Tungsten-Based Mesoporous Silicates W-MMM-E as Heterogeneous Catalysts for Liquid-Phase Oxidations with Aqueous H2O2

 ,
,  ,
, 
Abstract
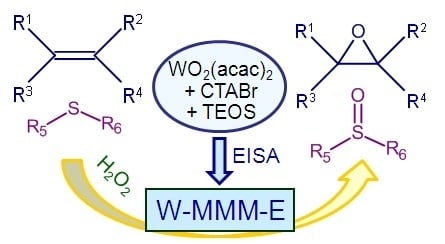
:
1. Introduction
2. Results and Discussion
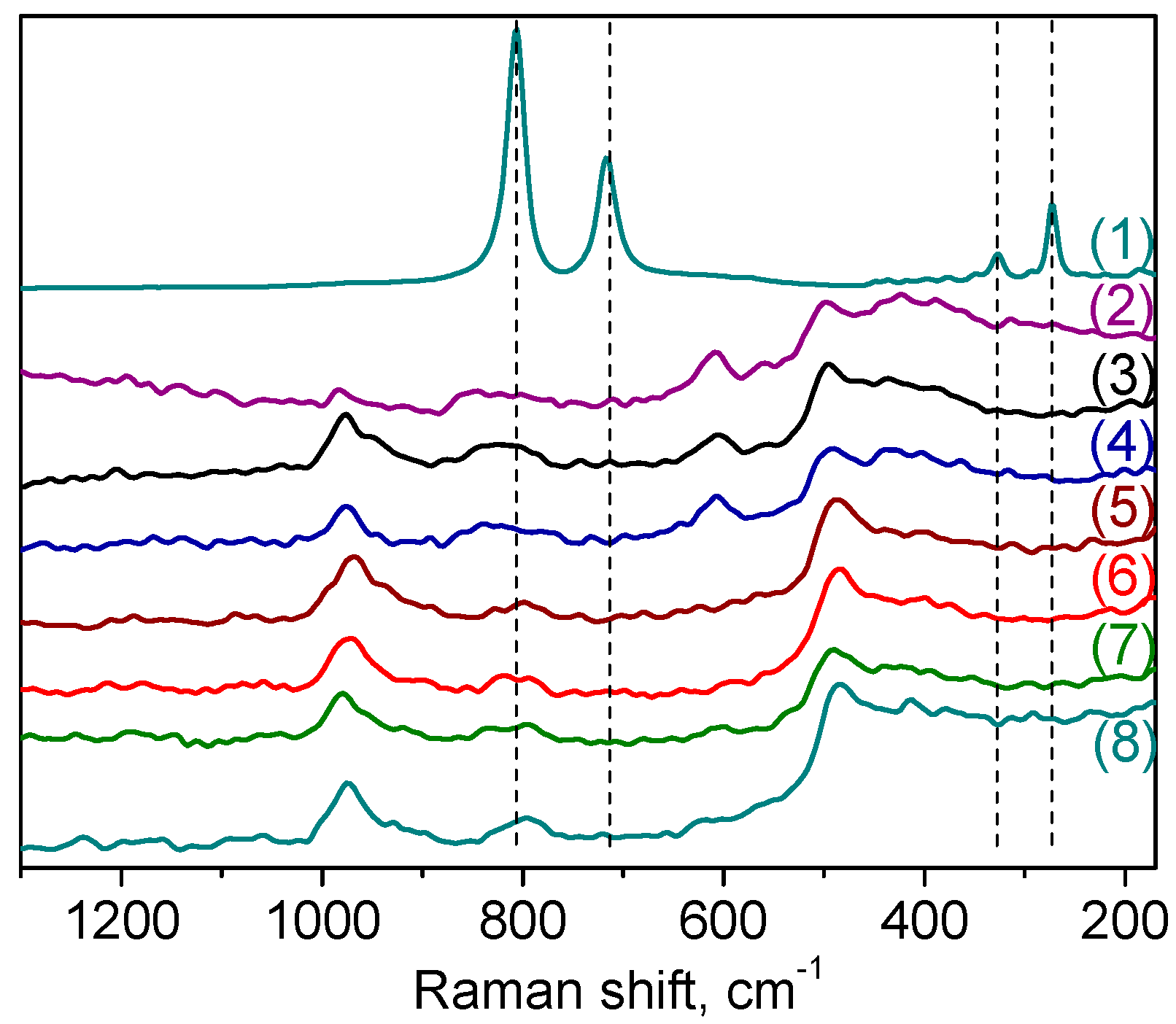
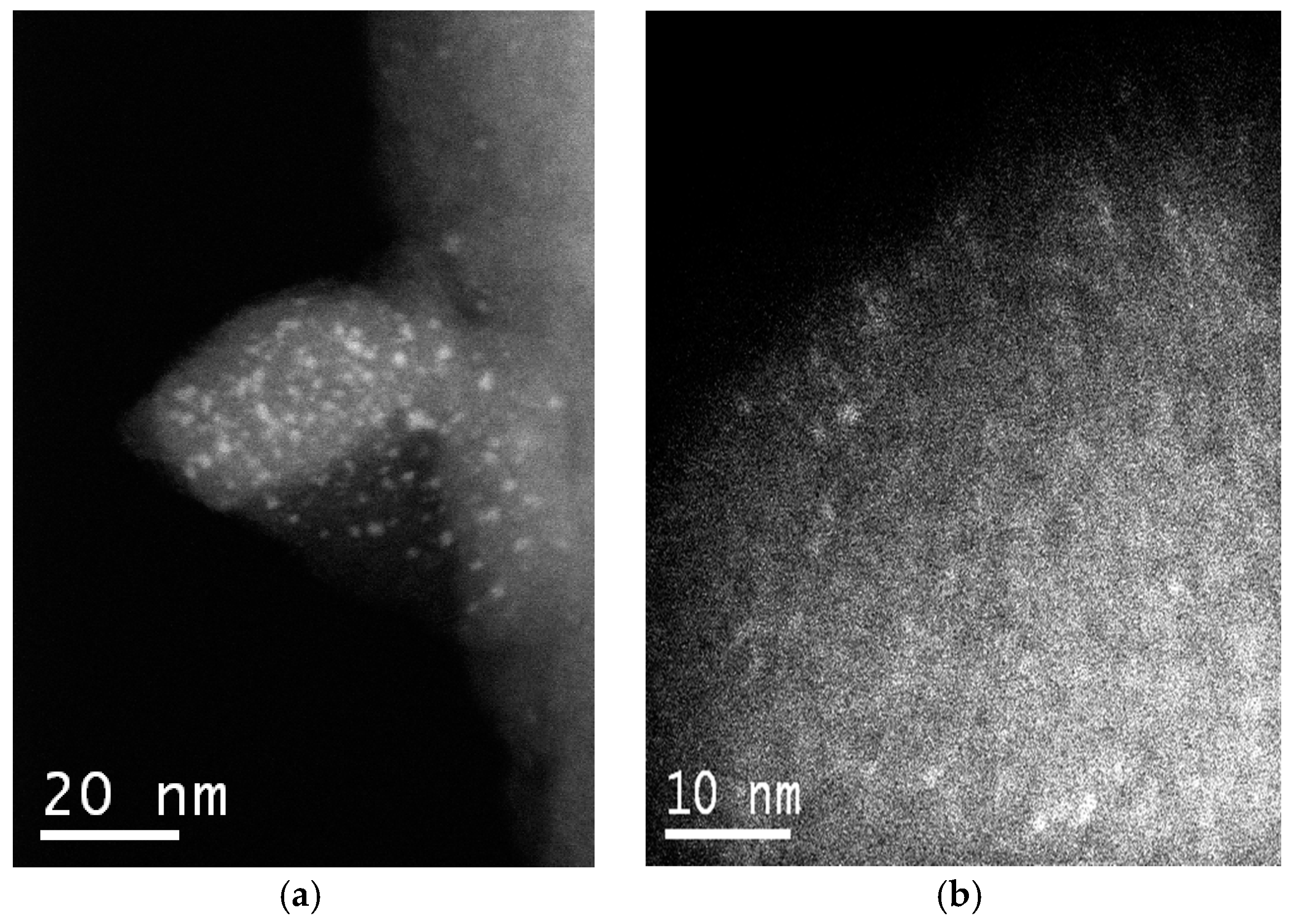
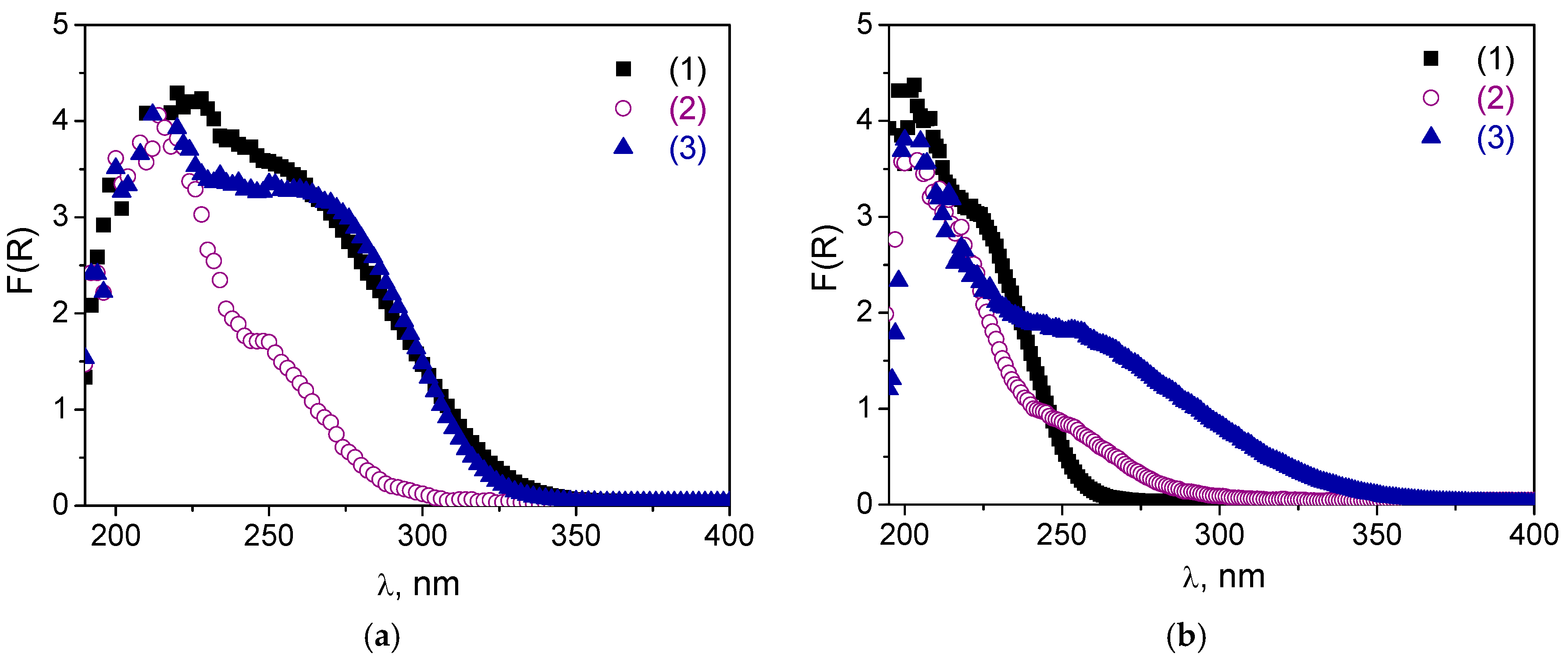
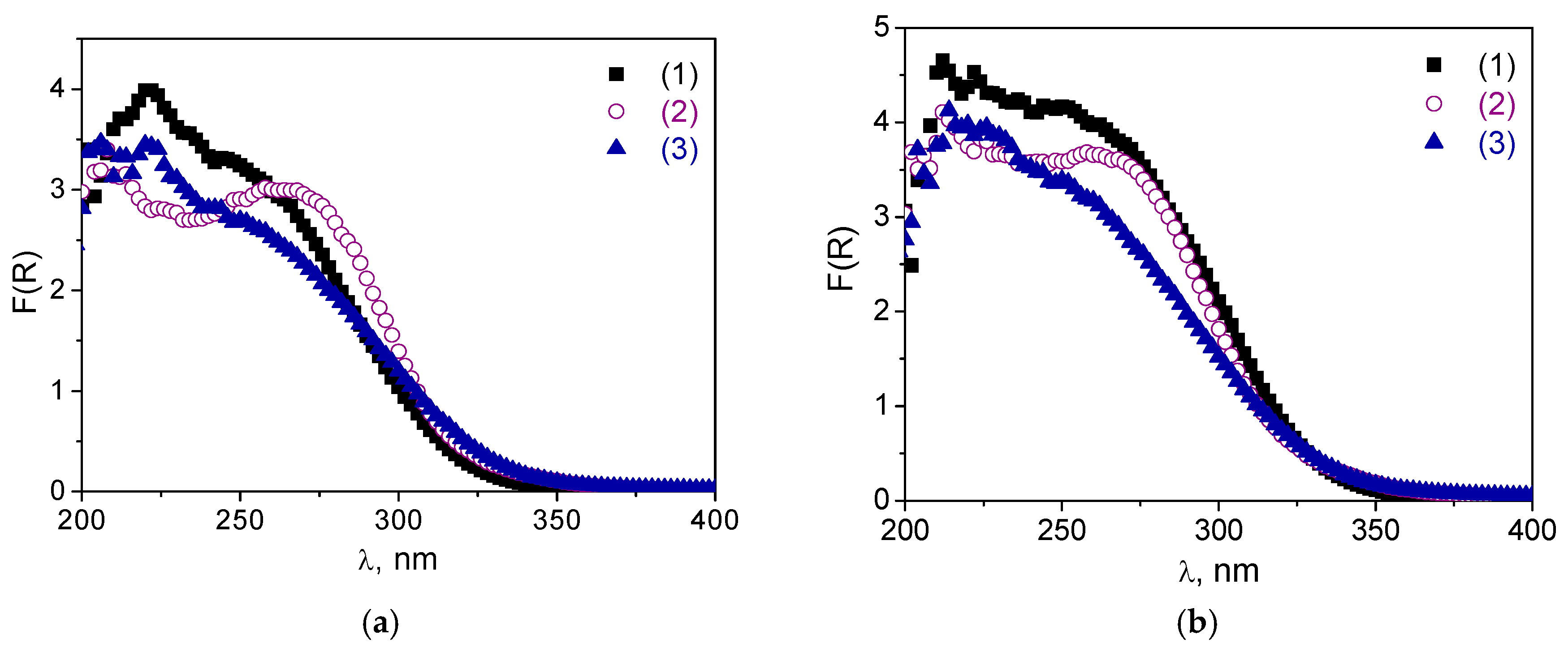
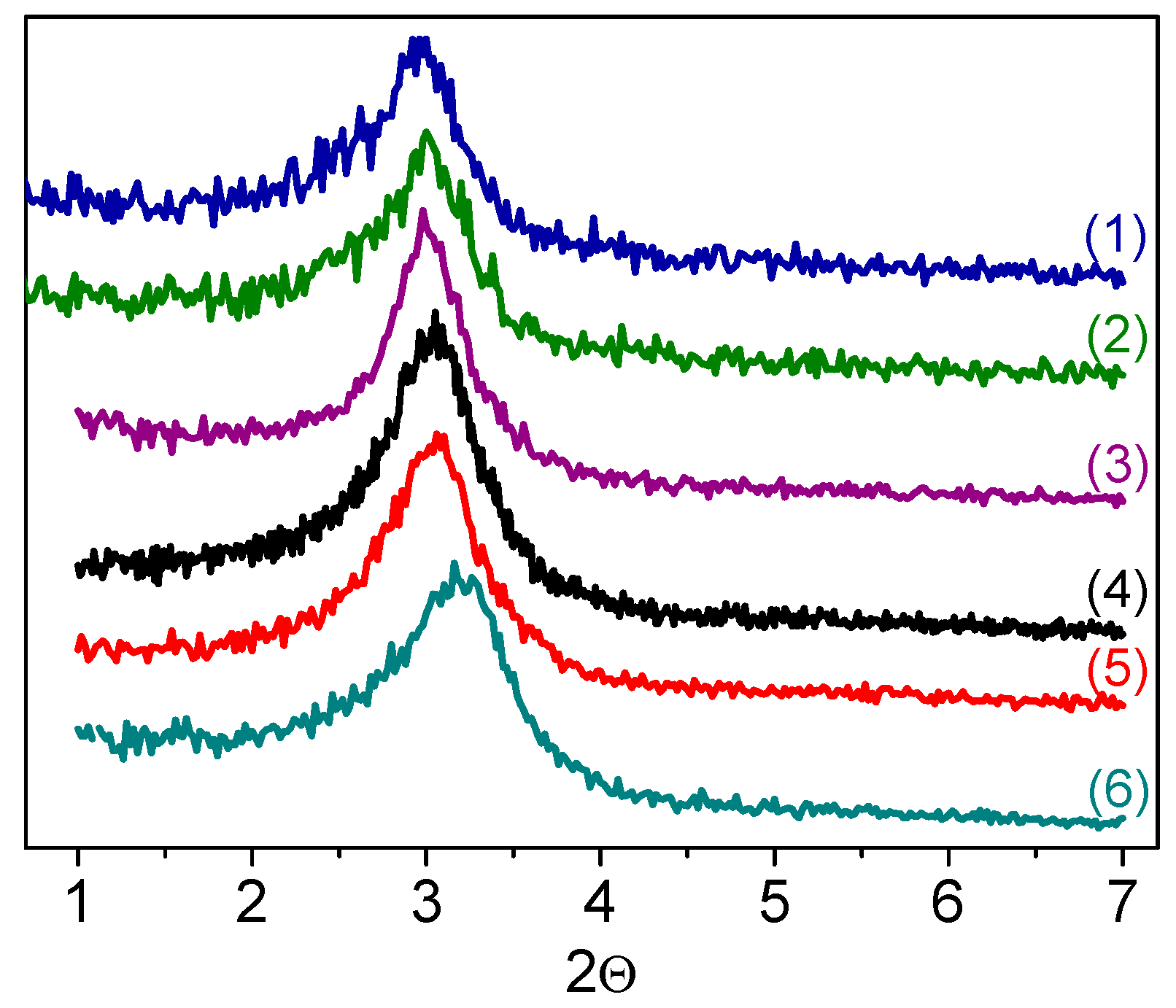
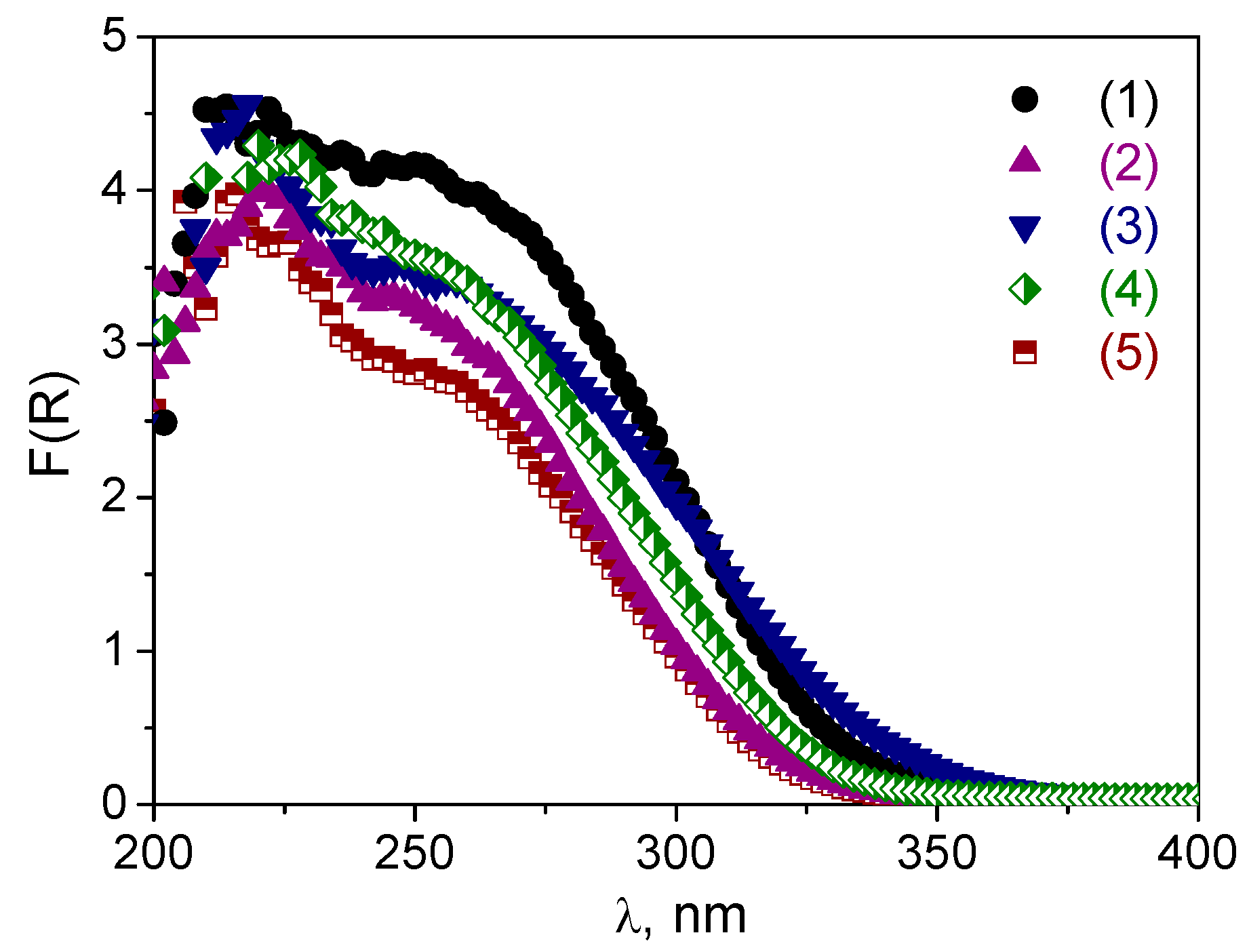
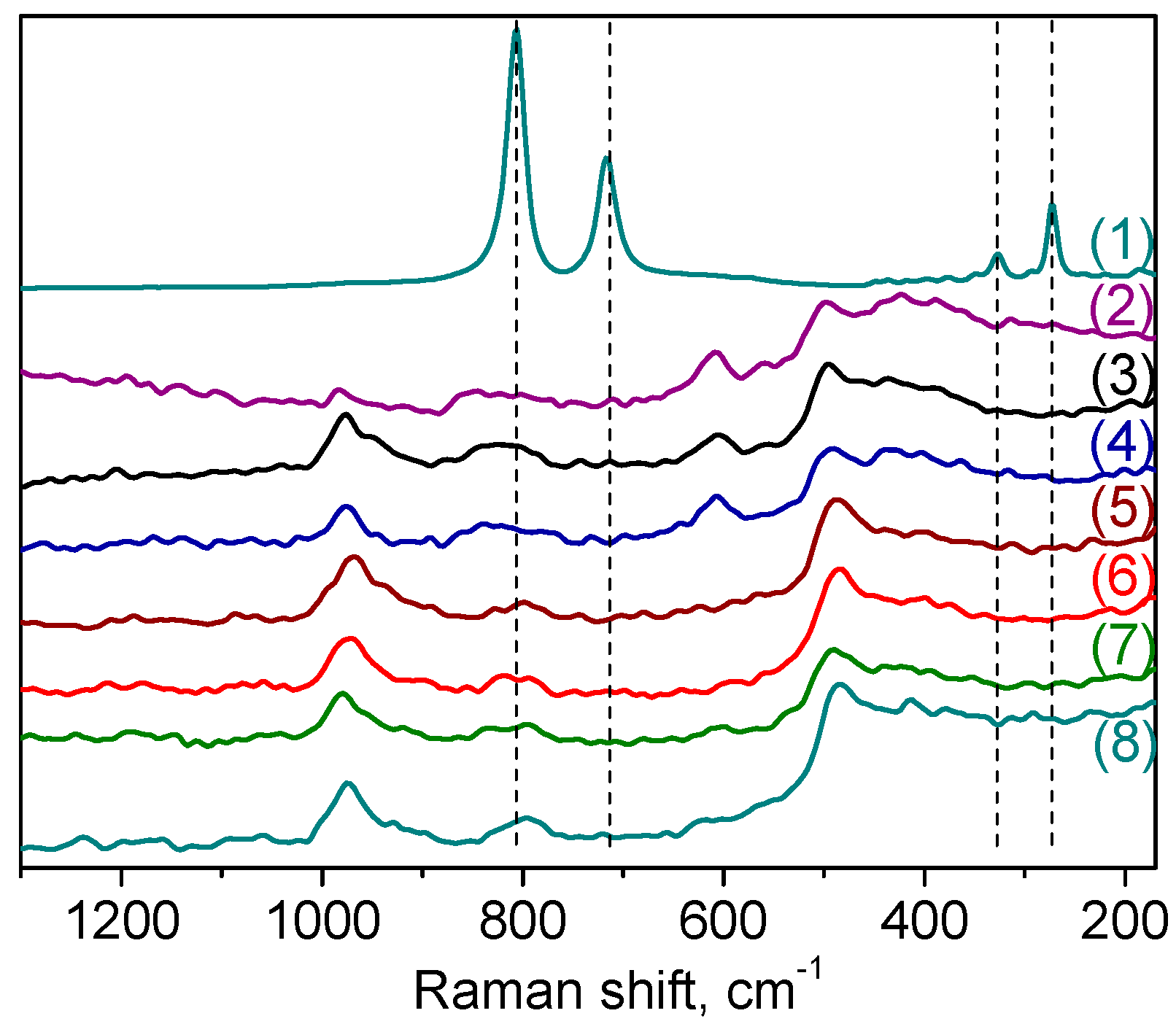
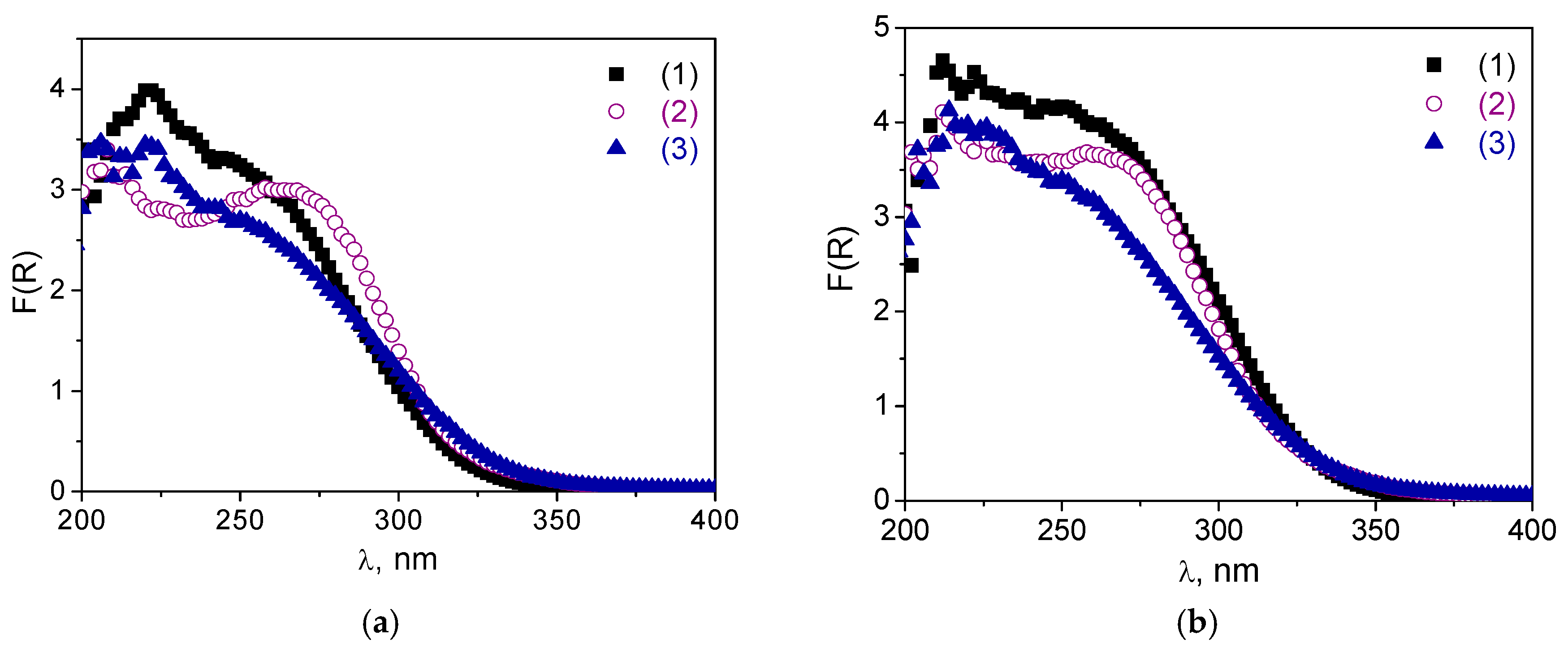
2.1. Catalyst Synthesis and Characterization
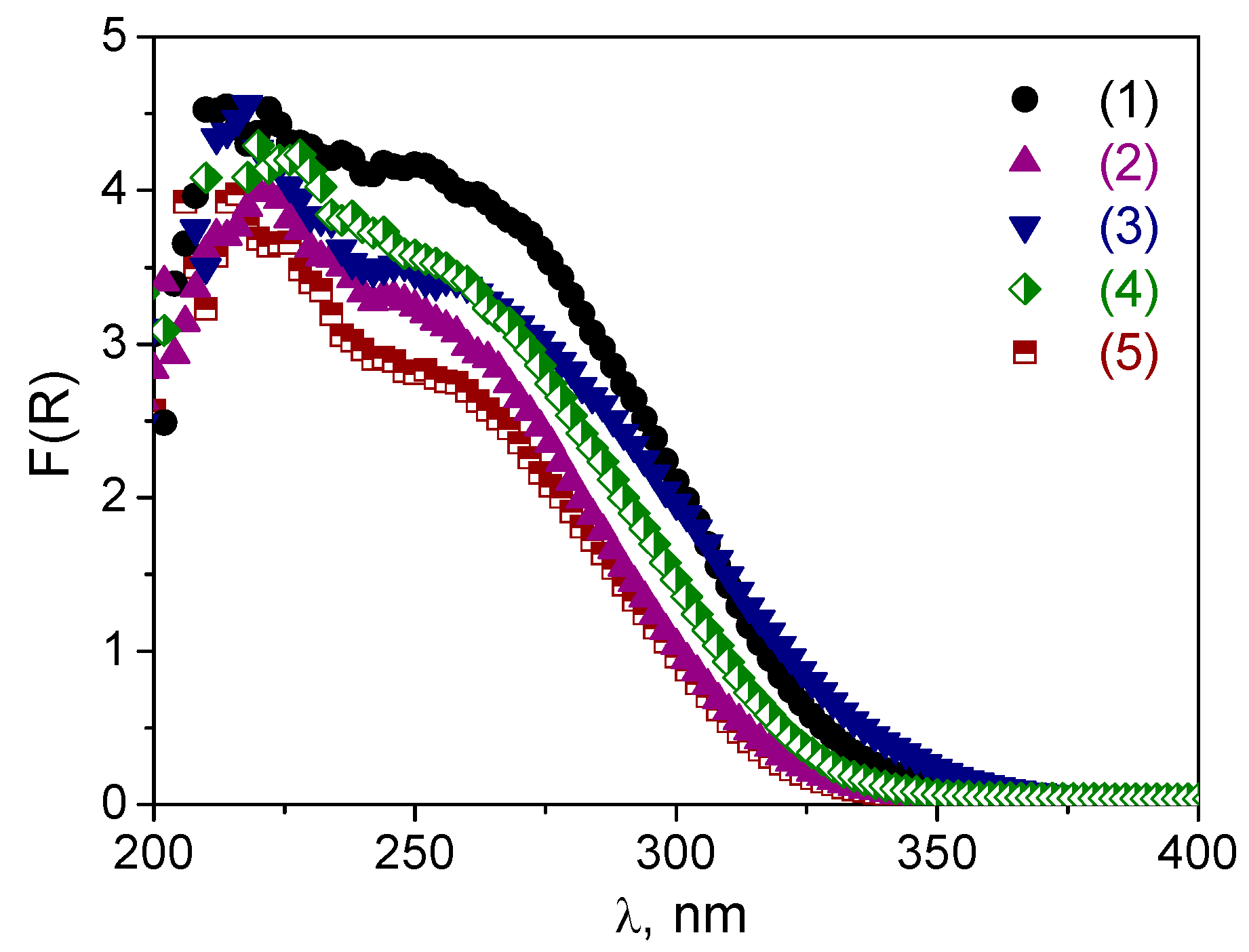
2.2. Hydrothermal Stability and Stability towards H2O2
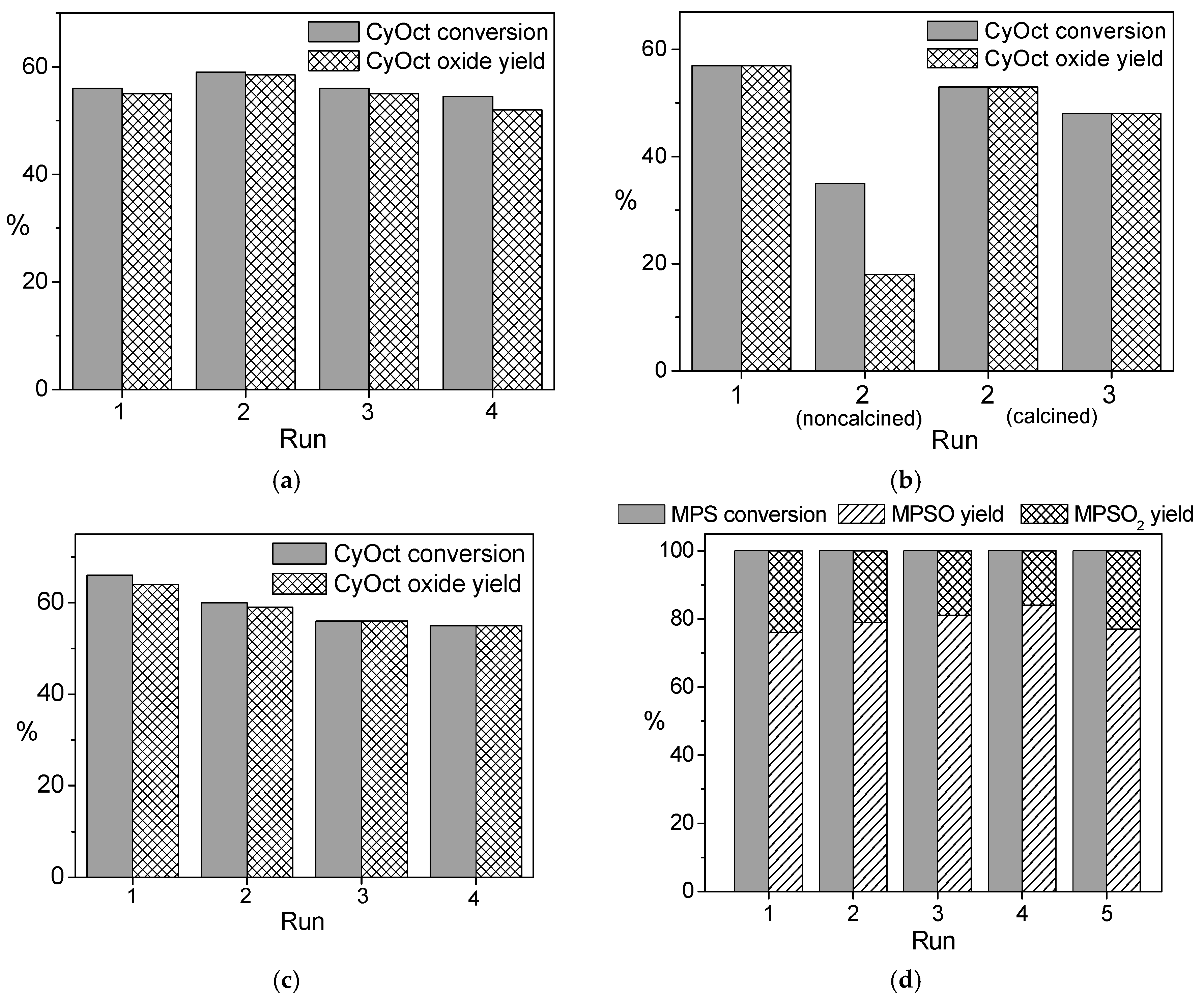
2.3. Catalytic Studies
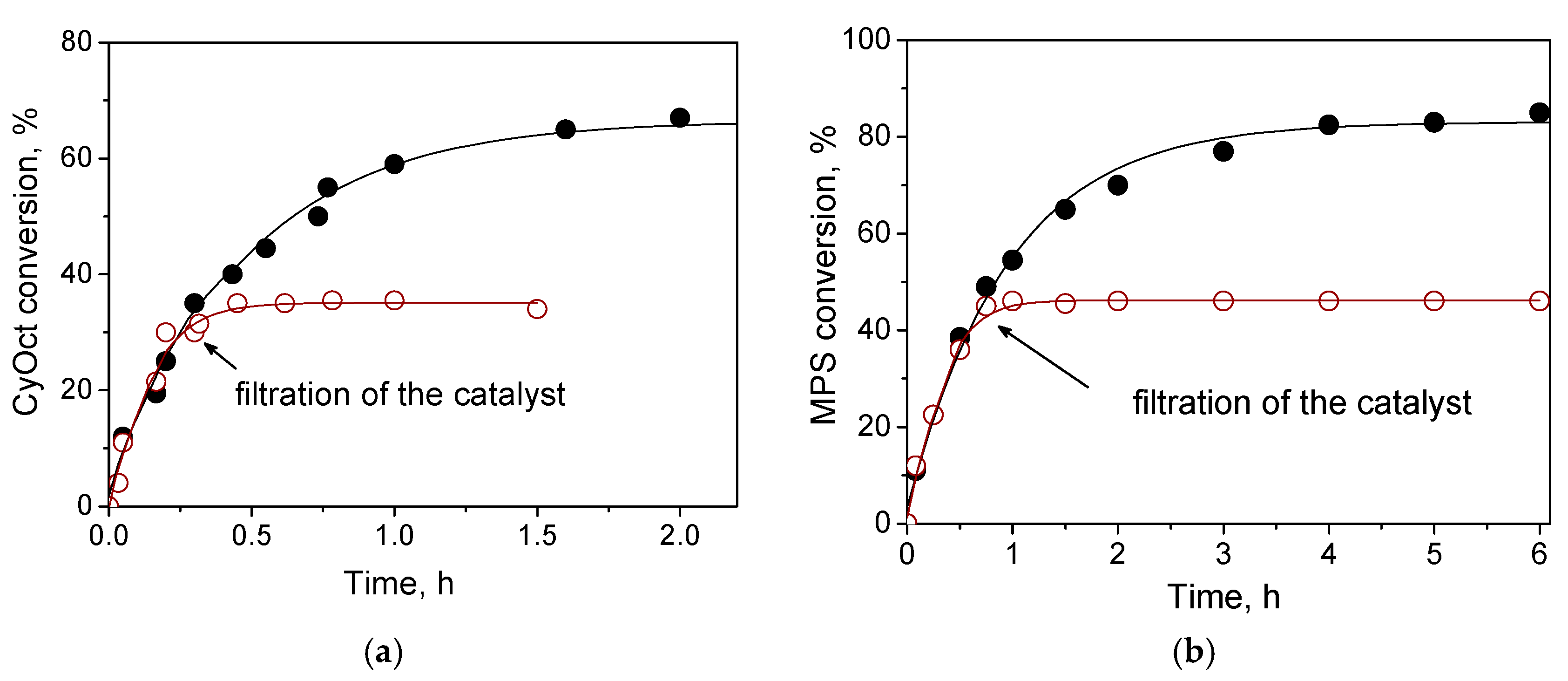
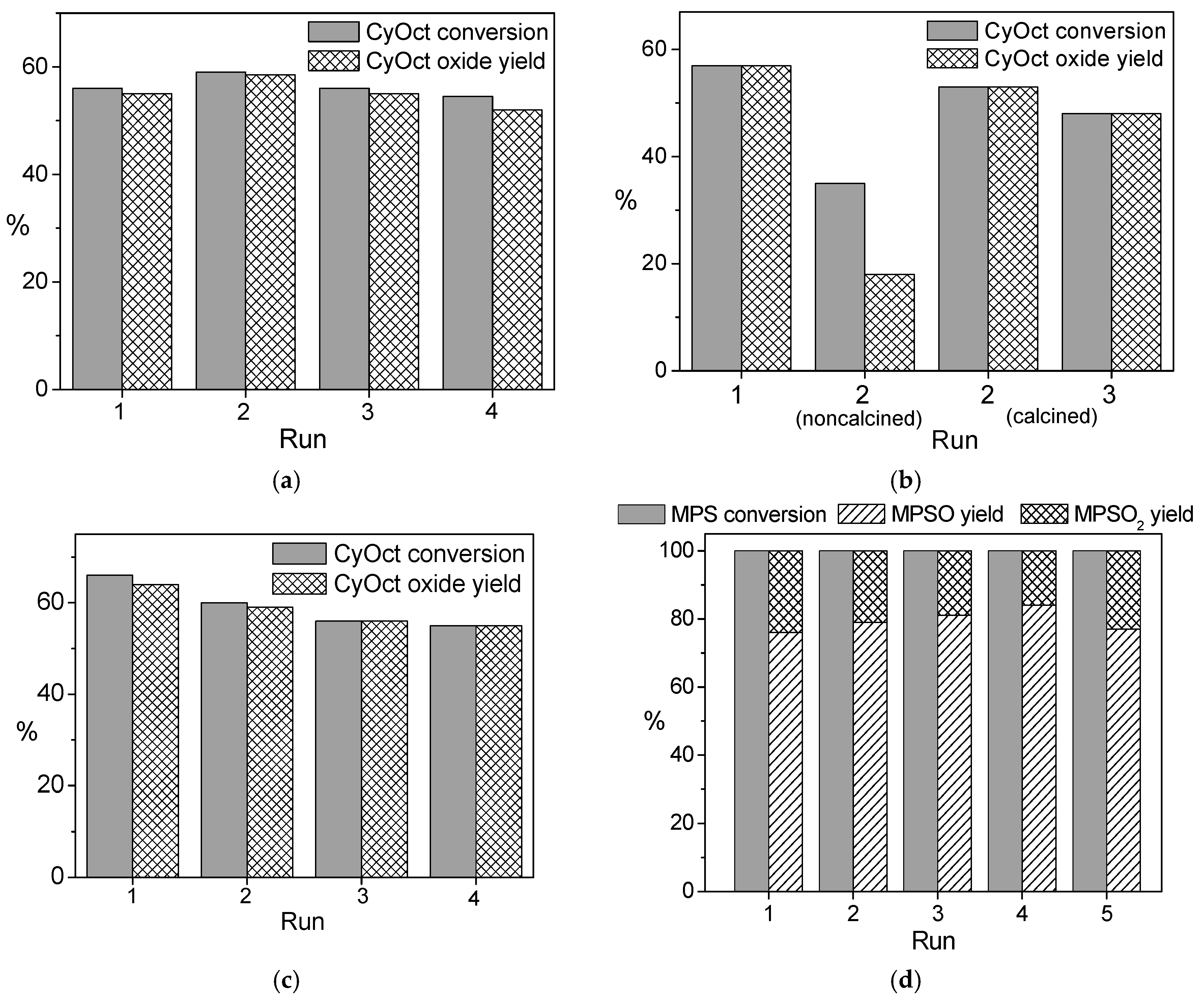
2.4. Catalyst Recyclability and Stability under Reaction Conditions
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation and Characterization
3.2.1. Method A
3.2.2. Method B
3.2.3. Method C
3.2.4. Preparation of Silica Matrix
3.3. Hydrothermal Stability and Stability towards Aqueous H2O2
3.4. Catalytic Oxidations
3.5. Instrumentation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lassner, E.; Schubert, W.-D.; Lüderitz, E.; Wolf, H.U. Tungsten, Tungsten alloys, and tungsten compounds. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar]
- Sienel, G.; Rieth, R.; Rowbottom, K.T. Epoxides. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar]
- Adolfsson, H. Transition metal-catalyzed epoxidation of alkenes. In Modern Oxidation Methods; Bäckvall, J.-E., Ed.; Wiley-VCH: Weinheim, Germany, 2004; pp. 21–49. [Google Scholar]
- Venturello, C.; D’Aloisio, R.; Bart, J.C.J.; Ricci, M. A New peroxotungsten heteropoly anion with special oxidizing properties: Synthesis and structure of tetrahexylammonium tetra(diperoxotungsto)phosphate(3-). J. Mol. Catal. 1985, 32, 107–110. [Google Scholar] [CrossRef]
- Ishii, Y.; Yamawaki, K.; Ura, T.; Yamada, H.; Yoshida, T.; Ogawa, M. Hydrogen peroxide oxidation catalyzed by heteropoly acids combined with cetylpyridinium chloride: Epoxidation of olefins and allylic acohols, ketonization of alcohols and diols, and oxidative cleavage of 1,2-diols and olefins. J. Org. Chem. 1988, 53, 3587–3593. [Google Scholar] [CrossRef]
- Hoegaerts, D.H.; Sels, B.F.; De Vos, D.E.; Verpoort, F.; Jacobs, P.A. Heterogeneous tungsten-based catalysts for the epoxidation of bulky olefins. Catal. Today 2000, 60, 209–218. [Google Scholar] [CrossRef]
- Grigoropoulou, G.; Clark, J.H.; Elings, J.A. Recent developments on the epoxidation of alkenes using hydrogen peroxide as an oxidant. Green Chem. 2003, 5, 1–7. [Google Scholar] [CrossRef]
- Brégeault, J.-M. Transition-metal complexes for liquid-phase catalytic oxidation: Some aspects of industrial reactions and of emerging technologies. Dalton Trans. 2003, 3289–3302. [Google Scholar] [CrossRef]
- Lane, B.S.; Burgess, K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 2003, 103, 2457–2474. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R.; Aoki, M.; Sato, K. Green oxidation with aqueous hydrogen peroxide. Chem. Commun. 2003, 1977–1986. [Google Scholar] [CrossRef]
- Kamata, K.; Yonehara, K.; Sumida, Y.; Yamaguchi, K.; Hikichi, S.; Mizuno, N. Efficient epoxidation of olefins with ≥99% selectivity and use of hydrogen peroxide. Science 2003, 300, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Yamaguchi, K.; Kamata, K. Epoxidation of olefins with hydrogen peroxide catalyzed by polyoxometalates. Coord. Chem. Rev. 2005, 249, 1944–1956. [Google Scholar] [CrossRef]
- Kim, D.S.; Ostromecki, M.; Wachs, I.E. Preparation and characterization of WO3/SiO2 catalysts. Catal. Lett. 1995, 33, 209–215. [Google Scholar] [CrossRef]
- Morey, M.; Bryan, J.; Schwarz, S.; Stucky, G. Pore Surface Functionalization of MCM-48 mesoporous silica with tungsten and molybdenum metal centers: Perspectives on catalytic peroxide activation. Chem. Mater. 2000, 12, 3435–3444. [Google Scholar] [CrossRef]
- Rhers, B.; Salameh, A.; Baudouin, A.; Quadrelli, E.A.; Taoufik, M.; Copéret, C.; Lefebvre, F.; Basset, J.-M.; Solans-Monfort, X.; Eisenstein, O.; et al. A well-defined, silica-supported tungsten imido alkylidene olefin metathesis catalyst. Organometallics 2006, 25, 3554–3557. [Google Scholar] [CrossRef]
- Herrera, J.E.; Kwak, J.H.; Hu, J.Z.; Wang, Y.; Peden, C.H.; Macht, J.; Iglesia, E. Synthesis, characterization, and catalytic function of novel highly dispersed tungsten oxide catalysts on mesoporous silica. J. Catal. 2006, 239, 200–211. [Google Scholar] [CrossRef]
- Le, Y.; Yang, X.; Dai, W.-L.; Gao, R.; Fan, K. Unexpected mononuclear W(VI) complexes containing phosphonate ligands anchored on mesoporous silica. Another strategy for immobilization. Catal. Commun. 2008, 9, 1838–1841. [Google Scholar] [CrossRef]
- Howell, J.G.; Li, Y.-P.; Bell, A.T. Propene metathesis over supported tungsten oxide catalysts: A study of active site formation. ACS Catal. 2016, 6, 7728–7738. [Google Scholar] [CrossRef]
- CRC Handbook of Chemistry and Physics, Internet Version 2005; Lide, D (Ed.) CRC Press: Boca Raton, FL, USA, 2005; Available online: http://www.hbcpnetbase.com (accessed on 23 June 2005).
- Kholdeeva, O.A. Selective oxidations catalyzed by mesoporous metal silicates. In Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; Clerici, M.G., Kholdeeva, O.A., Eds.; Wiley: Hoboken, NJ, USA, 2013; pp. 127–219. [Google Scholar]
- Briot, E.; Piquemal, J.-Y.; Vennat, M.; Bregeault, J.-M.; Chottard, G.; Manoli, J.-M. Aqueous acidic hydrogen peroxide as an efficient medium for tungsten insertion into MCM-41 mesoporous molecular sieves with high metal dispersion. J. Mater. Chem. 2000, 10, 953–958. [Google Scholar] [CrossRef]
- Hammond, C.; Straus, J.; Righettoni, M.; Pratsinis, S.E.; Hermans, I. Nanoparticulate tungsten oxide for catalytic epoxidations. ACS Catal. 2013, 3, 321–327. [Google Scholar] [CrossRef]
- Maheswari, R.; Pachamuthu, M.P.; Ramanathan, A.; Subramaniam, B. Synthesis, Characterization, and epoxidation activity of tungsten-incorporated SBA-16 (W-SBA-16). Ind. Eng. Chem. Res. 2014, 53, 18833–18839. [Google Scholar] [CrossRef]
- Yan, W.; Ramanathan, A.; Ghanta, M.; Subramaniam, B. Towards highly selective ethylene epoxidation catalysts using hydrogen peroxide and tungsten- or niobium-incorporated mesoporous silicate (KIT-6). Catal. Sci. Technol. 2014, 4, 4433–4439. [Google Scholar] [CrossRef]
- Brégeault, J.-M.; Piquemal, J.-Y.; Briot, E.; Duprey, E.; Launay, F.; Salles, L.; Vennat, M.; Legrand, A.-P. New approaches to anchoring or inserting highly dispersed tungsten oxo(peroxo) species in mesoporous silicates. Microporous Mesoporous Mater. 2001, 44–45, 409–417. [Google Scholar] [CrossRef]
- Zhang, Z.; Suo, J.; Zhang, X.; Li, S. Synthesis, characterization, and catalytic testing of W-MCM-41 mesoporous molecular sieves. Appl. Catal. A Gen. 1999, 179, 11–19. [Google Scholar] [CrossRef]
- Zhang, Z.; Suo, J.; Zhang, X.; Li, S. Synthesis of highly active tungsten-containing MCM-41 mesoporous molecular, sieve catalyst. Chem. Commun. 1998, 2, 241–242. [Google Scholar] [CrossRef]
- Dai, W.-L.; Chen, H.; Cao, Y.; Li, H.; Xie, S.; Fan, K. Novel economic and green approach to the synthesis of highly active W-MCM41 catalyst in oxidative cleavage of cyclopentene. Chem. Commun. 2003, 892–893. [Google Scholar] [CrossRef]
- Chen, H.; Dai, W.-L.; Deng, J.-F.; Fan, K. Novel Heterogeneous W-doped MCM-41 catalyst for highly selective oxidation of cyclopentene to glutaraldehyde by aqueous H2O2. Catal. Lett. 2002, 81, 131–136. [Google Scholar] [CrossRef]
- Trens, P.; Stathopoulos, V.; Hudson, M.J.; Pomonis, P. Synthesis and characterization of packed mesoporous tungsteno-silicates: Application to the catalytic dehydrogenation of 2-propanol. Appl. Catal. A Gen. 2004, 263, 103–108. [Google Scholar] [CrossRef]
- Klepel, O.; Bohlmann, W.; Ivanov, E.B.; Riede, V.; Papp, H. Incorporation of tungsten into MCM-41 framework. Microporous Mesoporous Mater. 2004, 76, 105–112. [Google Scholar] [CrossRef]
- Yang, X.-L.; Dai, W.-L.; Gao, R.; Chen, H.; Li, H.; Cao, Y.; Fan, K. Synthesis, characterization and catalytic application of mesoporous W-MCM-48 for the selective oxidation of cyclopentene to glutaraldehyde. J. Mol. Catal. A Chem. 2005, 241, 205–214. [Google Scholar] [CrossRef]
- Zhao, D.; Rodriguez, A.; Dimitrijevic, N.M.; Rajh, T.; Koodali, R.T. Synthesis, structural characterization, and photocatalytic performance of mesoporous W-MCM-48. J. Phys. Chem. C 2010, 114, 15728–15734. [Google Scholar] [CrossRef]
- Dimitrov, L.; Palcheva, R.; Spojakina, A.; Jiratova, K. Synthesis and characterization of W-SBA-15 and W-HMS as supports for HDS. J. Porous Mater. 2011, 18, 425–434. [Google Scholar] [CrossRef]
- Yang, X.-L.; Dai, W.-L.; Chen, H.; Cao, Y.; Li, H.; He, H.; Fan, K. Novel efficient and green approach to the synthesis of glutaraldehyde over highly active W-doped SBA-15 catalyst. J. Catal. 2005, 229, 259–263. [Google Scholar] [CrossRef]
- Bordoloi, A.; Vinu, A.; Halligudi, S.B. One-step synthesis of SBA-15 containing tungsten oxide nanoclusters: A chemoselective catalyst for oxidation of sulfides to sulfoxides under ambient conditions. Chem. Commun. 2007, 4806–4808. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-L.; Dai, W.-L.; Chen, H.; Xu, J.-H.; Cao, Y.; Li, H.; Fan, K. Novel tungsten-containing mesoporous HMS material: Its synthesis, characterization and catalytic application in the selective oxidation of cyclopentene to glutaraldehyde by aqueous H2O2. Appl. Catal. A Gen. 2005, 283, 1–8. [Google Scholar] [CrossRef]
- Ke, I.-S.; Liu, S.-T. Synthesis and catalysis of tungsten oxide in hexagonal mesoporous silicas (W-HMS). Appl. Catal. A Gen. 2007, 317, 91–96. [Google Scholar] [CrossRef]
- Gao, R.; Yanga, X.; Dai, W.-L.; Le, Y.; Li, H.; Fan, K. High-activity, single-site mesoporous WO3-MCF materials for the catalytic epoxidation of cycloocta-1,5-diene with aqueous hydrogen peroxide. J. Catal. 2008, 256, 259–267. [Google Scholar] [CrossRef]
- Su, Y.; Liu, Y.-M.; Wang, L.-C.; Chen, M.; Cao, Y.; Dai, W.-L.; He, H.-Y.; Fan, K.-N. Tungsten-containing MCF silica as active and recyclable catalysts for liquid-phase oxidation of 1,3-butanediol to 4-hydroxy-2-butanone. Appl. Catal. A Gen. 2006, 315, 91–100. [Google Scholar] [CrossRef]
- Ramanathan, A.; Maheswari, R.; Grady, B.P.; Moore, D.S.; Barich, D.H.; Subramaniam, B. Tungsten-incorporated cage-type mesoporous silicate: W-KIT-5. Microporous Mesoporous Mater. 2013, 175, 43–49. [Google Scholar] [CrossRef]
- Ramanathan, A.; Subramaniam, B.; Badloe, D.; Hanefeld, U.; Maheswari, R. Direct incorporation of tungsten into ultra-large-pore three-dimensional mesoporous silicate framework: W-KIT-6. J. Porous Mater. 2012, 19, 961–968. [Google Scholar] [CrossRef]
- Ten Dam, J.; Badloe, D.; Ramanathan, A.; Djanashvili, K.; Kapteijn, F.; Hanefeld, U. Synthesis, characterisation and catalytic performance of amesoporous tungsten silicate: W-TUD-1. Appl. Catal. A Gen. 2013, 468, 150–159. [Google Scholar] [CrossRef]
- Pope, M.T. Heteropoly- and Isopolyoxometalates; Springer: Berlin, Germany, 1983. [Google Scholar]
- Karakonstantis, L.; Kordulis, C.; Lycourghiotis, A. Mechanism of adsorption of tungstates on the interface of γ-alumina/electrolyte solutions. Langmuir 1992, 8, 1318–1324. [Google Scholar] [CrossRef]
- Salles, L.; Aubry, C.; Thouvenot, R.; Robert, F.; Doremieux-Morin, C.; Chottard, G.; Ledon, H.; Jeannin, Y.; Bregeault, J.-M. 31P and 183W NMR spectroscopic evidence for novel peroxo species in the “H3[PW12O40]∙yH2O/H202” system. Synthesis and X-ray structure of tetrabutylammonium (µ-Hydrogen phosphato)bis(µ-peroxo)bis(oxoperoxotungstate)(2-): A catalyst of olefin epoxidation in a biphase medium. Inorg. Chem. 1994, 33, 871–878. [Google Scholar]
- Ivanchikova, I.D.; Kovalev, M.K.; Mel’gunov, M.S.; Shmakov, A.N.; Kholdeeva, O.A. User-friendly synthesis of highly selective and recyclable mesoporous titanium-silicate catalysts for the clean production of substituted p-benzoquinones. Catal. Sci. Technol. 2014, 4, 200–207. [Google Scholar] [CrossRef]
- Ivanchikova, I.D.; Maksimchuk, N.V.; Skobelev, I.Y.; Kaichev, V.V.; Kholdeeva, O.A. Mesoporous niobium-silicates prepared by evaporation-induced self-assembly as catalysts for selective oxidations with aqueous H2O2. J. Catal. 2015, 332, 138–148. [Google Scholar] [CrossRef]
- Zhu, H.; Maheswari, R.; Ramanathan, A.; Subramaniam, B. Evaporation-induced self-assembly of mesoporous zirconium silicates with tunable acidity and facile catalytic dehydration activity. Microporous Mesoporous Mater. 2016, 223, 46–52. [Google Scholar] [CrossRef]
- Wu, J.-F.; Ramanathan, A.; Subramaniam, B. Novel tungsten-incorporated mesoporous silicates synthesized via evaporation-induced self-assembly: Enhanced metathesis performance. J. Catal. 2017, 350, 182–188. [Google Scholar] [CrossRef]
- Ramanathan, A.; Wu, J.-F.; Maheswari, R.; Hu, Y.; Subramaniam, B. Synthesis of molybdenum-incorporated mesoporous silicates by evaporation-induced self-assembly: Insights into surface oxide species and corresponding olefin metathesis activity. Microporous Mesoporous Mater. 2017, 245, 118–125. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Recent developments in liquid-phase selective oxidation using environmentally benign oxidants and mesoporous metal silicates. Catal. Sci. Technol. 2014, 4, 1869–1889. [Google Scholar] [CrossRef]
- Baxter, D.V.; Chisolm, M.H.; Doherty, S.; Gruhn, N.E. Chemical vapour deposition of electrochromic tungsten oxide films employing volatile tungsten(VI) oxo alkoxide/β-diketonate complexes. Chem. Commun. 1996, 1129–1130. [Google Scholar] [CrossRef]
- Choujaa, H.; Johnson, A.L.; Kociok-Köhn, G.; Molloy, K.C. Synthesis of heterobimetallic tungsten acetylacetonate/alkoxide complexes and their application as molecular precursors to metal tungstates. Polyhedron 2013, 59, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Kustova, G.N.; Chesalov, Y.A.; Plyasova, L.M.; Molina, I.Y.; Nizovskii, A.I. Vibrational spectra of WO3·nH2O and WO3 polymorphs. Vib. Spectrosc. 2011, 55, 235–240. [Google Scholar] [CrossRef]
- Graselli, J.G.; Bulkin, B.J. Analytical Raman Spectroscopy; Wiley: New York, NY, USA, 1991; p. 352. [Google Scholar]
- Griffith, W.P.; Wickins, T.D. Raman studies on species in aqueous solutions. Part II. Oxy-species of metals of Groups VIA, VA, and IVA. J. Chem. Soc. A 1967, 675–679. [Google Scholar] [CrossRef]
- Mączka, M.; Hanuza, J.; Waśkowska, A. Vibrational studies of alkali metal hexatungstates. J. Raman Spectrosc. 2003, 34, 432–437. [Google Scholar] [CrossRef]
- Ivanchikova, I.D.; Skobelev, I.Y.; Maksimchuk, N.V.; Paukshtis, E.A.; Shashkov, M.V.; Kholdeeva, O.A. Toward understanding the unusual reactivity of mesoporous niobium silicates in epoxidation of C=C bonds with hydrogen peroxide. J. Catal. 2017, 356, 85–99. [Google Scholar] [CrossRef]
- Maurya, M.R.; Kumar, M.; Sikarwar, S. Polymer-anchored oxoperoxo complexes of vanadium(V), molybdenum(VI) and tungsten(VI) as catalyst for the oxidation of phenol and styrene using hydrogen peroxide as oxidant. React. Funct. Polym. 2006, 66, 808–818. [Google Scholar] [CrossRef]
- Adam, F.; Iqbal, A. The liquid phase oxidation of styrene with tungsten modified silica as a catalyst. Chem. Eng. J. 2011, 171, 1379–1386. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Wallau, M.; Arends, I.W.C.E.; Schuchardt, U. Heterogeneous Catalysts for Liquid-Phase Oxidations: Philosophers’ Stones or Trojan Horses? Acc. Chem. Res. 1998, 31, 485–493. [Google Scholar] [CrossRef]
- Adachi, K.; Toyomura, S.; Miyakuni, Y.; Yamazaki, S.; Honda, K.; Hirano, T. Dioxomolybdenum(VI) and dioxotungsten(VI) complexes: Efficient catalytic activity for crosslinking reaction in ethylene-vinyl acetate copolymer/alkoxysilane composites. Polym. Adv. Technol. 2015, 26, 597–605. [Google Scholar] [CrossRef]
- Buono-Core, G.E.; Klahn, A.H.; Castillo, C.; Bustamante, M.J.; Munoz, E.; Cabello, G.; Chornik, B. Synthesis and evaluation of bis-β-diketonate dioxotungsten(VI) complexes as precursors for the photodeposition of WO3 films. Polyhedron 2011, 30, 201–206. [Google Scholar] [CrossRef]
- Yu, S.; Holm, R.H. Aspects of the oxygen atom transfer chemistry of tungsten. Inorg. Chem. 1989, 28, 4385–4391. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Si/W a | Wcalc b, wt % | Wexp c, wt % | SBET, m2/g | Vp d, cm3/g | Dp e, nm |
|---|---|---|---|---|---|---|
| A-1 | 278 | 1.0 | 0.9 | 1130 | 0.57 | 2.48 |
| 1180 f | 0.50 | 2.49 f | ||||
| A-2 | 143 | 2.0 | 1.6 | 1440 | 0.61 | 2.51 |
| B-1 | 294 | 1.0 | 0.9 | 1240 | 0.51 | 2.54 |
| B-1.4 | 208 | 1.4 | 1.4 | 1140 | 0.49 | 2.56 |
| C-1 | 294 | 1.0 | 0.8 | 1170 | 0.46 | 2.41 |
| 1180 f | 0.49 | 2.45 f |
| Entry | Catalyst | Oxidant | Time, h | Substrate Conversion, % | TOF a, h−1 | CyOct Oxide Selectivity, % | W Leaching, ppm |
|---|---|---|---|---|---|---|---|
| 1 | - | 30% H2O2 | 1.5 | 3 | - | 17 | - |
| 2 | A-2 | 30% H2O2 | 2 | 74 | 53 | >99 | 55 |
| 3 | 50% H2O2 | 2 | 61 | 54 | >99 | 36 | |
| 4 | A-1 | 30% H2O2 | 2 | 66 | 53 | 98 b | 55 |
| 5 | 50% H2O2 | 2 | 56 | 54 | 98 b | 24 | |
| 6 | B-1.4 | 30% H2O2 | 2 | 65 | 63 | 94 b | 50 |
| 7 | 50% H2O2 | 2 | 60 | 65 | 93 b | 52 | |
| 8 | B-1 | 30% H2O2 | 2 | 62 | 58 | 98 b | 45 |
| 9 | 50% H2O2 | 2 | 57 | 65 | >99 | 17 | |
| 10 | C-1 | 30% H2O2 | 2 | 68 | 66 | 97 b | 58 |
| 11 | 50% H2O2 | 2 | 66 | 64 | 96 b | 20 | |
| 12 | WO2(acac)2 | 30% H2O2 | 2 | 85 | 95 | 99 b | - |
| 13 | WO3 | 30% H2O2 | 2.5 | 14 | 13 | 43 | - |
| 14 | SiO2-EISA c | 30% H2O2 | 2 | 12 | - | 8 | - |
| Entry | Substrate | Catalyst | Time, h | Substrate Conversion, % | Epoxide Selectivity, % |
|---|---|---|---|---|---|
| 1 |  | A-1 | 5 | 28 | 82 |
| 2 | C-1 | 5 | 29 | 85 | |
| 3 | 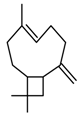 | A-1 | 3 | 77 | 99 |
| 4 | C-1 | 3 | 80 | >99 | |
| 5 | 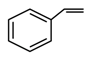 | A-1 a | 3 | 23 | 45 b |
| 6 | C-1 a | 3 | 22 | 53 c | |
| 7 | Nb-MMM-E d | 3 | 26 | 39 e | |
| 8 | Ti-MMM-E d | 4 | 33 | 46 f | |
| 9 | 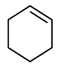 | A-1 a | 1.5 | 32 | 81 g |
| 10 | C-1 a | 1 | 30 | 83 h | |
| 11 | Nb-MMM-E d | 0.5 | 30 | 70 i | |
| 12 | Ti-MMM-E d | 2.5 | 35 | 65 i |
| Entry | Catalyst | MPS Conversion, % | MPSO Yield a, % | MPSO2 Yield a, % | H2O2 Efficiency, % |
|---|---|---|---|---|---|
| 1 | A-1 | 95 | 81 (85) | 14 (15) | 91 |
| 2 | B-1.4 | 99 | 80 (82) | 19 (18) | 98 |
| 3 | B-1 | 98 | 80 (82) | 18 (18) | 97 |
| 4 | C-1 | 96 | 85 (89) | 11 (11) | 89 |
| 5 | WO2(acac)2 | 100 | 79 (79) | 21 (21) | 100 |
| 6 | SiO2-EISAb | 7 | 7 (100) | 0 (0) | - |
| Entry | Sulfide | Conversion, % | Sulfoxide | Selectivity a, % | H2O2 efficiency, % |
|---|---|---|---|---|---|
| 1 | 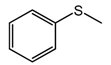 | 91 | 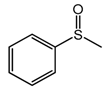 | 91 | 99 |
| 2 b | | 96 | | 89 | 89 |
| 3 | 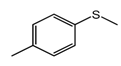 | 84 |  | 88 | 94 |
| 4 |  | 85 | 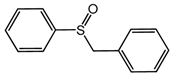 | 84 | 97 |
| 5 c |  | 68 | 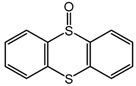 | 59 | 100 |
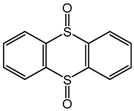 | 35 | ||||
| 6 d | | 86 | | 95 | 90 |
| Entry | Catalyst | MPS Conversion, % | MPSO Yield a, % | MPSO2 Yield a, % | H2O2 Efficiency, % | TOF, h−1 |
|---|---|---|---|---|---|---|
| 1 | C-1 | 91 | 83 (91) | 8 (9) | 99 | 120 |
| 2 | Ti-MMM-E | 82 | 67 (82) | 15 (18) | 97 | 45 |
| 3 | Nb-MMM-E | 83 | 67 (81) | 16 (19) | 99 | 90 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maksimchuk, N.; Ivanchikova, I.; Zalomaeva, O.; Chesalov, Y.; Shmakov, A.; Zaikovskii, V.; Kholdeeva, O. Tungsten-Based Mesoporous Silicates W-MMM-E as Heterogeneous Catalysts for Liquid-Phase Oxidations with Aqueous H2O2. Catalysts 2018, 8, 95. https://doi.org/10.3390/catal8030095
Maksimchuk N, Ivanchikova I, Zalomaeva O, Chesalov Y, Shmakov A, Zaikovskii V, Kholdeeva O. Tungsten-Based Mesoporous Silicates W-MMM-E as Heterogeneous Catalysts for Liquid-Phase Oxidations with Aqueous H2O2. Catalysts. 2018; 8(3):95. https://doi.org/10.3390/catal8030095
Chicago/Turabian StyleMaksimchuk, Nataliya, Irina Ivanchikova, Olga Zalomaeva, Yurii Chesalov, Alexandr Shmakov, Vladimir Zaikovskii, and Oxana Kholdeeva. 2018. "Tungsten-Based Mesoporous Silicates W-MMM-E as Heterogeneous Catalysts for Liquid-Phase Oxidations with Aqueous H2O2" Catalysts 8, no. 3: 95. https://doi.org/10.3390/catal8030095
APA StyleMaksimchuk, N., Ivanchikova, I., Zalomaeva, O., Chesalov, Y., Shmakov, A., Zaikovskii, V., & Kholdeeva, O. (2018). Tungsten-Based Mesoporous Silicates W-MMM-E as Heterogeneous Catalysts for Liquid-Phase Oxidations with Aqueous H2O2. Catalysts, 8(3), 95. https://doi.org/10.3390/catal8030095