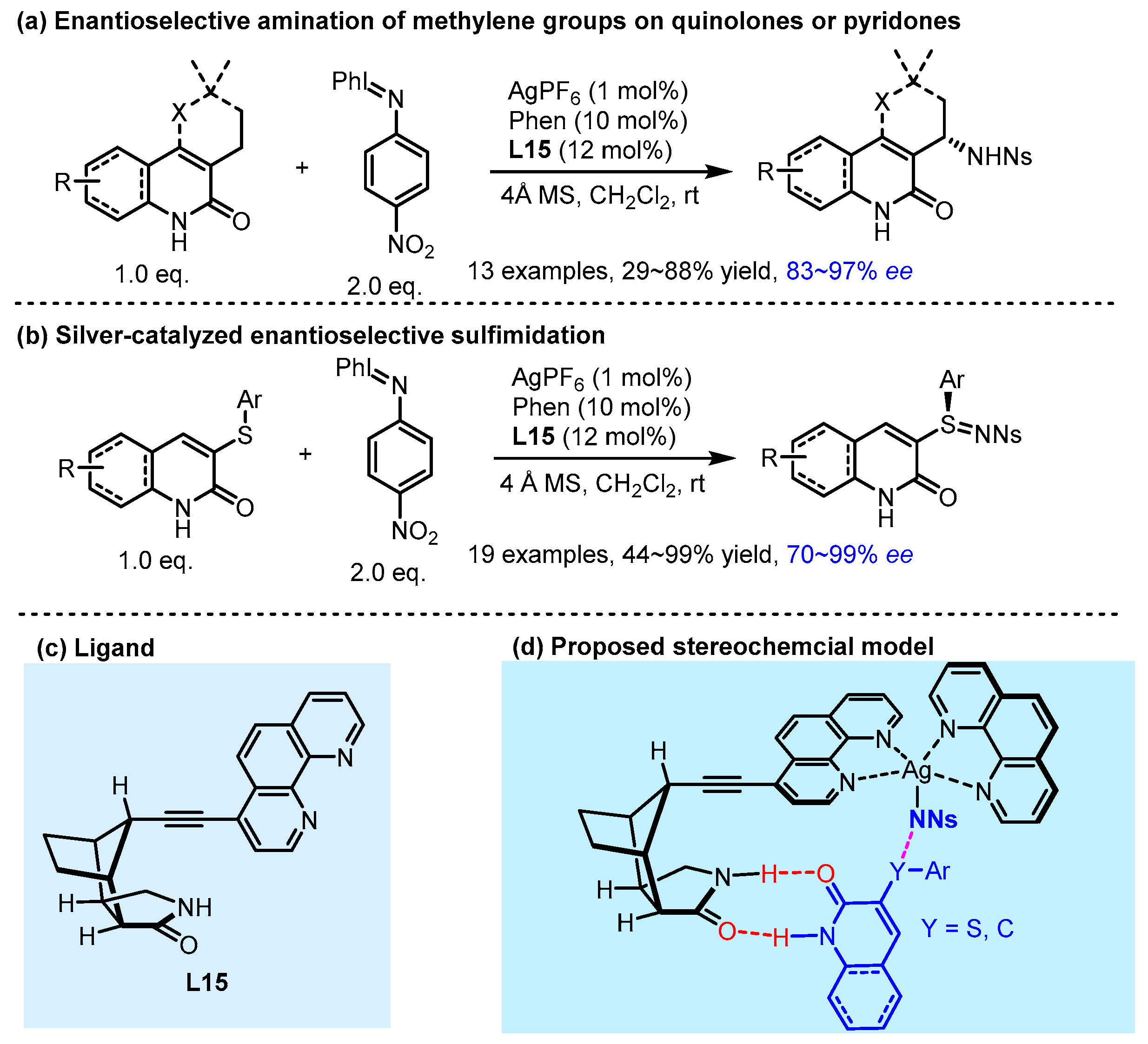
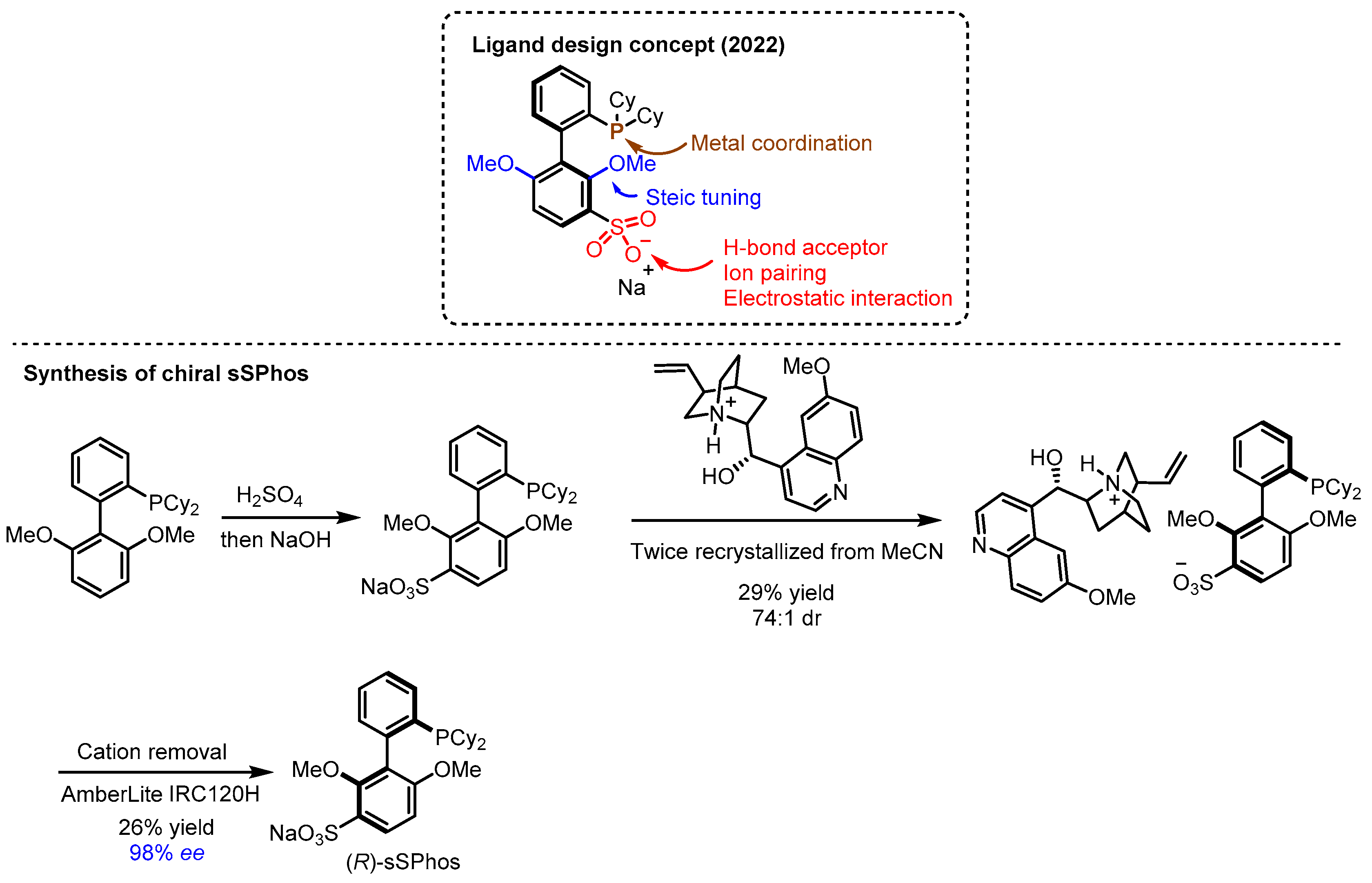
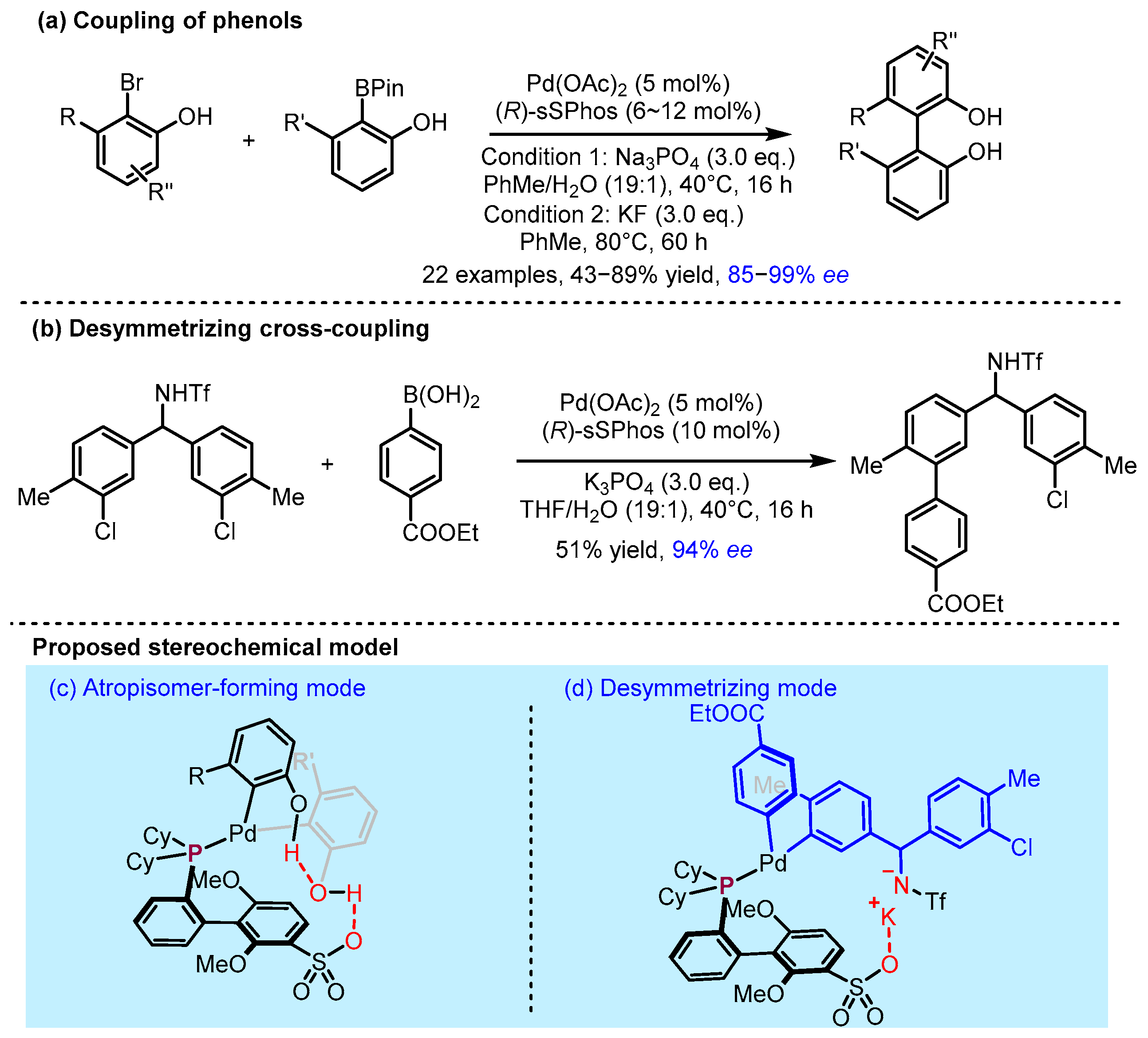
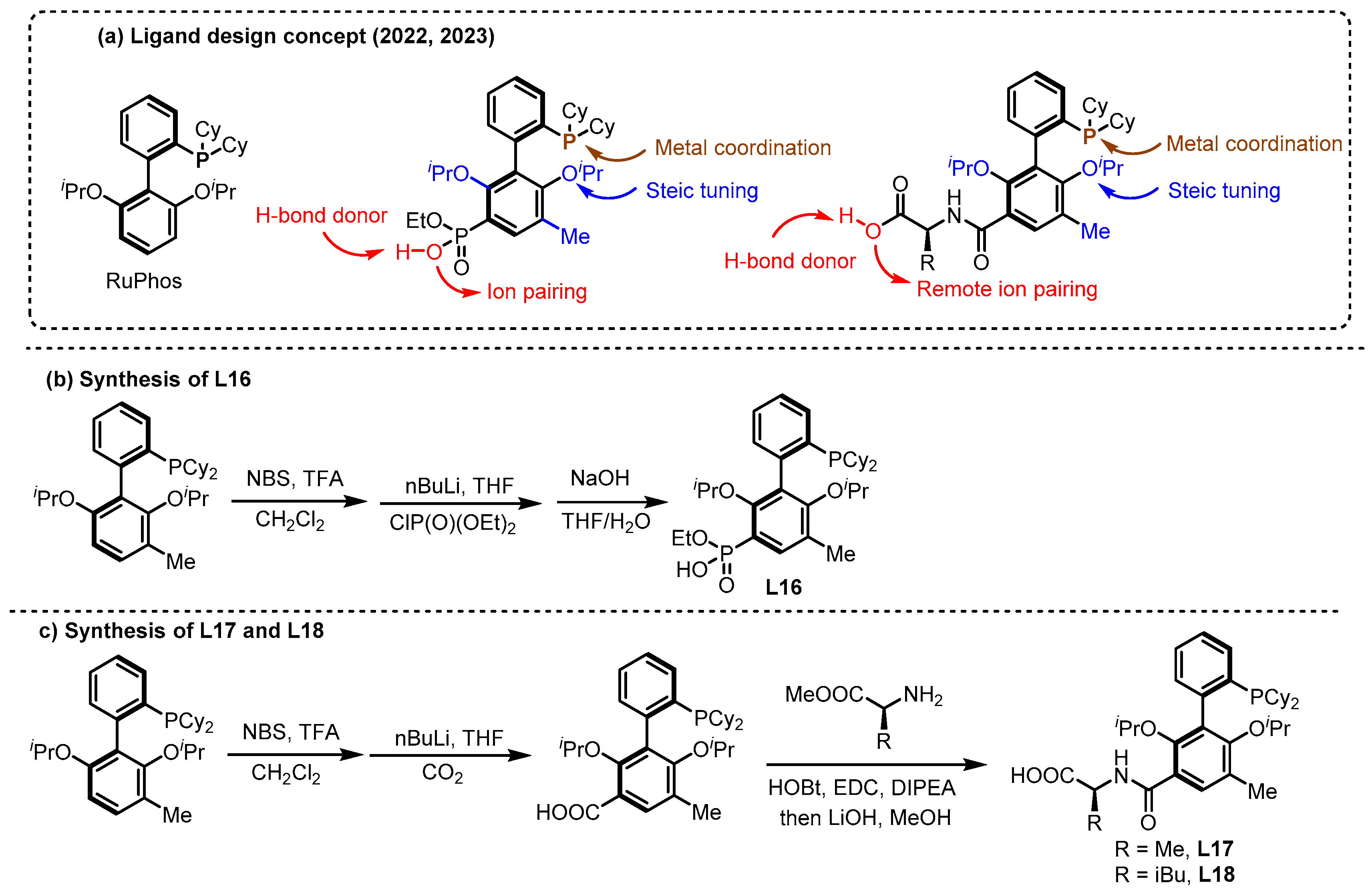
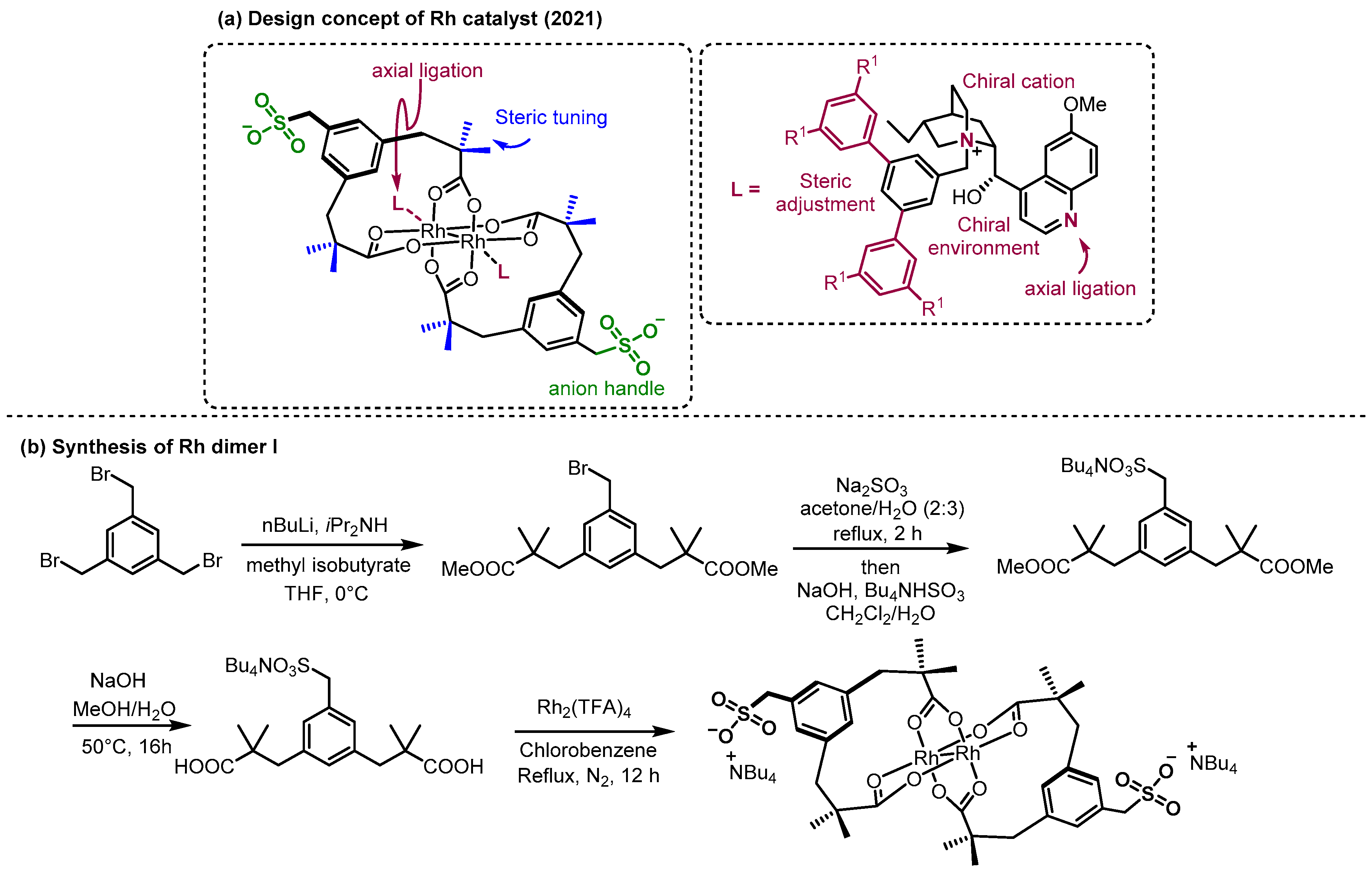
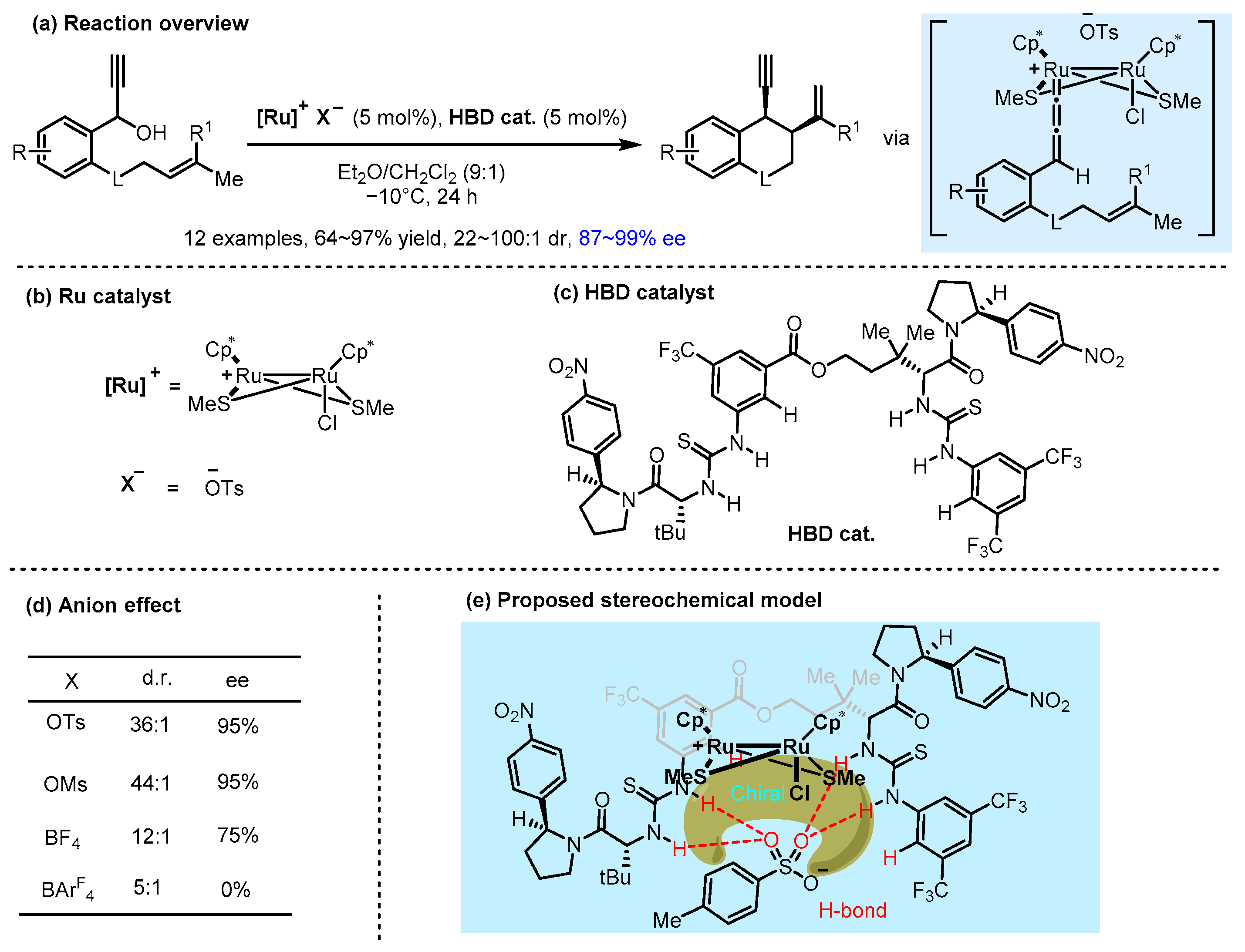
2.2. Hydrogen Bonding and Ion Pairing Involved Enantioselective Transition Metal Catalysis
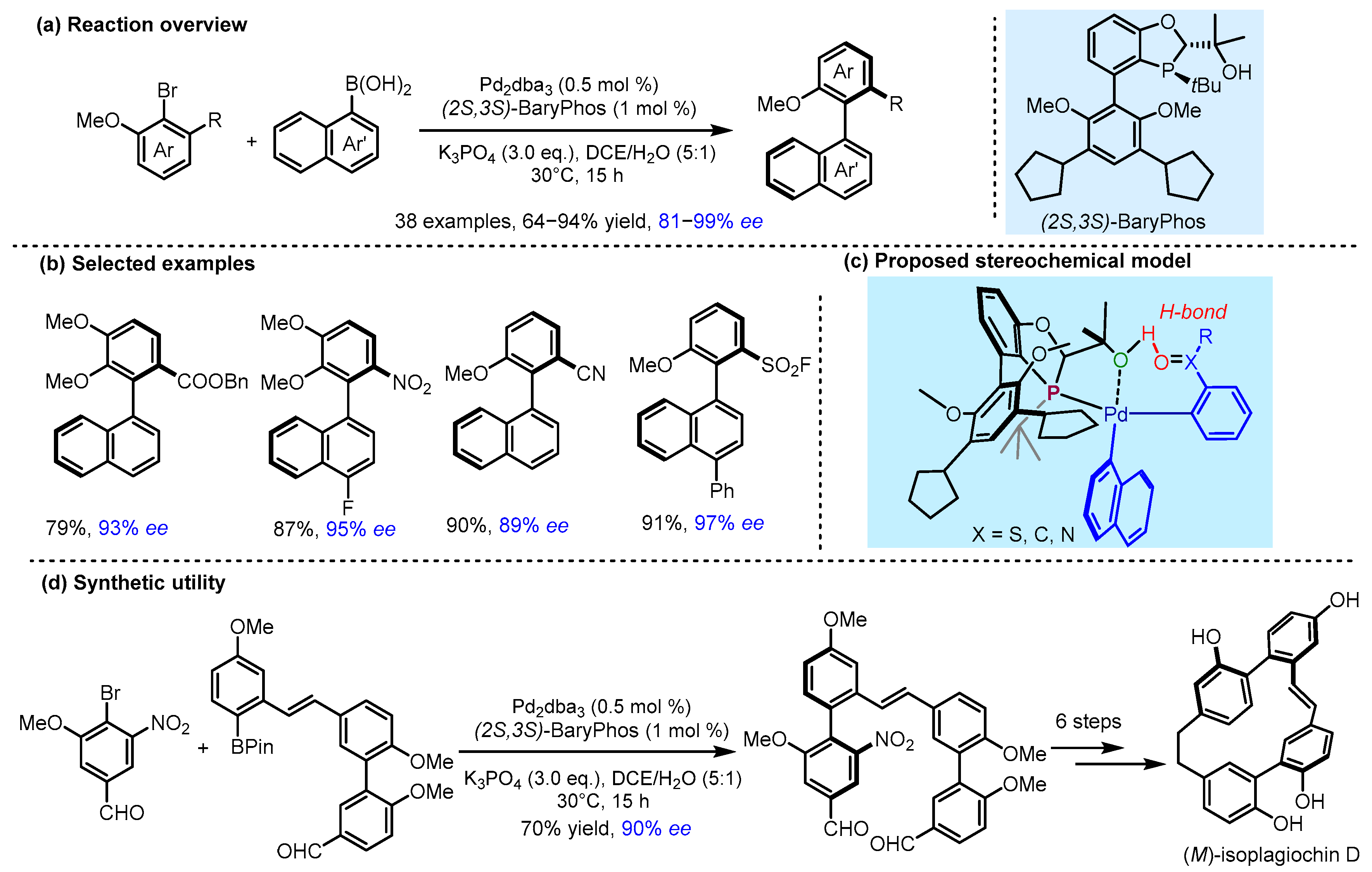
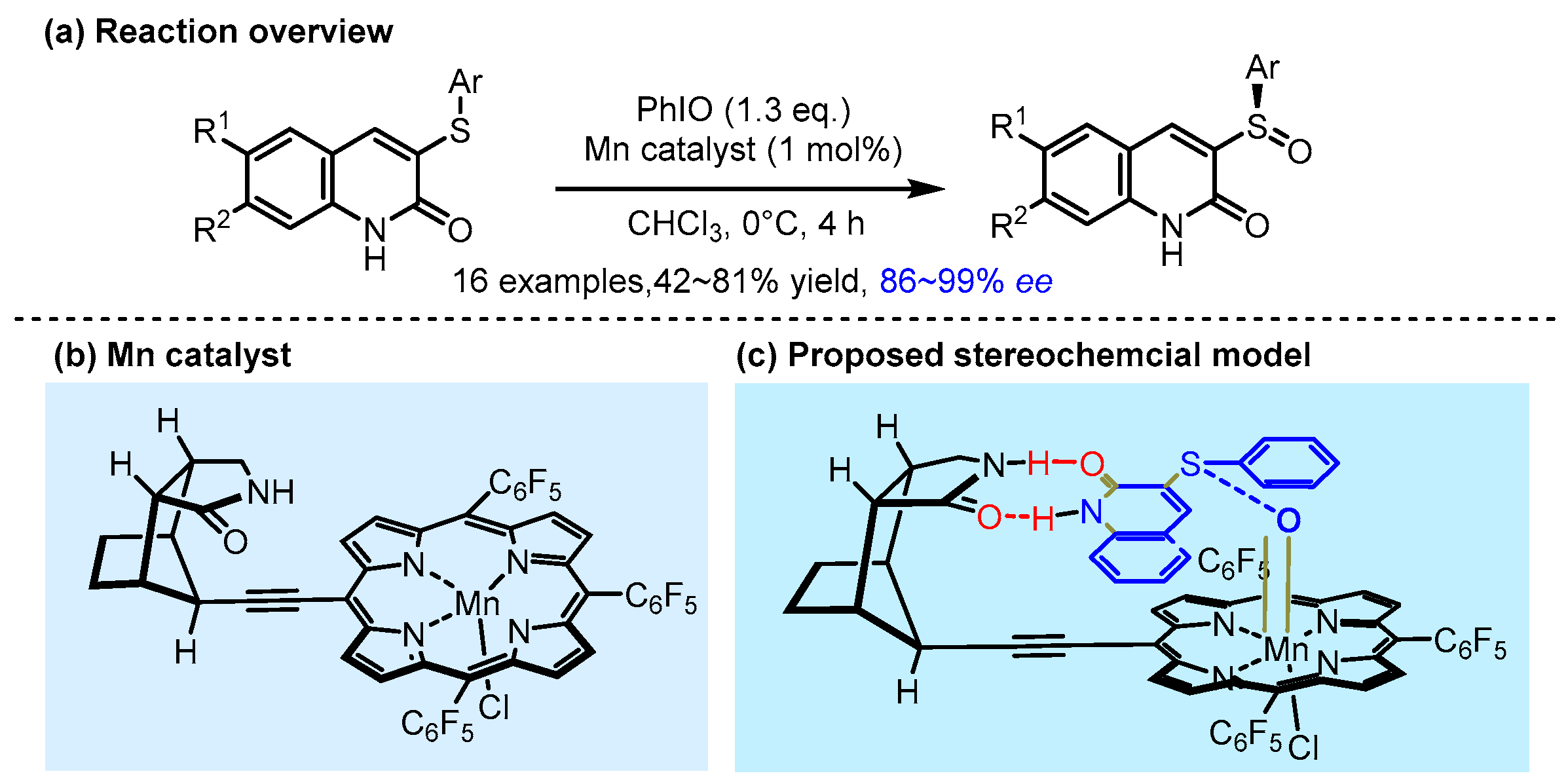
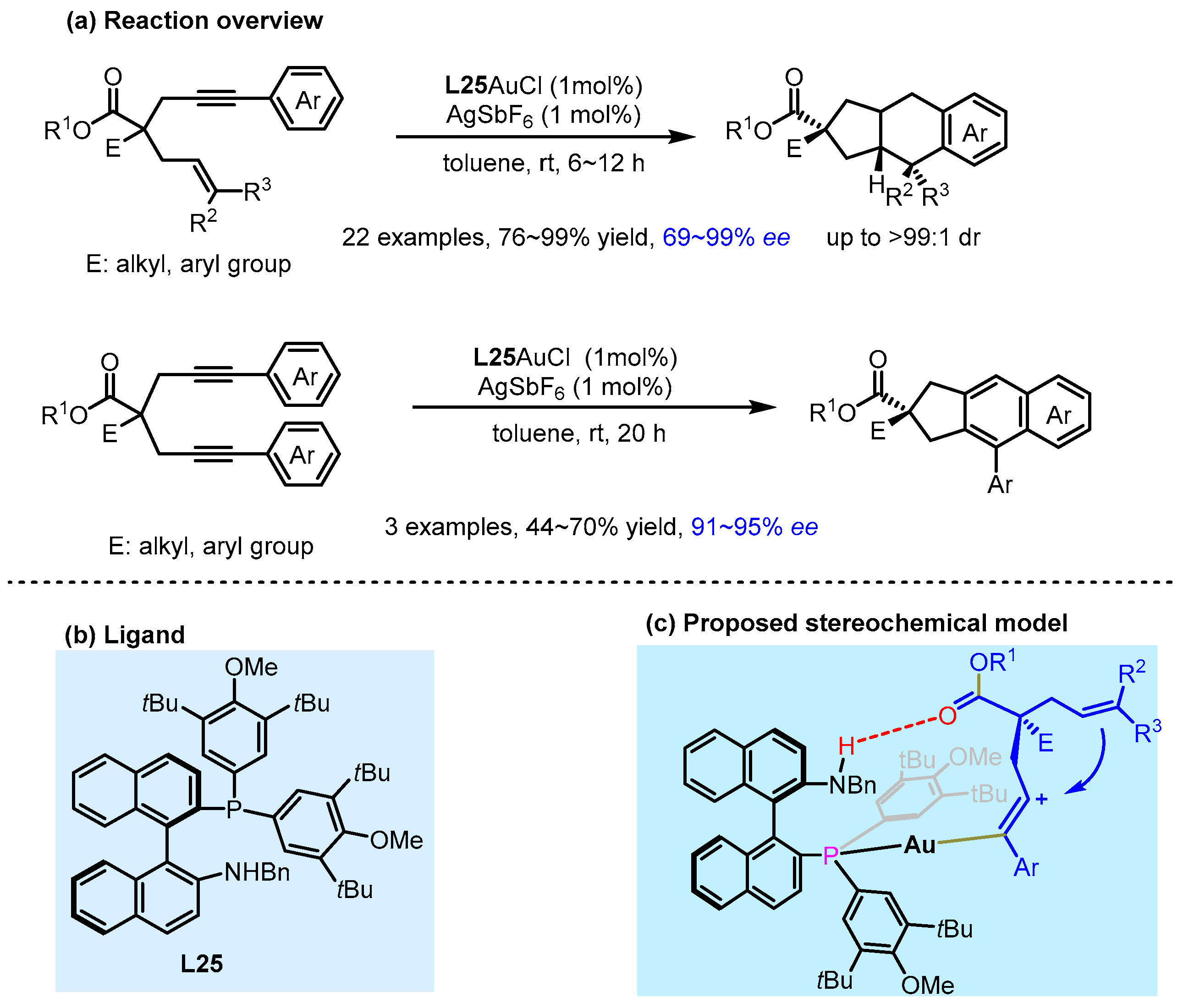
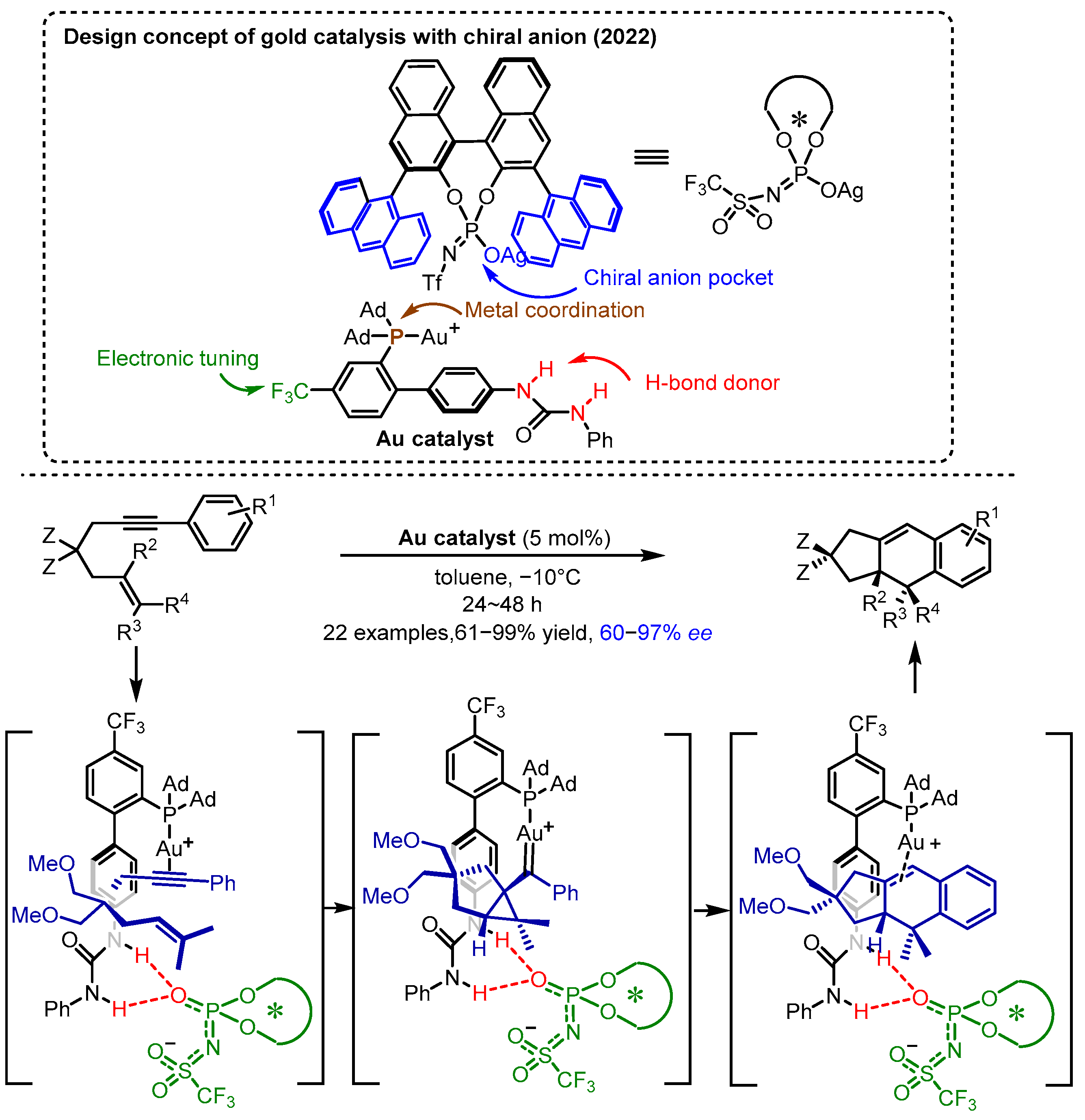
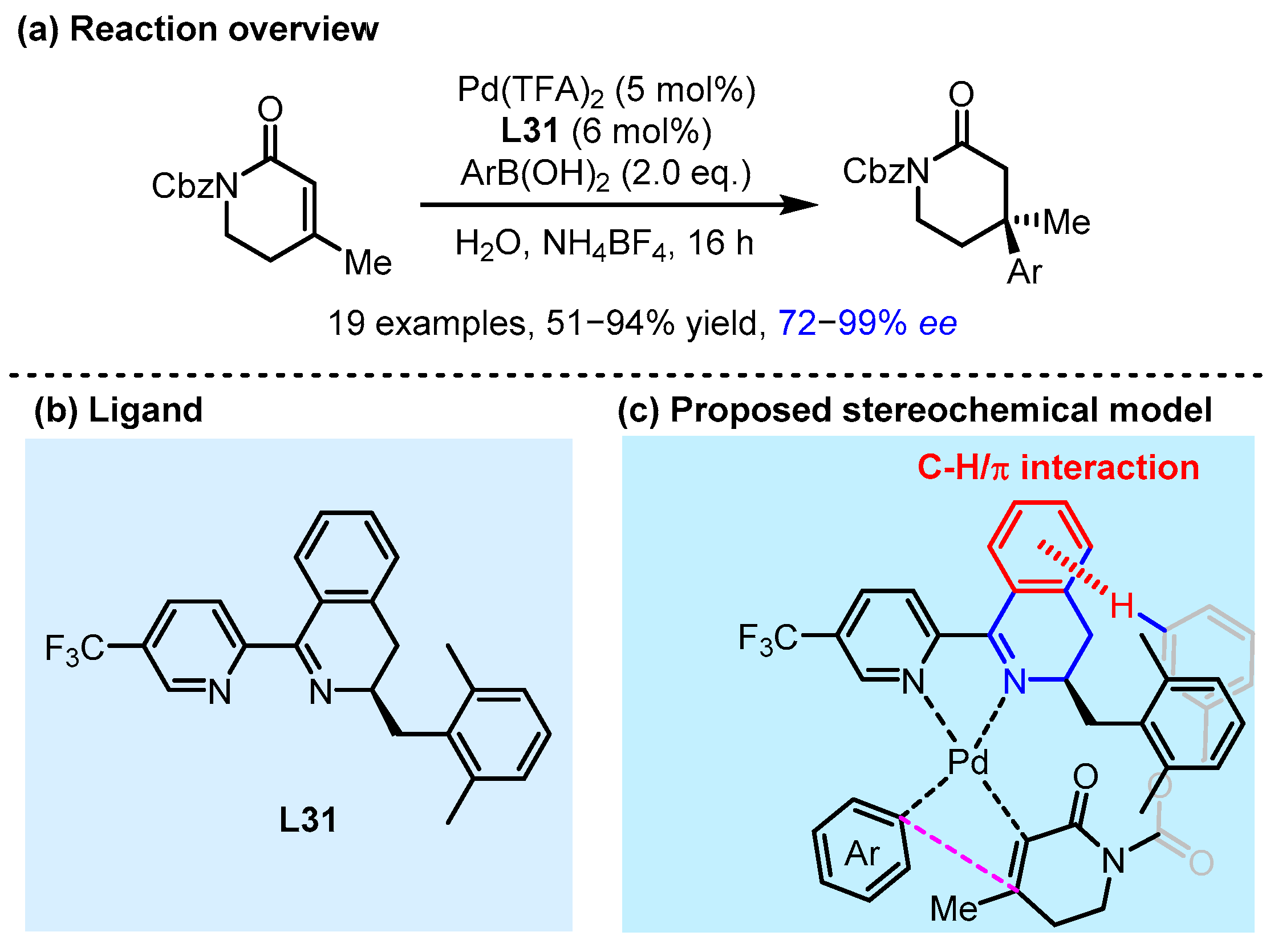
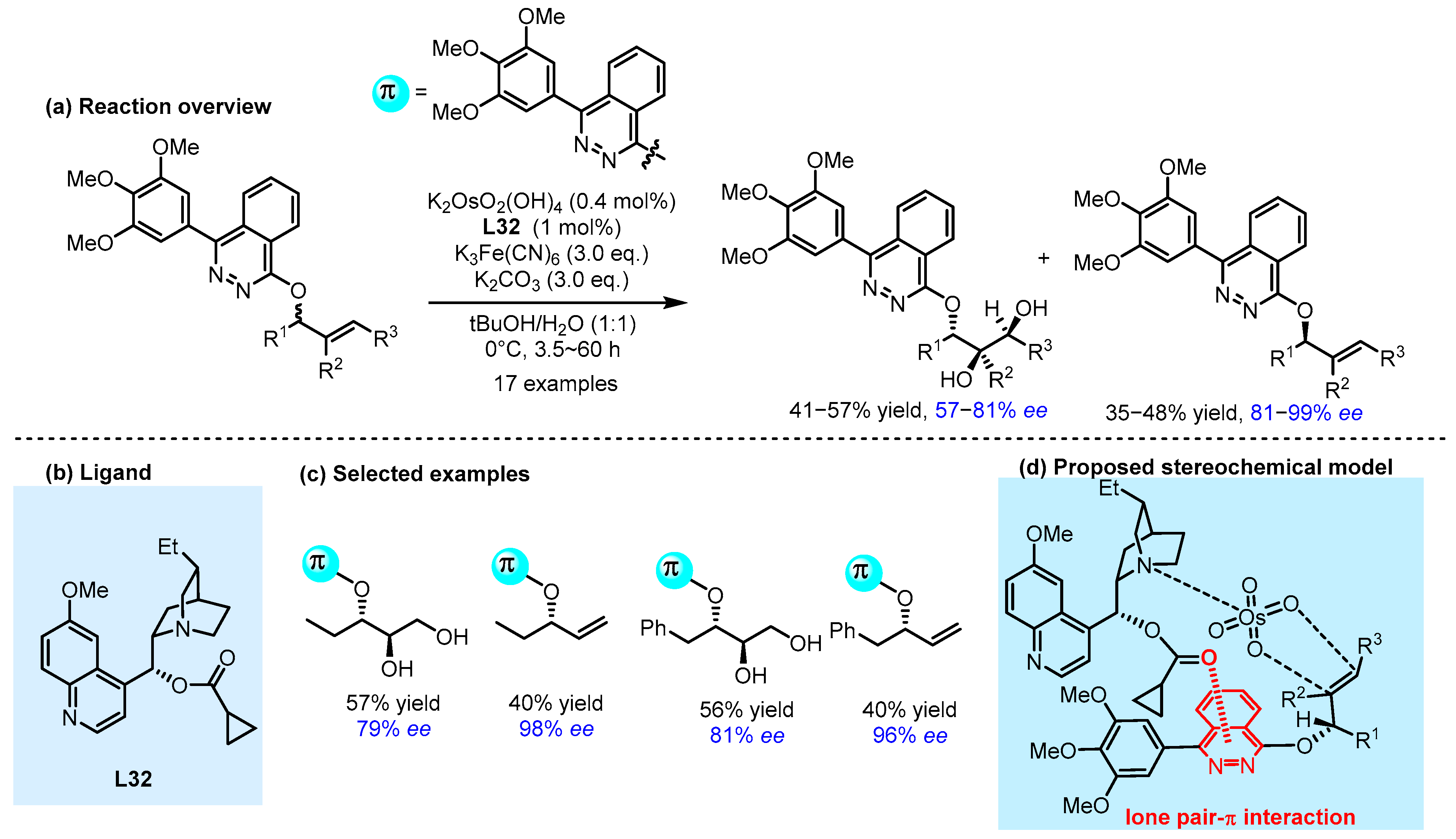
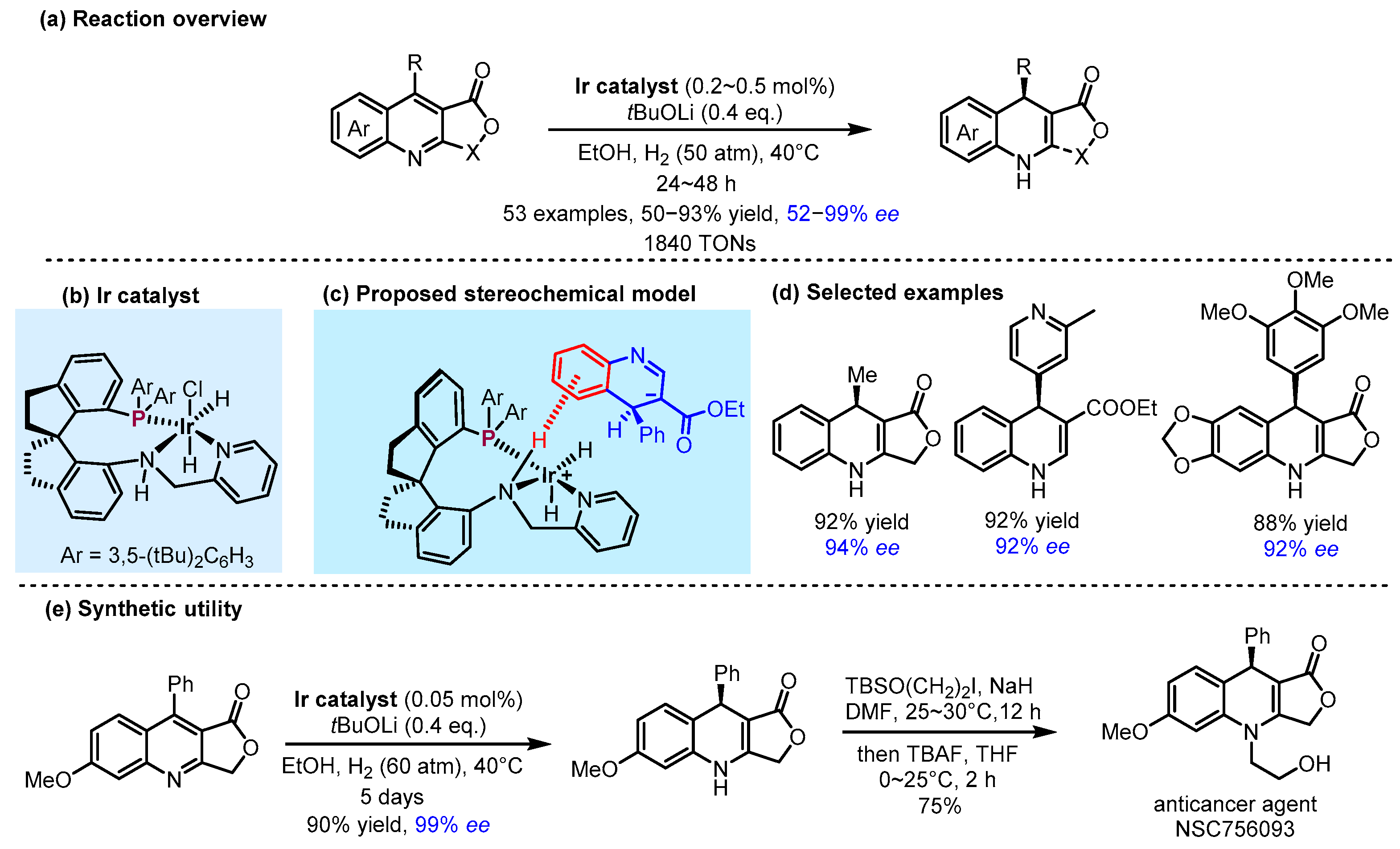
2.3. π Interactions Involved Enantioselective Transition Metal Catalysis
3. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Seki, M. A New Catalytic System for Ru-Catalyzed C–H Arylation Reactions and Its Application in the Practical Syntheses of Pharmaceutical Agents. Org. Process Res. Dev. 2016, 20, 867–877. [Google Scholar] [CrossRef]
- Mercs, L.; Albrecht, M. Beyond Catalysis: N-Heterocyclic Carbene Complexes as Components for Medicinal, Luminescent, and Functional Materials Applications. Chem. Soc. Rev. 2010, 39, 1903–1912. [Google Scholar] [CrossRef]
- Jenck, J.F.; Agterberg, F.; Droescher, M.J. Products and Processes for a Sustainable Chemical Industry: A Review of Achievements and Prospects. Green. Chem. 2004, 6, 544–556. [Google Scholar] [CrossRef]
- He, Y.-M.; Cheng, Y.-Z.; Duan, Y.; Zhang, Y.-D.; Fan, Q.-H.; You, S.-L.; Luo, S.; Zhu, S.-F.; Fu, X.-F.; Zhou, Q.-L. Recent Progress of Asymmetric Catalysis from a Chinese Perspective. CCS Chem. 2023, 5, 2685–2716. [Google Scholar] [CrossRef]
- Piou, T.; Rovis, T. Electronic and Steric Tuning of a Prototypical Piano Stool Complex: Rh(III) Catalysis for C–H Functionalization. Acc. Chem. Res. 2018, 51, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-M.; Lackner, A.D.; Toste, F.D. Development of Catalysts and Ligands for Enantioselective Gold Catalysis. Acc. Chem. Res. 2014, 47, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Reek, J.N.H.; de Bruin, B.; Pullen, S.; Mooibroek, T.J.; Kluwer, A.M.; Caumes, X. Transition Metal Catalysis Controlled by Hydrogen Bonding in the Second Coordination Sphere. Chem. Rev. 2022, 122, 12308–12369. [Google Scholar] [CrossRef]
- Peluso, P.; Chankvetadze, B. Recognition in the Domain of Molecular Chirality: From Noncovalent Interactions to Separation of Enantiomers. Chem. Rev. 2022, 122, 13235–13400. [Google Scholar] [CrossRef]
- Sawamura, M.; Ito, Y. Catalytic Asymmetric Synthesis by Means of Secondary Interaction between Chiral Ligands and Substrates. Chem. Rev. 1992, 92, 857–871. [Google Scholar] [CrossRef]
- Pachisia, S.; Gupta, R. Supramolecular Catalysis: The Role of H-Bonding Interactions in Substrate Orientation and Activation. Dalton Trans. 2021, 50, 14951–14966. [Google Scholar] [CrossRef]
- Fanourakis, A.; Docherty, P.J.; Chuentragool, P.; Phipps, R.J. Recent Developments in Enantioselective Transition Metal Catalysis Featuring Attractive Noncovalent Interactions between Ligand and Substrate. ACS Catal. 2020, 10, 10672–10714. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Sawamura, M.; Hayashi, T. Catalytic Asymmetric Aldol Reaction: Reaction of Aldehydes with Isocyanoacetate Catalyzed by a Chiral Ferrocenylphosphine-Gold(I) Complex. J. Am. Chem. Soc. 1986, 108, 6405–6406. [Google Scholar] [CrossRef]
- Carboni, S.; Gennari, C.; Pignataro, L.; Piarulli, U. Supramolecular Ligand–Ligand and Ligand–Substrate Interactions for Highly Selective Transition Metal Catalysis. Dalton Trans. 2011, 40, 4355–4373. [Google Scholar] [CrossRef]
- Zhao, Q.; Chen, C.; Wen, J.; Dong, X.-Q.; Zhang, X. Noncovalent Interaction-Assisted Ferrocenyl Phosphine Ligands in Asymmetric Catalysis. Acc. Chem. Res. 2020, 53, 1905–1921. [Google Scholar] [CrossRef]
- Yao, Q.-J.; Shi, B.-F. Cobalt(III)-Catalyzed Enantioselective C–H Functionalization: Ligand Innovation and Reaction Development. Acc. Chem. Res. 2025, 26, 1895–1899. [Google Scholar] [CrossRef] [PubMed]
- Krueger, C.A.; Kuntz, K.W.; Dzierba, C.D.; Wirschun, W.G.; Gleason, J.D.; Snapper, M.L.; Hoveyda, A.H. Ti-Catalyzed Enantioselective Addition of Cyanide to Imines. A Practical Synthesis of Optically Pure α-Amino Acids. J. Am. Chem. Soc. 1999, 121, 4284–4285. [Google Scholar] [CrossRef]
- Yang, T.; Sun, Y.; Wang, H.; Lin, Z.; Wen, J.; Zhang, X. Iridium-Catalyzed Enantioselective Hydrogenation of Oxocarbenium Ions: A Case of Ionic Hydrogenation. Angew. Chem. Int. Ed. 2020, 59, 6108–6114. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, Z.; Jin, S.; Fan, X.; Geng, M.; Zhou, Y.; Wen, S.; Wang, X.; Chung, L.W.; Dong, X.-Q.; et al. Enzyme-Inspired Chiral Secondary-Phosphine-Oxide Ligand with Dual Noncovalent Interactions for Asymmetric Hydrogenation. Angew. Chem. Int. Ed. 2017, 56, 6808–6812. [Google Scholar] [CrossRef]
- Wen, J.; Tan, R.; Liu, S.; Zhao, Q.; Zhang, X. Strong Brønsted Acid Promoted Asymmetric Hydrogenation of Isoquinolines and Quinolines Catalyzed by a Rh–Thiourea Chiral Phosphine Complex Via Anion Binding. Chem. Sci. 2016, 7, 3047–3051. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, S.; Huang, K.; Wang, R.; Zhang, X. A Novel Chiral Bisphosphine-Thiourea Ligand for Asymmetric Hydrogenation of Β,Β-Disubstituted Nitroalkenes. Org. Lett. 2013, 15, 4014–4017. [Google Scholar] [CrossRef]
- Du, L.; Cao, P.; Xing, J.; Lou, Y.; Jiang, L.; Li, L.; Liao, J. Hydrogen-Bond-Promoted Palladium Catalysis: Allylic Alkylation of Indoles with Unsymmetrical 1,3-Disubstituted Allyl Acetates Using Chiral Bis(Sulfoxide) Phosphine Ligands. Angew. Chem. Int. Ed. 2013, 52, 4207–4211. [Google Scholar] [CrossRef]
- Wang, Z.; Nicolini, C.; Hervieu, C.; Wong, Y.-F.; Zanoni, G.; Zhang, L. Remote Cooperative Group Strategy Enables Ligands for Accelerative Asymmetric Gold Catalysis. J. Am. Chem. Soc. 2017, 139, 16064–16067. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Li, W.; Li, Z.; Zhang, J. P-Chiral Phosphines Enabled by Palladium/Xiao-Phos-Catalyzed Asymmetric P–C Cross–Coupling of Secondary Phosphine Oxides and Aryl Bromides. J. Am. Chem. Soc. 2019, 141, 20556–20564. [Google Scholar] [CrossRef] [PubMed]
- Fackler, P.; Berthold, C.; Voss, F.; Bach, T. Hydrogen-Bond-Mediated Enantio- and Regioselectivity in a Ru-Catalyzed Epoxidation Reaction. J. Am. Chem. Soc. 2010, 132, 15911–15913. [Google Scholar] [CrossRef] [PubMed]
- Sladojevich, F.; Trabocchi, A.; Guarna, A.; Dixon, D.J. A New Family of Cinchona-Derived Amino Phosphine Precatalysts: Application to the Highly Enantio- and Diastereoselective Silver-Catalyzed Isocyanoacetate Aldol Reaction. J. Am. Chem. Soc. 2011, 133, 1710–1713. [Google Scholar] [CrossRef]
- Lang, K.; Park, J.; Hong, S. Urea/Transition-Metal Cooperative Catalyst for Anti-Selective Asymmetric Nitroaldol Reactions. Angew. Chem. Int. Ed. 2012, 51, 1620–1624. [Google Scholar] [CrossRef]
- Hayashi, M.; Iwanaga, M.; Shiomi, N.; Nakane, D.; Masuda, H.; Nakamura, S. Direct Asymmetric Mannich-Type Reaction of A-Isocyanoacetates with Ketimines Using Cinchona Alkaloid/Copper(II) Catalysts. Angew. Chem. Int. Ed. 2014, 53, 8411–8415. [Google Scholar] [CrossRef]
- Ma, J.; Ding, X.; Hu, Y.; Huang, Y.; Gong, L.; Meggers, E. Metal-Templated Chiral Brønsted Base Organocatalysis. Nat. Commun. 2014, 5, 4531. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, P.; Di, X.; Dai, Q.; Zhang, Z.-M.; Zhang, J. Gold-Catalyzed Asymmetric Intramolecular Cyclization of N-Allenamides for the Synthesis of Chiral Tetrahydrocarbolines. Angew. Chem. Int. Ed. 2017, 56, 15905–15909. [Google Scholar] [CrossRef]
- Yang, H.; Sun, J.; Gu, W.; Tang, W. Enantioselective Cross-Coupling for Axially Chiral Tetra-Ortho-Substituted Biaryls and Asymmetric Synthesis of Gossypol. J. Am. Chem. Soc. 2020, 142, 8036–8043. [Google Scholar] [CrossRef]
- Tang, W.; Capacci, A.G.; Wei, X.; Li, W.; White, A.; Patel, N.D.; Savoie, J.; Gao, J.J.; Rodriguez, S.; Qu, B.; et al. A General and Special Catalyst for Suzuki–Miyaura Coupling Processes. Angew. Chem. Int. Ed. 2010, 49, 5879–5883. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tang, W. Enantioselective Construction of Ortho-Sulfur- or Nitrogen-Substituted Axially Chiral Biaryls and Asymmetric Synthesis of Isoplagiochin D. Nat. Commun. 2022, 13, 4577. [Google Scholar] [CrossRef]
- Howell, G.P.; Agnew, L.R.; Bauer, C.; Bell, F.J.; Campbell, A.D.; Dai, K.; Dave, D.; Ellis, S.R.; Foulkes, M.J.; Gall, M.A.Y.; et al. Comprehensive Synthetic Route Redesign of AZD5991: A High-Complexity Atropisomeric Macrocycle. Org. Process Res. Dev. 2025, 29, 804–827. [Google Scholar] [CrossRef]
- Das, S.; Incarvito, C.D.; Crabtree, R.H.; Brudvig, G.W. Molecular Recognition in the Selective Oxygenation of Saturated C–H Bonds by a Dimanganese Catalyst. Science 2006, 312, 1941–1943. [Google Scholar] [CrossRef] [PubMed]
- Burg, F.; Buchelt, C.; Kreienborg, N.M.; Merten, C.; Bach, T. Enantioselective Synthesis of Diaryl Sulfoxides Enabled by Molecular Recognition. Org. Lett. 2021, 23, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Ghosh, B.; Breitenlechner, S.; Feßner, M.; Merten, C.; Bach, T. Intermolecular Enantioselective Amination Reactions Mediated by Visible Light and a Chiral Iron Porphyrin Complex. Angew. Chem. Int. Ed. 2024, 63, e202407003. [Google Scholar] [CrossRef]
- Annapureddy, R.R.; Jandl, C.; Bach, T. A Chiral Phenanthroline Ligand with a Hydrogen-Bonding Site: Application to the Enantioselective Amination of Methylene Groups. J. Am. Chem. Soc. 2020, 142, 7374–7378. [Google Scholar] [CrossRef]
- Annapureddy, R.R.; Burg, F.; Gramüller, J.; Golub, T.P.; Merten, C.; Huber, S.M.; Bach, T. Silver-Catalyzed Enantioselective Sulfimidation Mediated by Hydrogen Bonding Interactions. Angew. Chem. Int. Ed. 2021, 60, 7920–7926. [Google Scholar] [CrossRef]
- Burg, F.; Breitenlechner, S.; Jandl, C.; Bach, T. Enantioselective Oxygenation of Exocyclic Methylene Groups by a Manganese Porphyrin Catalyst with a Chiral Recognition Site. Chem. Sci. 2020, 11, 2121–2129. [Google Scholar] [CrossRef]
- Anderson, K.W.; Buchwald, S.L. General Catalysts for the Suzuki–Miyaura and Sonogashira Coupling Reactions of Aryl Chlorides and for the Coupling of Challenging Substrate Combinations in Water. Angew. Chem. Int. Ed. 2005, 44, 6173–6177. [Google Scholar] [CrossRef]
- Pearce-Higgins, R.; Hogenhout, L.N.; Docherty, P.J.; Whalley, D.M.; Chuentragool, P.; Lee, N.; Lam, N.Y.S.; McGuire, T.M.; Valette, D.; Phipps, R.J. An Enantioselective Suzuki–Miyaura Coupling to Form Axially Chiral Biphenols. J. Am. Chem. Soc. 2022, 144, 15026–15032. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, S.; García-Fortanet, J.; Del Aguila Sanchez, M.A.; Buchwald, S.L. Palladium(0)-Catalyzed Arylative Dearomatization of Phenols. J. Am. Chem. Soc. 2011, 133, 9282–9285. [Google Scholar] [CrossRef] [PubMed]
- Kadarauch, M.; Whalley, D.M.; Phipps, R.J. Ssphos: A General Ligand for Enantioselective Arylative Phenol Dearomatization Via Electrostatically-Directed Palladium Catalysis. J. Am. Chem. Soc. 2023, 145, 25553–25558. [Google Scholar] [CrossRef]
- Lou, Y.; Wei, J.; Li, M.; Zhu, Y. Distal Ionic Substrate–Catalyst Interactions Enable Long-Range Stereocontrol: Access to Remote Quaternary Stereocenters through a Desymmetrizing Suzuki–Miyaura Reaction. J. Am. Chem. Soc. 2022, 144, 123–129. [Google Scholar] [CrossRef]
- Wei, J.; Gandon, V.; Zhu, Y. Amino Acid-Derived Ionic Chiral Catalysts Enable Desymmetrizing Cross-Coupling to Remote Acyclic Quaternary Stereocenters. J. Am. Chem. Soc. 2023, 145, 16796–16811. [Google Scholar] [CrossRef] [PubMed]
- Roizen, J.L.; Harvey, M.E.; Du Bois, J. Metal-Catalyzed Nitrogen-Atom Transfer Methods for the Oxidation of Aliphatic C–H Bonds. Acc. Chem. Res. 2012, 45, 911–922. [Google Scholar] [CrossRef]
- Fanourakis, A.; Williams, B.D.; Paterson, K.J.; Phipps, R.J. Enantioselective Intermolecular C–H Amination Directed by a Chiral Cation. J. Am. Chem. Soc. 2021, 143, 10070–10076. [Google Scholar] [CrossRef] [PubMed]
- Paterson, K.J.; Dahiya, A.; Williams, B.D.; Phipps, R.J. Tertiary Amides as Directing Groups for Enantioselective C−H Amination Using Ion-Paired Rhodium Complexes. Angew. Chem. Int. Ed. 2024, 63, e202317489. [Google Scholar] [CrossRef]
- Hodson, N.J.; Takano, S.; Fanourakis, A.; Phipps, R.J. Enantioselective Nitrene Transfer to Hydrocinnamyl Alcohols and Allylic Alcohols Enabled by Systematic Exploration of the Structure of Ion-Paired Rhodium Catalysts. J. Am. Chem. Soc. 2024, 146, 22629–22641. [Google Scholar] [CrossRef]
- Dorel, R.; Echavarren, A.M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, L. Designed Bifunctional Ligands in Cooperative Homogeneous Gold Catalysis. CCS Chem. 2020, 3, 1989–2002. [Google Scholar] [CrossRef]
- Zhao, K.; Kohnke, P.; Yang, Z.; Cheng, X.; You, S.-L.; Zhang, L. Enantioselective Dearomative Cyclization Enabled by Asymmetric Cooperative Gold Catalysis. Angew. Chem. Int. Ed. 2022, 61, e202207518. [Google Scholar] [CrossRef] [PubMed]
- Kohnke, P.; Zhang, L. Bifunctional Phosphine-Enabled Regioselective Cycloisomerization of Enynyl Esters En Route to Bicyclo[2.2.1]Heptenes. Org. Lett. 2023, 25, 1295–1298. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, C.D.; Zhao, K.; Zhang, L. Gold-Catalyzed Asymmetric Transformation of Hydroxylated Propargylic Esters. ChemPlusChem 2023, 88, e202300314. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, K.; Li, X.; Quintanilla, C.D.; Zhang, L. Asymmetric Dearomatization of Phenols Via Ligand-Enabled Cooperative Gold Catalysis. Angew. Chem. Int. Ed. 2023, 62, e202309256. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Yang, Z.; Yang, J.; Li, X.; Quintanilla, C.D.; Zhang, L. Desymmetrization and Parallel Kinetic Resolution of 1-Ethynylcyclobutanols Via Asymmetric Cooperative Gold Catalysis. J. Am. Chem. Soc. 2023, 145, 27205–27210. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, L. Gold-Catalyzed Homo/Heterodimerization of Terminal Alkynes Facilitated by Metal–Ligand Cooperation. Org. Lett. 2024, 26, 5736–5740. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, K.; Quintanilla, C.D.; Zhang, L. Chiral Bifunctional Phosphine Ligand Enables Asymmetric Trapping of Catalytic Vinyl Gold Carbene Species. J. Am. Chem. Soc. 2024, 146, 2308–2312. [Google Scholar] [CrossRef]
- Zuccarello, G.; Mayans, J.G.; Escofet, I.; Scharnagel, D.; Kirillova, M.S.; Pérez-Jimeno, A.H.; Calleja, P.; Boothe, J.R.; Echavarren, A.M. Enantioselective Folding of Enynes by Gold(I) Catalysts with a Remote C2-Chiral Element. J. Am. Chem. Soc. 2019, 141, 11858–11863. [Google Scholar] [CrossRef]
- Ji, K.; Zheng, Z.; Wang, Z.; Zhang, L. Enantioselective Oxidative Gold Catalysis Enabled by a Designed Chiral P,N-Bidentate Ligand. Angew. Chem. Int. Ed. 2015, 54, 1245–1249. [Google Scholar] [CrossRef]
- Zhang, J.-Q.; Liu, Y.; Wang, X.-W.; Zhang, L. Synthesis of Chiral Bifunctional Nhc Ligands and Survey of Their Utilities in Asymmetric Gold Catalysis. Organometallics 2019, 38, 3931–3938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Smal, V.; Retailleau, P.; Voituriez, A.; Frison, G.; Marinetti, A.; Guinchard, X. Tethered Counterion-Directed Catalysis: Merging the Chiral Ion-Pairing and Bifunctional Ligand Strategies in Enantioselective Gold(I) Catalysis. J. Am. Chem. Soc. 2020, 142, 3797–3805. [Google Scholar] [CrossRef]
- Li, X.; Liao, S.; Wang, Z.; Zhang, L. Ligand-Accelerated Gold-Catalyzed Addition of in Situ Generated Hydrazoic Acid to Alkynes under Neat Conditions. Org. Lett. 2017, 19, 3687–3690. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Xiao, Y.; Yang, T.; Chen, G.-Q.; Zhang, X.; Che, C.-M. Gold-Catalyzed Highly Enantioselective Cycloadditions of 1,6-Enynes and 1,6-Diynes Assisted by Remote Hydrogen Bonding Interaction. iScience 2024, 27, 110876. [Google Scholar] [CrossRef] [PubMed]
- Franchino, A.; Martí, À.; Echavarren, A.M. H-Bonded Counterion-Directed Enantioselective Au(I) Catalysis. J. Am. Chem. Soc. 2022, 144, 3497–3509. [Google Scholar] [CrossRef]
- Huang, B.; Khanh Mai, B.; Warzok, U.; Liu, P.; Dean Toste, F. On the Gold(I)-Catalyzed Enantioselective Addition of Indole to Diphenylallene Via Anion-Binding Catalysis. Tetrahedron Lett. 2024, 149, 155247. [Google Scholar] [CrossRef]
- Ovian, J.M.; Vojáčková, P.; Jacobsen, E.N. Enantioselective Transition-Metal Catalysis Via an Anion-Binding Approach. Nature 2023, 616, 84–89. [Google Scholar] [CrossRef]
- Li, J.-Y.; Xie, P.-P.; Zhou, T.; Qian, P.-F.; Zhou, Y.-B.; Li, H.-C.; Hong, X.; Shi, B.-F. Ir(III)-Catalyzed Asymmetric C–H Activation/Annulation of Sulfoximines Assisted by the Hydrogen-Bonding Interaction. ACS Catal. 2022, 12, 9083–9091. [Google Scholar] [CrossRef]
- Zhou, T.; Qian, P.-F.; Li, J.-Y.; Zhou, Y.-B.; Li, H.-C.; Chen, H.-Y.; Shi, B.-F. Efficient Synthesis of Sulfur-Stereogenic Sulfoximines Via Ru(II)-Catalyzed Enantioselective C–H Functionalization Enabled by Chiral Carboxylic Acid. J. Am. Chem. Soc. 2021, 143, 6810–6816. [Google Scholar] [CrossRef]
- Sakai, S.; Fujioka, A.; Imai, K.; Uchiyama, K.; Shimizu, Y.; Higashida, K.; Sawamura, M. Silver-Catalyzed Asymmetric Aldol Reaction of Isocyanoacetic Acid Derivatives Enabled by Cooperative Participation of Classical and Nonclassical Hydrogen Bonds. Adv. Synth. Catal. 2022, 364, 2333–2339. [Google Scholar] [CrossRef]
- Tsutsumi, R.; Taguchi, R.; Yamanaka, M. Chiral Bipyridine Ligand with Flexible Molecular Recognition Site: Development and Application to Copper-Catalyzed Asymmetric Borylation of α,β-Unsaturated Ketones. ChemCatChem 2022, 14, e202101278. [Google Scholar] [CrossRef]
- Cardoso, F.S.P.; Abboud, K.A.; Aponick, A. Design, Preparation, and Implementation of an Imidazole-Based Chiral Biaryl P,N-Ligand for Asymmetric Catalysis. J. Am. Chem. Soc. 2013, 135, 14548–14551. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Patel, N.D.; Xu, G.; Xu, X.; Savoie, J.; Ma, S.; Hao, M.-H.; Keshipeddy, S.; Capacci, A.G.; Wei, X.; et al. Efficient Chiral Monophosphorus Ligands for Asymmetric Suzuki–Miyaura Coupling Reactions. Org. Lett. 2012, 14, 2258–2261. [Google Scholar] [CrossRef]
- Rokade, B.V.; Guiry, P.J. Axially Chiral P,N-Ligands: Some Recent Twists and Turns. ACS Catal. 2018, 8, 624–643. [Google Scholar] [CrossRef]
- Changotra, A.; Bhaskararao, B.; Hadad, C.M.; Sunoj, R.B. Insights on Absolute and Relative Stereocontrol in Stereodivergent Cooperative Catalysis. J. Am. Chem. Soc. 2020, 142, 9612–9624. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Ji, Y.; Liu, H.; Pang, Y.; Zhou, B.; Cheng, M.; Liu, Y.; Lin, B.; Liu, Y. Silver Triflate/N-Fluorobenzenesulfonimide-Catalyzed Cycloisomerization of Tryptamine-Ynamide to Spiro[Indoline-3,4’-Piperidine] Induced by Cation-π-π Interactions between Substrate and Metal Ligand. Adv. Synth. Catal. 2020, 362, 192–205. [Google Scholar] [CrossRef]
- Zhao, X.-J.; Li, Z.-H.; Ding, T.-M.; Tian, J.-M.; Tu, Y.-Q.; Wang, A.-F.; Xie, Y.-Y. Enantioselective Synthesis of 3,3’-Disubstituted 2-Amino-2’-Hydroxy-1,1’-Binaphthyls by Copper-Catalyzed Aerobic Oxidative Cross-Coupling. Angew. Chem. Int. Ed. 2021, 60, 7061–7065. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Xie, P.-P.; Liu, L.; Fan, J.; Zhang, Z.-Z.; Hong, X.; Shi, B.-F. Cp*Co(III)-Catalyzed Enantioselective Hydroarylation of Unactivated Terminal Alkenes Via C–H Activation. J. Am. Chem. Soc. 2021, 143, 19112–19120. [Google Scholar] [CrossRef]
- Baek, D.; Ryu, H.; Hahm, H.; Lee, J.; Hong, S. Palladium Catalysis Featuring Attractive Noncovalent Interactions Enabled Highly Enantioselective Access to β-Quaternary δ-Lactams. ACS Catal. 2022, 12, 5559–5564. [Google Scholar] [CrossRef]
- Jin, M.Y.; Zhen, Q.; Xiao, D.; Tao, G.; Xing, X.; Yu, P.; Xu, C. Engineered Non-Covalent π Interactions as Key Elements for Chiral Recognition. Nat. Commun. 2022, 13, 3276. [Google Scholar] [CrossRef]
- Zhen, Q.; Liu, C.; Lv, X.; Jin, M.Y.; Xu, C. Asymmetric Dihydroxylation-Based Desymmetrization Enabled by Lone-Pair−π Interaction. ACS Catal. 2023, 13, 9745–9752. [Google Scholar] [CrossRef]
- Zhu, C.-L.; Yan, X.; Bin, H.-Y.; Wu, X.; Huang, Z.-Y.; Yan, P.-C.; Huang, G.; Xie, J.-H.; Zhou, Q.-L. Enantioselective Synthesis of Chiral 1,4-Dihydroquinolines Via Iridium-Catalyzed Asymmetric Partial Hydrogenation of Quinolines. J. Am. Chem. Soc. 2025, 147, 5725–5735. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Z.; He, D.; Luo, L.; Tang, W. Recent Advances in Enantioselective Transition Metal Catalysis Mediated by Ligand–Substrate Noncovalent Interactions. Catalysts 2025, 15, 395. https://doi.org/10.3390/catal15040395
Cao Z, He D, Luo L, Tang W. Recent Advances in Enantioselective Transition Metal Catalysis Mediated by Ligand–Substrate Noncovalent Interactions. Catalysts. 2025; 15(4):395. https://doi.org/10.3390/catal15040395
Chicago/Turabian StyleCao, Zhen, Dongyang He, Lin Luo, and Wenjun Tang. 2025. "Recent Advances in Enantioselective Transition Metal Catalysis Mediated by Ligand–Substrate Noncovalent Interactions" Catalysts 15, no. 4: 395. https://doi.org/10.3390/catal15040395
APA StyleCao, Z., He, D., Luo, L., & Tang, W. (2025). Recent Advances in Enantioselective Transition Metal Catalysis Mediated by Ligand–Substrate Noncovalent Interactions. Catalysts, 15(4), 395. https://doi.org/10.3390/catal15040395