
MIKC-Type MADS-Box Gene Family Discovery and Evolutionary Investigation in Rosaceae Plants

Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Family Identification
2.2. Sequence Analysis
2.3. Phylogenetic and Conservative Analyses
2.4. Selective Evolutionary Pressure Analysis
2.5. Functional Difference Analyses
3. Results
3.1. Phylogenetic and Sequence Analyses
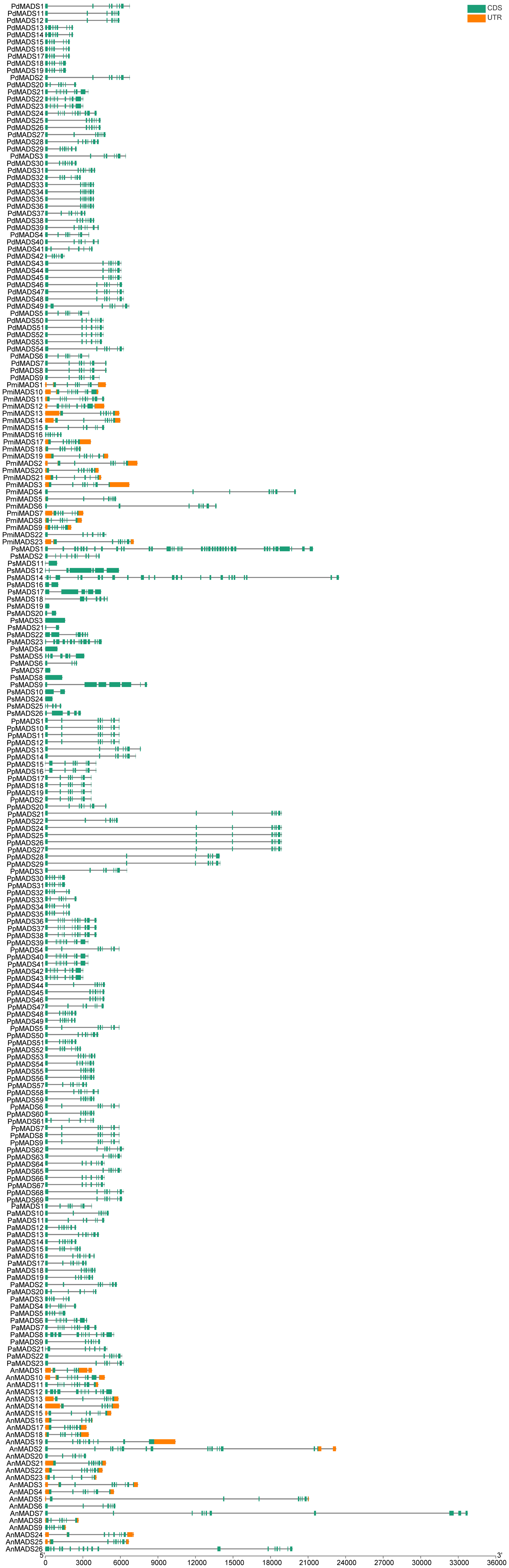
3.2. Motif and Structure Analyses
3.3. Selective Pressure Analysis
3.4. Functional Divergence Analysis
3.5. Functional Distance Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.; Mu, Y.X.; Wang, J. Research progress of floral development regulation by MADS-box gene family. Acta Agric. Zhejiangensis 2021, 33, 1149–1158. [Google Scholar]
- Kim, S.H.; Mizuno, K.; Fujimura, T. Isolation of MADS-box genes from sweet potato [Ipomoea batatas (L.) Lam.] expressed specifically in vegetative tissues. Plant Cell Physiol. 2002, 43, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.B.; Jia, H.M.; Wang, Y.; Wang, G.Y.; Zhou, C.C.; Jia, H.J.; Gao, Z.S. Genome-wide identification and analysis of the MADS-box gene family and its potential role in fruit development and ripening in red bayberry (Morella rubra). Gene 2019, 717, 144045. [Google Scholar] [CrossRef]
- Yanofsky, M.F.; Ma, H.; Bowman, J.L.; Drews, G.N.; Feldmann, K.A.; Meyerowitz, E.M. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature 1990, 346, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Schwarzsommer, Z.; Hue, I.; Huijser, P.; Flor, P.J.; Hansen, R.; Tetens, F.; Lonnig, W.E.; Saedler, H.; Sommer, H. Characterization of the antirrhinum floral homeotic MADS-box gene deficiens-evidence for DNA-bingding and autoregulation of its persistent expression throughout flower development. EMBO J. 1992, 11, 251–263. [Google Scholar] [CrossRef]
- Dreni, L.; Zhang, D. Flower development: The evolutionary history and functions of the AGL6 subfamily MADS-box genes. J. Exp. Bot. 2016, 67, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Buylla, E.R.; Pelaz, S.; Liljegren, S.J.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Pouplana, L.R.D.; Martinez-Castilla, L.; Yanofsky, M.F. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. USA 2000, 97, 5328–5333. [Google Scholar] [CrossRef] [Green Version]
- Riechmann, J.L.; Meyerowttz, E.M. MADS domain proteins in plant development. Biol. Chem. 1997, 378, 1079–1101. [Google Scholar]
- Zhao, X.Y.; Xian, D.Y.; Song, M.; Tang, Q.L. Research progress of MIKC-type MADS-box protein regulation on flowering. Biotechnol. Bull. 2014, 7, 8–15. [Google Scholar]
- Susanne, S.; Alice, K.; Sirui, P.; Lars, S.J.; Rainer, M. Genome-wide analysis of MIKC-type MADS-box genes in wheat: Pervasive duplications, functional conservation and putative neofunctionalization. New Phytol. 2020, 225, 511–529. [Google Scholar]
- Chunmei, H.; Can, S.; Jaime, A.T.S.; Li, M.Z.; Duan, J. Genome-wide identification and classification of MIKC-type MADS-box genes in Streptophyte lineages and expression analyses to reveal their role in seed germination of orchid. BMC Plant Biol. 2019, 19, 223. [Google Scholar]
- Jiang, S.C.; Pang, C.Y.; Song, M.Z.; Wei, H.L.; Fan, S.-l.; Yu, S.X. Analysis of MIKCc-type MADS-box gene family in Gossypium hirsutum. J. Integr. Agric. 2014, 13, 1239–1249. [Google Scholar] [CrossRef]
- Gao, W.; Zhang, L.; Xue, C.L.; Zhang, Y.; Liu, M.J.; Zhao, J. Expression of E-type MADS-box genes in flower and fruits and protein interaction analysis in Chineses Jujube. Acta Hortic. Sin. 2022, 49, 739–748. [Google Scholar]
- Huang, H.J.; Wang, S.; Jiang, J.; Liu, G.; Li, H.; Chen, S.; Xu, H. Overexpression of BpAP1 induces early flowering and produces dwarfism in Betula plantyphylla × Betula pendula. Physiol. Plant 2014, 151, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Qi, T.; Ma, J.; Ma, L.; Liu, X. Cloning and functional analysis of SEP-like gene from Phyllostachys violascens. J. Nucl. Agric. Sci. 2016, 30, 1453–1459. [Google Scholar]
- Parenicová, L.; Folter, S.D.; Kieffer, M.; Horner, D.S.; Colombo, L. Molecular and phylogenetic Analyses of the complete MADS-box transcription factor family in Arabidopsis new openings to the MADS World. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L. Sequencing of allotetraploid cotton(Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Prakash, A.; Jeffryes, M.; Bateman, A.; Finn, R.D. The HMMER Web server for protein sequence similarity search. Curr. Protoc. Bioinform. 2017, 60, 3–15. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. A simple method to control over-alignment in the MAFFT multiple sequence alignment program. Bioinformatics 2016, 32, 1933–1942. [Google Scholar] [CrossRef] [Green Version]
- Timothy, L.B.; James, J.; Charles, E.G.; William, S.N. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. Prottest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhir, K.; Glen, S.; Michael, L.; Christina, K.; Koichiro, T. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar]
- Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Yu, G.; Lam, T.T.Y.; Zhu, H.; Guan, Y. Two methods for mapping and visualizing associated data on phylogeny using ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.H. PAML4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.H.; Nielsen, R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J. Mol. Evol. 1998, 46, 409–418. [Google Scholar] [CrossRef]
- Yang, Z.H.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual siyes along specific lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.H.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Zou, Y.Y.; Su, Z.X.; Huang, W.; Zhou, Z.; Arendsee, Z.; Zeng, Y.W. An update of DIVERGE software for functional divergence analysis of protein family. Mol. Biol. Evol. 2013, 30, 1713–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Velden, K.V. DIVERGE: Phylogeny-based analysis for functional-structural divergence of a protein family. Bioinformatics 2002, 18, 500–501. [Google Scholar] [CrossRef] [Green Version]
- Gu, X. A simple statistical method for estimating type-II(cluster-specific) functional divergence of protein sequences. Mol. Biol. Evol. 2006, 23, 1937–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.F.; Gu, X. Functional divergence in the caspase gene family and altered funcional constrains: Statistical analysis and prediction. Genetics 2001, 158, 1311–1320. [Google Scholar] [CrossRef]
- Christina, E.W.; Elisa, V.; Sergio, J.T.; Lgnazio, V.; Douglas, G.B. A genome-wide analysis of MADS-box genes in peach [Prunus persica (L.) Batsch]. BMC Plant Biol. 2015, 15, 41. [Google Scholar]
- Hisayo, Y.; Tomomi, O.; Hiroaki, J.; Yukari, H.; Ryuta, S.; Ryutaro, T. Expressional regulation of PpDAM5 and PpDAM6, peach (Prunus persica) dormancy-associated MADS-box genes, by low temperature and dormancy-breaking reagent treatmen. J. Exp. Bot. 2011, 62, 3481–3488. [Google Scholar]
- Liu, X.Y.; He, Z.J.; Qiu, Y.M. Screening and Bioinformatics analysis of almond MADS-box Gene family. Mol. Plant Breed. 2022, 20, 1477–1486. [Google Scholar]
- Chen, C.; Zhu, G.P.; Zhao, H.; Liu, H.M.; Luo, Y.; Xu, W.Y.; Huang, M.Z.; Wu, Y.T.N.; Wang, L. Genome-wide identification of MADS-box Gene family and expression analysis in Prunus sibirica. Mol. Plant Breed. 2020, 18, 6575–6585. [Google Scholar]
- Zhang, Y.; Wang, J.; Yu, Z.; Xu, Q.; Zhang, L.; Pan, Y. Bioinformatics analysis of MIKC-type MADS-box gene family in legumes. Chin. J. Oil Crop Sci. 2022, 44, 798–809. [Google Scholar]
- Wu, K.L.; Guo, Z.J.; Wang, H.H.; Li, J. The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res. 2005, 12, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yv, T.; Lu, L.; Ku, T.; Li, C.; Chen, S. Flora Reipublicae Popularis Sinicae; Science Press: Beijing, China, 1986; Volume 38, pp. 11–40. [Google Scholar]
- Vallender, E.J.; Lahn, B.T. Positive selection on the human genome. Hum. Mol. Genet. 2004, 13, R245–R254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.Q.; Zhu, K.Y.; Shi, X.H. Adaptive evolution and identification analysis of the MADS-box gene family in Paeonia lactiflora. Mol. Plant Breed. 2019, 17, 6959–6966. [Google Scholar]
- Tian, Y.X.; Wang, Q.G.; Zhang, H.; Zhou, N.N.; Yan, H.J.; Jian, H.Y.; Li, G.S.; Tang, K.X.; Qiu, X.Q. Genome-wide identification and evolutionary analysis of MLO gene family in Rosaceae plants. Hortic. Plant J. 2022, 8, 110–122. [Google Scholar] [CrossRef]
- Ma, S.; Wang, C.; Sun, F.; Wei, B.; Nie, Y. Genetic diversity of an endangered plant Amygdalus ledebouriana in Xinjiang. Sci. Silvae Sin. 2019, 9, 71–80. [Google Scholar]
- Yan, G.; Xu, Z. Study on the wild fruit tree diseases of Tianshan mountains and their distribution in Xinjiang. Arid. Zone Res. 2001, 18, 47–49. [Google Scholar]
- Wu, Y.M.; Wu, Y.A.; Wu, Y.X.; Cao, Z. The advances of the studies on almond (Amygdalus communis L.) A literature review. J. Gansu Agric. Univ. 1996, 1, 86–92. [Google Scholar]

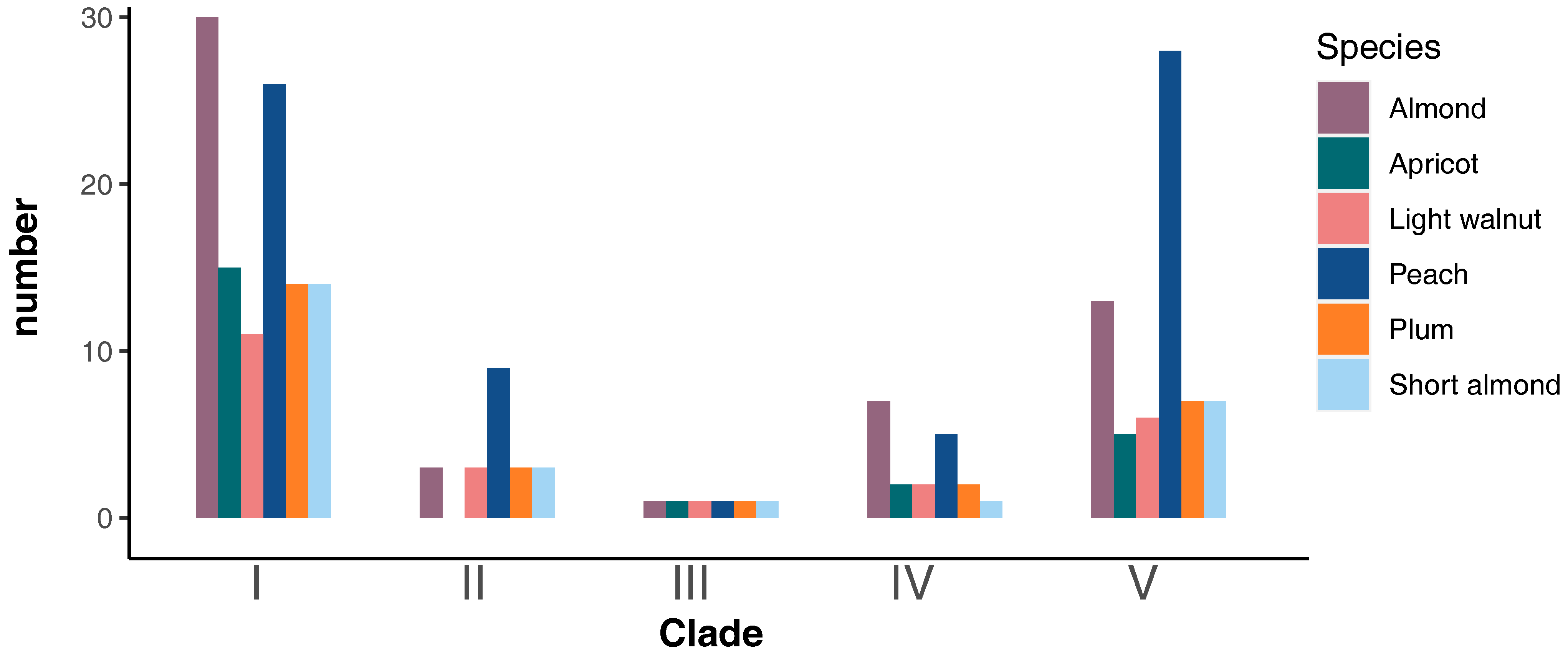



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Common Name | MIKC-Type MADS-Box Gene Numbers | Clade | ||||
|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | |||
| Amygdalus nana | Short almond | 26 | 14 | 3 | 1 | 1 | 7 |
| Prunus mira | Light walnut | 23 | 11 | 3 | 1 | 2 | 6 |
| Prunus persica | Peach | 69 | 26 | 9 | 1 | 5 | 28 |
| Prunus armeniaca | Apricot | 23 | 15 | 0 | 1 | 2 | 5 |
| Prunus salicina | Plum | 27 | 14 | 3 | 1 | 2 | 7 |
| Prunus dulcis | Almond | 54 | 30 | 3 | 1 | 7 | 13 |
| Total | 222 | 110 | 21 | 6 | 19 | 66 | |
| Model | np/aa | ln L | Parameters Estimates | Compared Model | p-Value | ||
|---|---|---|---|---|---|---|---|
| Two ratio Model 2 | 448 | −3172.993 | ω: | ω0 = 1.533 | ω1 = 0.768; ω2 = 999.000; ω3 = 0.862; ω4 = 2.208; ω5 = 2.452 | Model 0 vs. Two ratio Model 2 | >0.05 |
| Model 0 | 443 | −3175.884 | ω= | 1.558 | |||
| Model | np/aa | ln L | Parameters Estimates | Compared Model | p-Value | Positive Sites | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Model A | 446 | −3166.858 | Site class | 0 | 1 | 2a | 2b | Model A vs. Model A null | >0.05 | Not allowed |
| f | 0.000 | 0.000 | 0.156 | 0.844 | ||||||
| ω0 | 0.229 | 1.000 | 0.229 | 1.000 | ||||||
| ω1 | 0.229 | 1.000 | 999.000 | 999.000 | ||||||
| Model A null | 445 | −3165.206 | 1 | Not allowed | ||||||
| Model | np | ln L | Parameters Estimates | Compared Model | p-Value | Positive Selected Sites |
|---|---|---|---|---|---|---|
| M0 | 443 | −3144.848 | ω = 0.318 | M0 vs. M3 | <0.001 | Not allowed |
| M3 | 447 | −3054.877 | p: 0.266, 0.519, 0.216 | Not allowed | ||
| ω: 0.095, 0.432, 0.955 | ||||||
| M1a | 444 | −3071.511 | p: 0.330, 0.670 | M1a vs. M2a | <0.001 | Not allowed |
| ω: 0.193, 1.000 | ||||||
| M2a | 446 | −3062.176 | p: 0.289, 0.528, 0.183 | Not allowed | ||
| ω: 0.204, 1.000, 2.045 | ||||||
| M7 | 444 | −3055.573 | p = 0.760, q = 1.042 | M7 vs. M8 | >0.05 | Not allowed |
| M8 | 446 | −3053.688 | p0 = 0.778 | 11 E 0.616 | ||
| p = 0.852 |
| Clade | θ ± SE | MFE z-Scores | p-Value | Amino Acid Position with Q(k) > 0.9 | ||
|---|---|---|---|---|---|---|
| Type I | Type II | Type I | Type II | |||
| I/II | 0.579 ± 0.156 | −0.112 ±0.189 | −4.115715 | <0.001 | >0.05 | — |
| I/III | 0.871 ± 0.158 | 0.005 ± 0.182 | −6.191108 | <0.001 | >0.05 | 193,299,307,316.332,341,343, 346,359,370,371,374,386,397,400,403,407 |
| I/IV | 0.327 ± 0.225 | −0.081 ± 0.207 | −1.505518 | >0.001 | >0.05 | 190,301,302,310,311,316.346,349.361, 388,403 |
| I/V | −0.002 ± 0.263 | −0.660 ± 0.379 | 0.009046 | >0.001 | >0.05 | — |
| II/III | 1.058 ± 0.159 | 0.299 ± 0.050 | −7.305373 | <0.001 | >0.05 | 190,192,193,195,272,273,290,292,295, 297,298,299,300,301,303,305,307,314, 316,318,320,321,332,342,343,345,347, 349,356,358,359,366,370,371,373,374, 385,386,387,395,396,397,403,404,405, 406,409,411 |
| II/IV | 0.874 ± 0.239 | 0.193 ± 0.070 | −3.826513 | <0.001 | >0.05 | 193,271,272,290,296,298,299,301,302, 305,310,311,313,316,319,320,331,341, 347,350,352,361,370,371,373,382,388, 394,397,403,409 |
| II/V | 0.831 ± 0.288 | −0.221 ± 0.203 | −2.974702 | >0.001 | >0.05 | — |
| III/IV | 1.085 ± 0.232 | 0.232 ± 0.063 | −4.882459 | <0.001 | >0.05 | 190,193,195,271,292,296,301,302,307, 310,311,313,314,319,321,331,332,341, 343,345,347,349,350,352,358,359,361, 374,382,386,387,388,394,395,404,406 |
| III/V | 1.150 ± 0.279 | −0.233 ± 0.205 | −4.220686 | <0.001 | >0.05 | — |
| IV/V | 0.205 ± 0.410 | −0.425 ± 0.248 | −0.50314 | >0.001 | >0.05 | — |
| Clade | bF |
|---|---|
| I | 0.405 |
| II | −1.270 |
| III | −2.453 |
| IV | −0.801 |
| V | −0.403 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, Y.; Zhu, G.; Li, F.; Wang, L.; Chen, C.; Zhao, H. MIKC-Type MADS-Box Gene Family Discovery and Evolutionary Investigation in Rosaceae Plants. Agronomy 2023, 13, 1695. https://doi.org/10.3390/agronomy13071695
Qin Y, Zhu G, Li F, Wang L, Chen C, Zhao H. MIKC-Type MADS-Box Gene Family Discovery and Evolutionary Investigation in Rosaceae Plants. Agronomy. 2023; 13(7):1695. https://doi.org/10.3390/agronomy13071695
Chicago/Turabian StyleQin, Yue, Gaopu Zhu, Fangdong Li, Lin Wang, Chen Chen, and Han Zhao. 2023. "MIKC-Type MADS-Box Gene Family Discovery and Evolutionary Investigation in Rosaceae Plants" Agronomy 13, no. 7: 1695. https://doi.org/10.3390/agronomy13071695