

Comparative Transcriptomic Insights into the Mechanisms Underlying Maize (Zea mays L.) Embryogenic Callus Differentiation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Trait Evaluation
2.2. Transcriptome Sequencing
2.3. Transcriptome Data Analysis
2.4. Quantitative Real-Time PCR Validation
3. Results
3.1. Phenotypic Variation
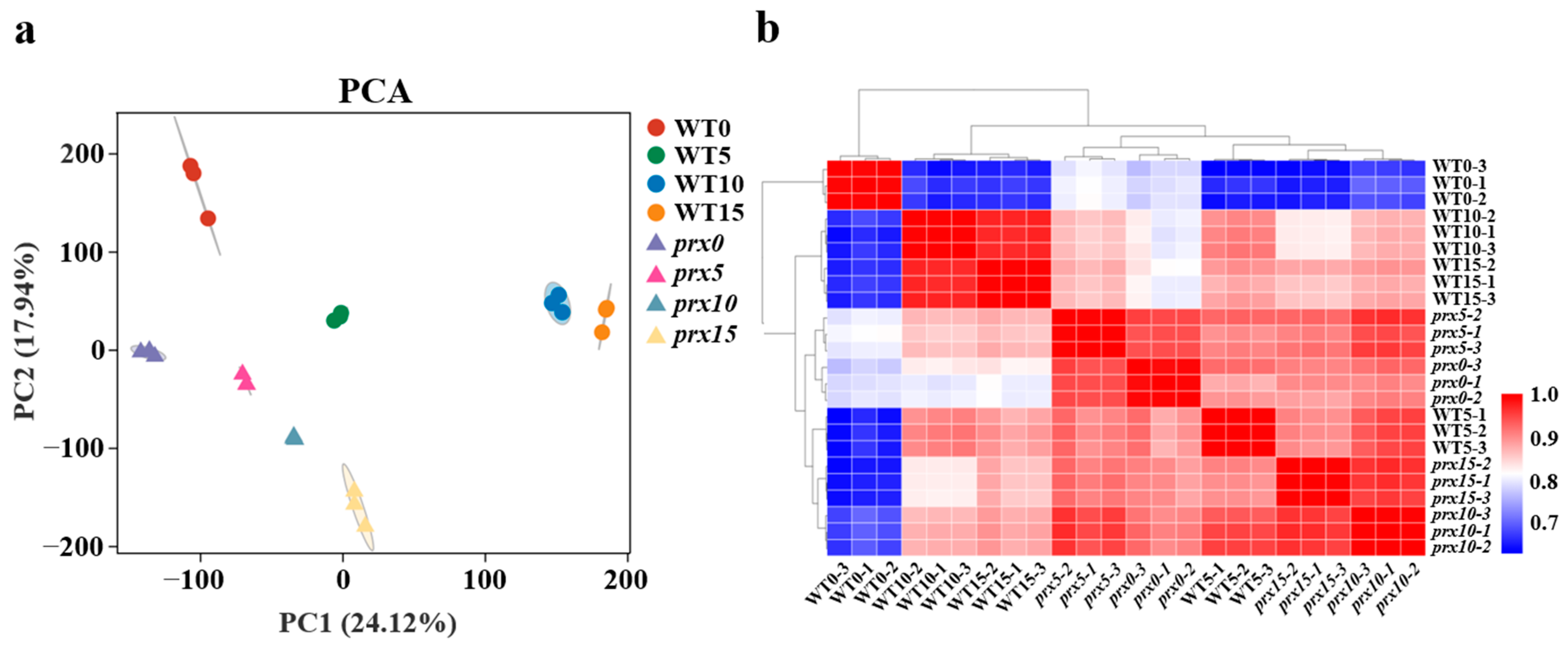
3.2. Transcriptome Sequencing of the Cultures
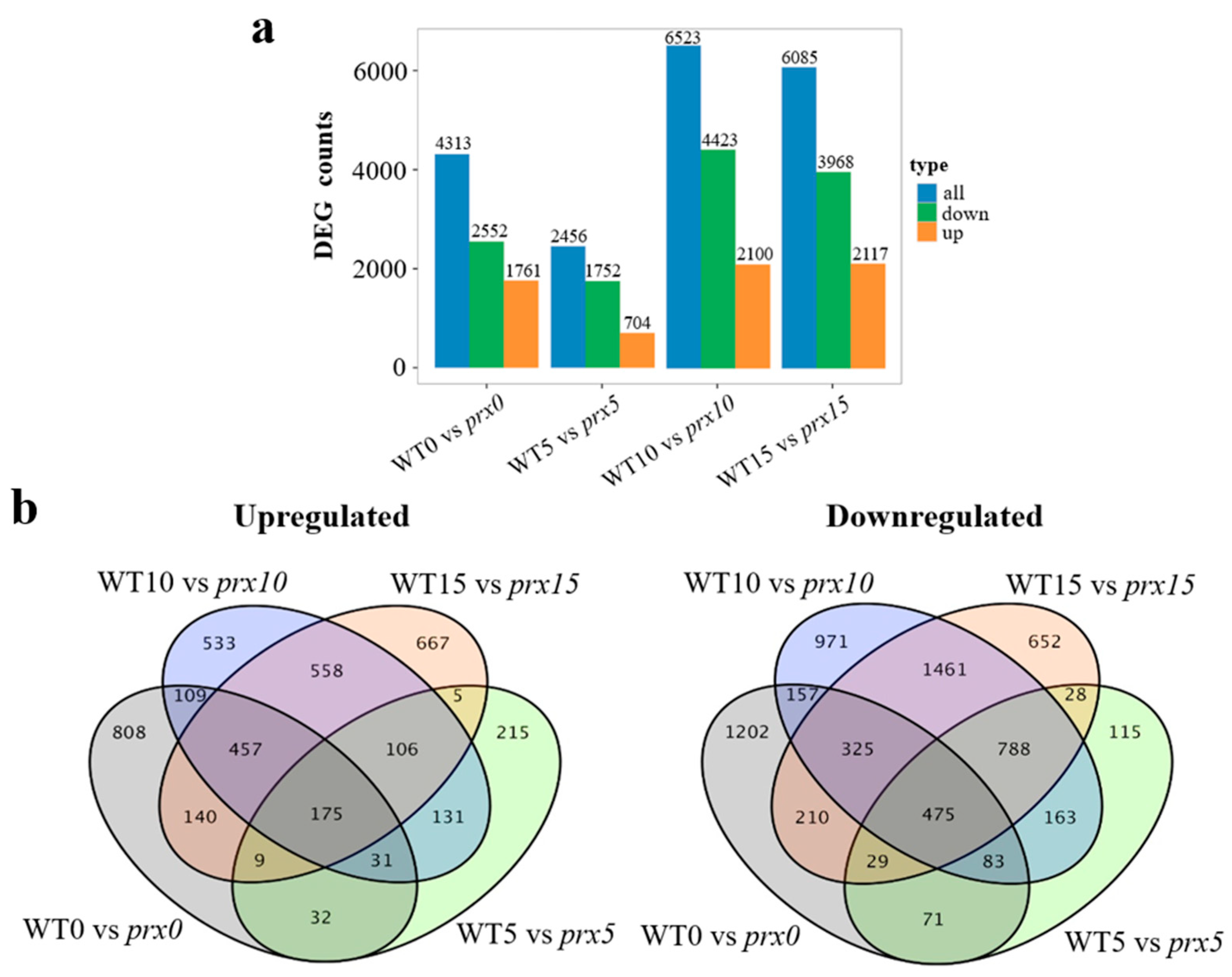
3.3. DEGs in Genotypes with Different Regenerative Capacities
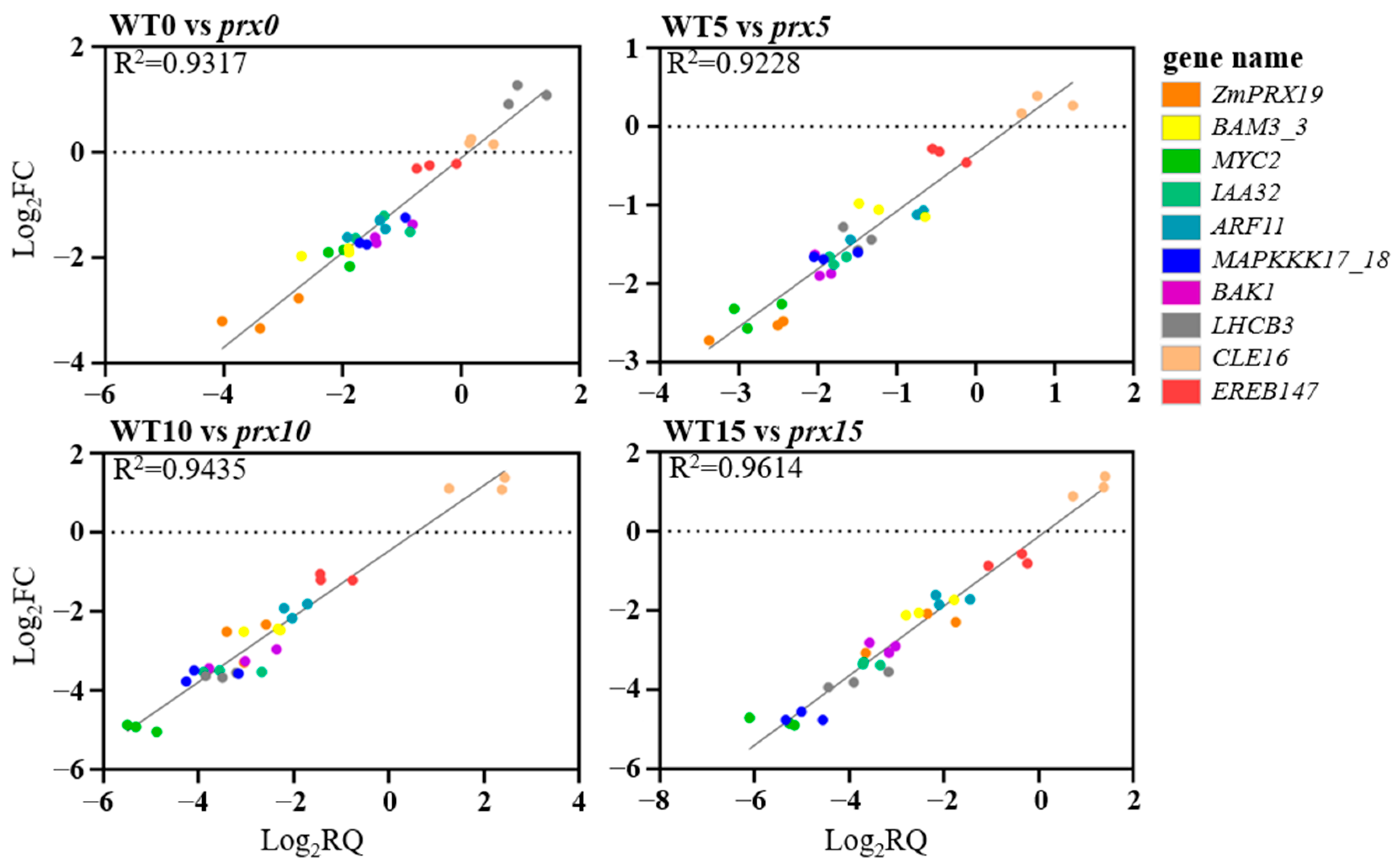
3.4. Reliability Validation of DEG Expression via qPCR
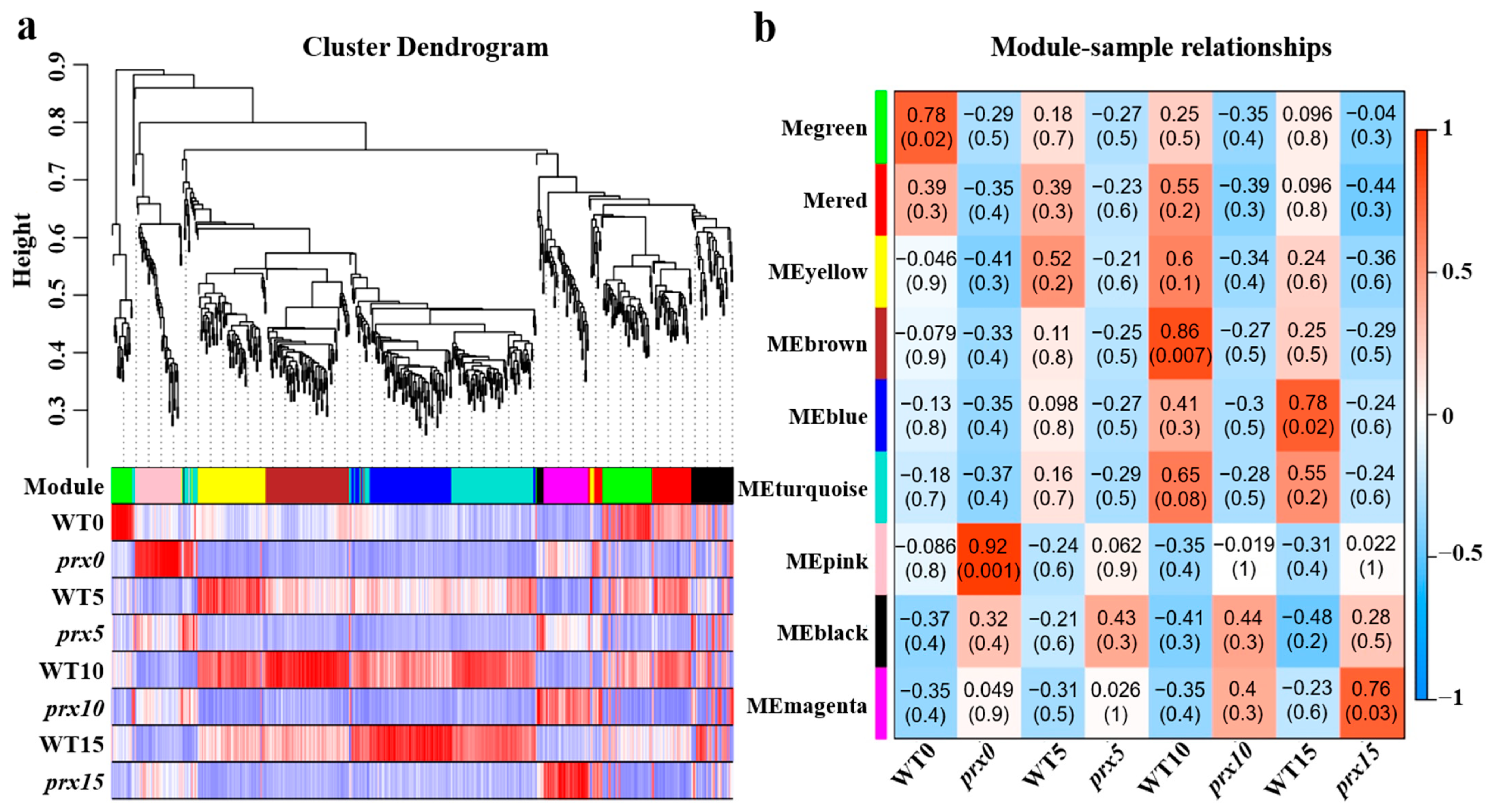
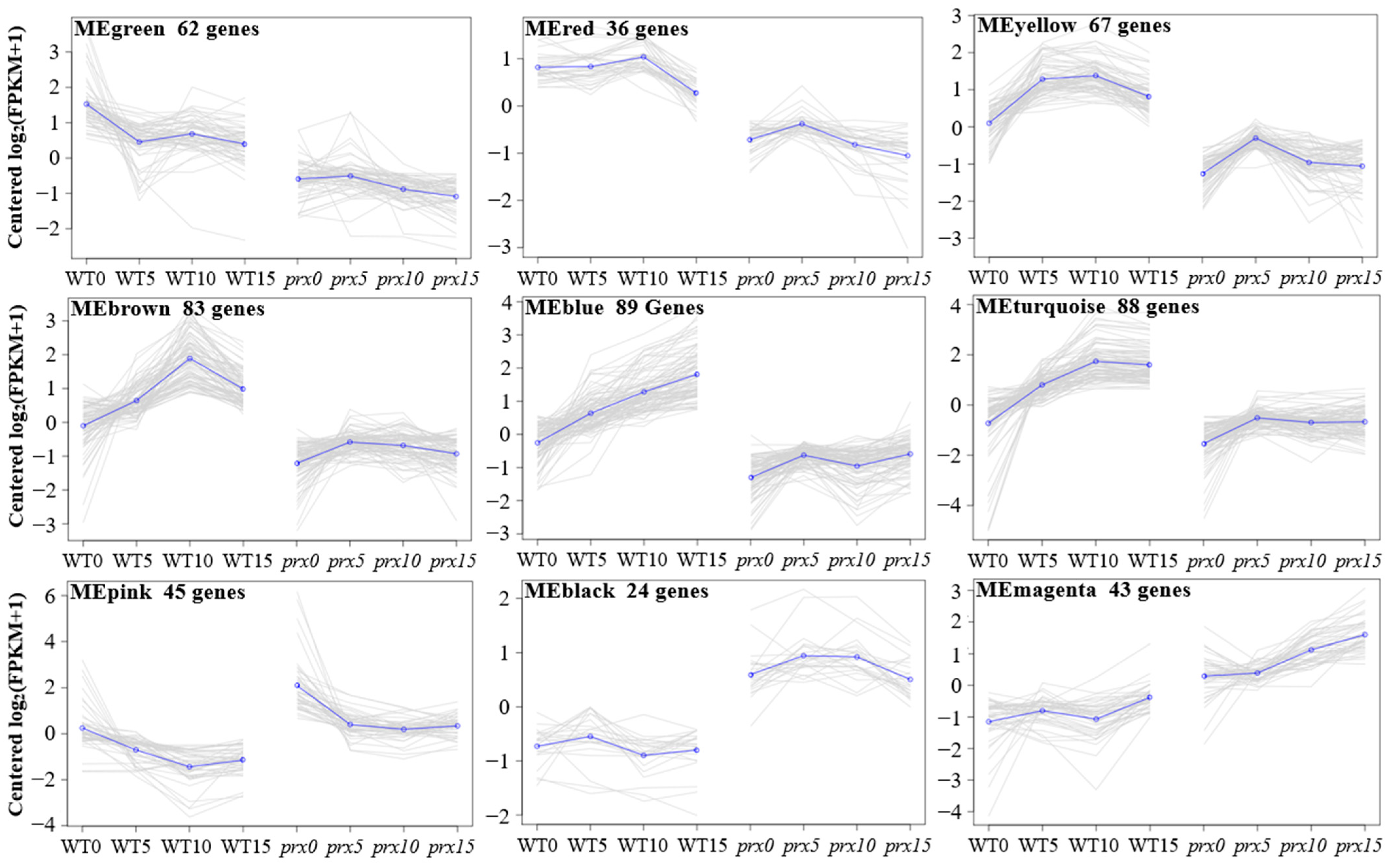
3.5. Weighted Gene Coexpression Network Analysis
3.6. GO Analysis of the Module Genes
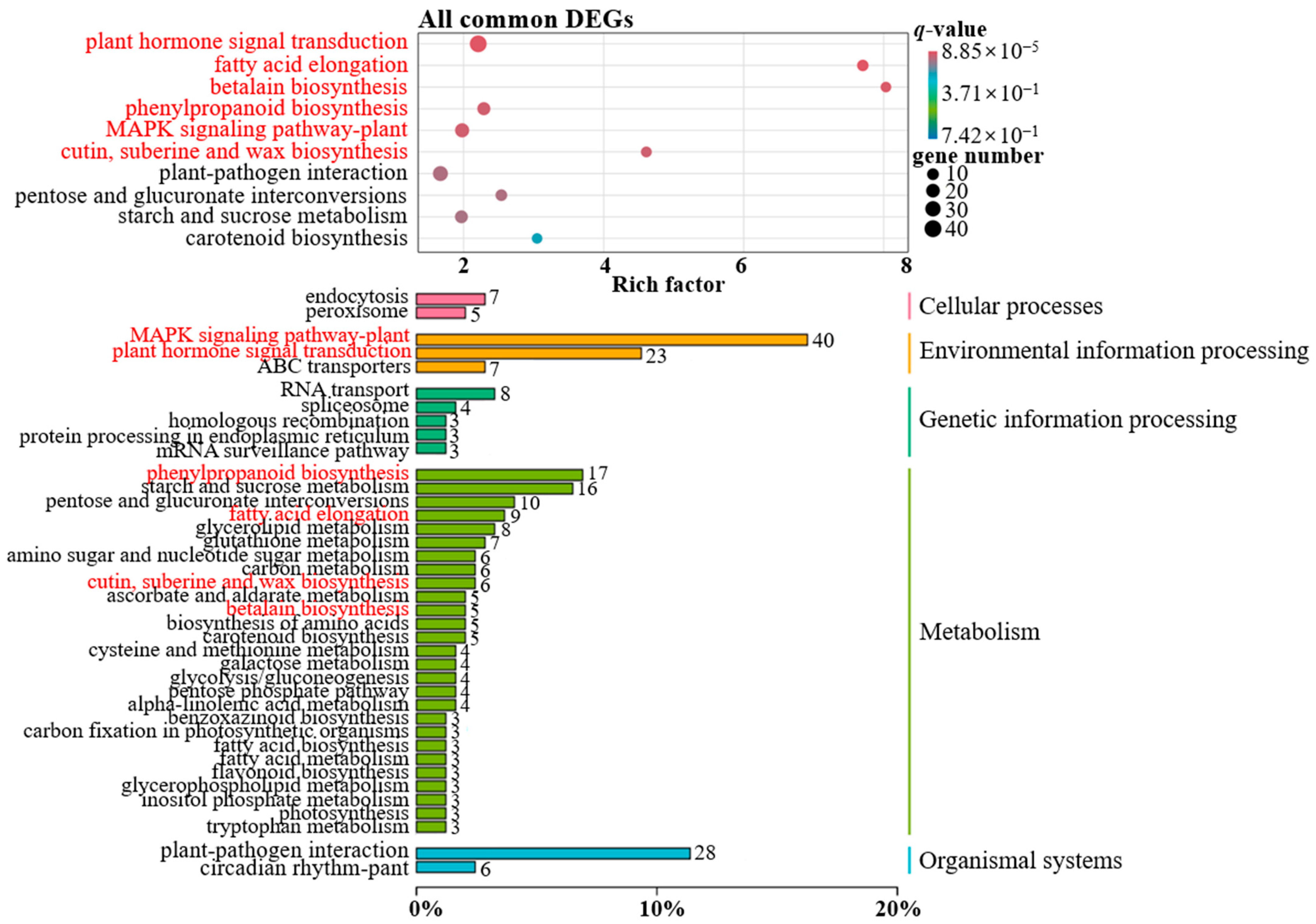
3.7. KEGG Pathway Enrichment Analysis
3.8. KEGG Pathway Enrichment Analysis
3.9. PPI Network Analysis
4. Discussion
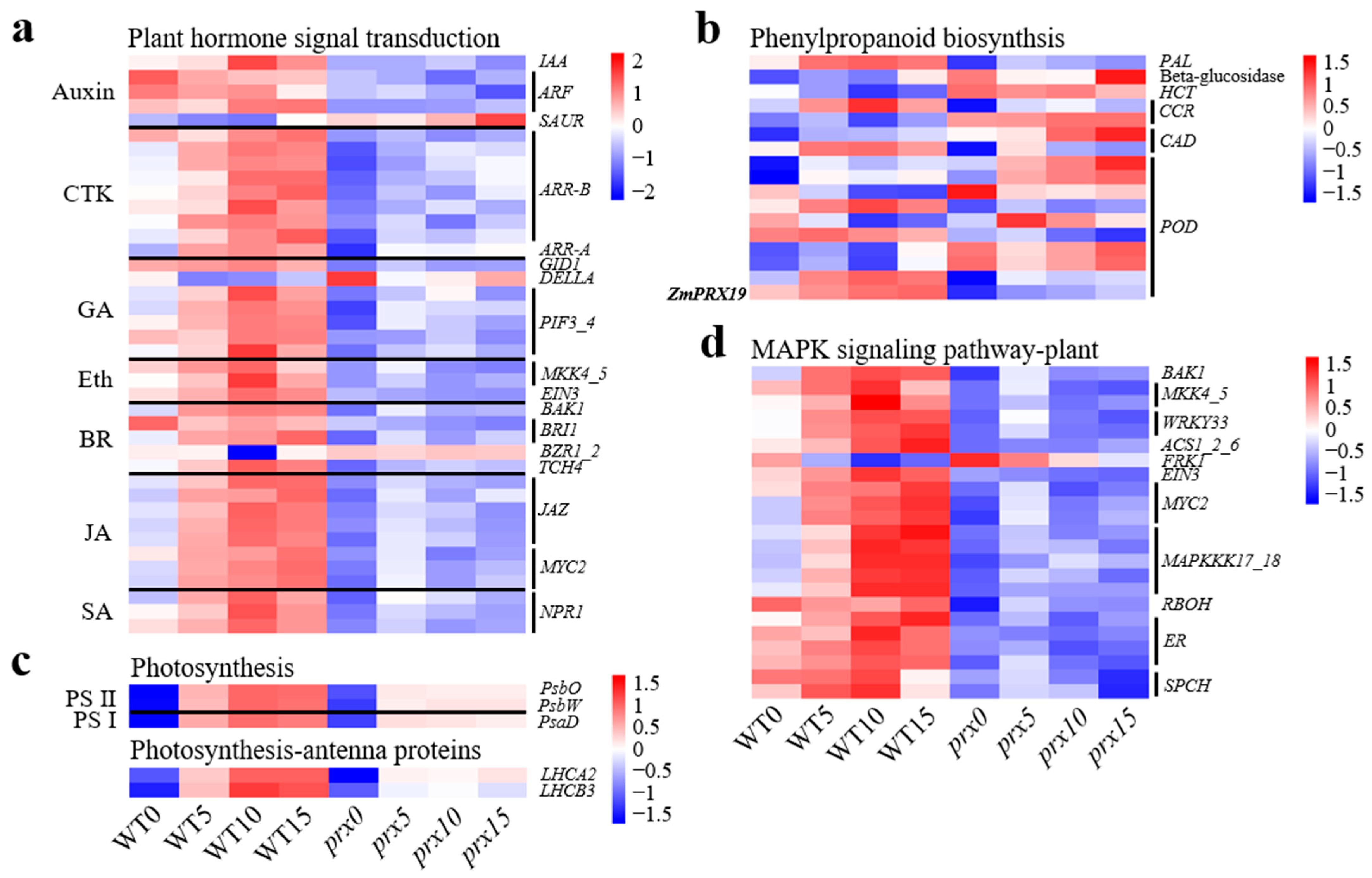
4.1. Signaling Pathways Involved in EC Differentiation
4.2. Phenylpropanoid Biosynthesis in EC Differentiation
4.3. Photosynthesis in Plant Regeneration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bidabadi, S.S.; Jain, S.M. Cellular, molecular, and physiological aspects of in vitro plant regeneration. Plants 2020, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Haberlandt, G. Culturversuche mit isolierten Pflanzenzellen. In Sitzungsberichte der Kaiserlichen Akademie der Wissenschaften; Mathematisch-Naturwissenschaftliche Classe; Springer: Wien, Austria, 1902; Volume 111, pp. 69–92. [Google Scholar]
- Ishida, Y.; Hiei, Y.; Komari, T. Agrobacterium-mediated transformation of maize. Nat. Protoc. 2007, 2, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Luo, W.; Linghu, Q.; Abe, F.; Hisano, H.; Sato, K.; Kamiya, Y.; Kanako, K.; Onishi, K.; Endo, M.; et al. In planta genome editing in commercial wheat varieties. Front. Plant Sci. 2021, 12, 648841. [Google Scholar] [CrossRef] [PubMed]
- Lardon, R.; Geelen, D. Natural variation in plant pluripotency and regeneration. Plants 2020, 9, 1261. [Google Scholar] [CrossRef] [PubMed]
- Nagle, M.F.; Yuan, J.; Kaur, D.; Ma, C.; Peremyslova, E.; Jiang, Y.; Rivera, A.N.; Jawdt, S.; Chen, J.G.; Feng, K.; et al. GWAS supported by computer vision identifies large numbers of candidate regulators of in planta regeneration in Populus trichocarpa. G3-Genes Genomes Genet. 2024, 14, jkae026. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Yang, Y.; Pan, G.; Shen, Y. New insights into tissue culture plant-regeneration mechanisms. Front. Plant Sci. 2022, 13, 926752. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Liu, M.; Yan, Y.; Qing, C.; Zhang, X.; Zhang, Y.; Long, Y.; Wang, L.; Pan, L.; Zou, C.; et al. Genetic dissection of maize embryonic callus regenerative capacity using multi-locus genome-wide association studies. Front. Plant Sci. 2018, 9, 561. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Solanki, M.; Shukla, L.I. Evidences for differential expression of miR167d-5p, target, positional nucleotide preference, and its role in somatic and different stages of regenerating calli of Oryza sativa. Plant Cell Tissue Organ Cult. 2019, 136, 537–548. [Google Scholar] [CrossRef]
- Islam, M.K.; Mummadi, S.T.; Liu, S.; Wei, H. Regulation of regeneration in Arabidopsis thaliana. aBIOTECH 2023, 4, 332–351. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Q.; Zhu, G.; Wang, S.; Ma, Y.; Miao, H.; Zhang, S.; Huang, S.; Zhang, Z.; Gu, X. Genetic analysis and identifcation of a candidate gene associated with in vitro regeneration ability of cucumber. Theor. Appl. Genet. 2018, 131, 2663–2675. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, H.; Cao, L.; Wang, S.; Li, Y.; Zhang, Y.; Jiang, W.; Zhou, Y.; Wang, H. Down-regulation of multiple CDK inhibitor ICK/KRP genes promotes cell proliferation, callus induction and plant regeneration in Arabidopsis. Front. Plant Sci. 2015, 6, 825. [Google Scholar] [CrossRef]
- Shim, S.; Lee, H.G.; Seo, P.J. MET1-dependent DNA methylation represses light signaling and influences plant regeneration in Arabidopsis. Mol. Cells 2021, 44, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Lopes, F.L.; Galvan-Ampudia, C.; Landrein, B. WUSCHEL in the shoot apical meristem: Old player, new tricks. J. Exp. Bot. 2021, 72, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Guo, H.; Zhang, L.; Fan, Y.; Fan, Y.; Zeng, F. SELTP-assembled battery drives totipotency of somatic plant cell. Plant Biotechnol. J. 2019, 17, 1188–1190. [Google Scholar] [CrossRef]
- Luján-Soto, E.; Juárez-González, V.T.; Reyes, J.L.; Dinkova, T.D. MicroRNA Zma-miR528 versatile regulation on target mRNAs during maize somatic embryogenesis. Int. J. Mol. Sci. 2021, 22, 5310. [Google Scholar] [CrossRef]
- Wójcik, A.M.; Wójcikowska, B.; Gaj, M.D. Current perspectives on the auxin-mediated genetic network that controls the induction of somatic embryogenesis in plants. Int. J. Mol. Sci. 2020, 21, 1333. [Google Scholar] [CrossRef]
- Dai, L.; Zhang, Y.; Han, S.; Hao, D. QTL mapping of somatic regeneration-related traits in maize. Agriculture 2022, 12, 393. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Baren, M.J.; Salzberg, S.L.; Word, B.J.; Pachter, L. Transcript assembly and quantification by RNA Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Kaufman, L.; Rousseeuw, P.J. Finding Groups in Data: An Introduction to Cluster Analysis; John Wiley & Sons: Hoboken, NJ, USA, 2009; Volume 344. [Google Scholar]
- Yu, G.; Wang, L.; Han, Y.; He, Q. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Becker, R.A.; Chambers, J.M.; Wilks, A.R. The New S Language; Wadsworth & Brooks; Chapman & Hall: London, UK, 1988. [Google Scholar]
- Rychlik, W. OLIGO 7 primer analysis software. Methods Mol. Biol. 2007, 402, 35–39. [Google Scholar]
- Schefe, J.H.; Lehmann, K.E.; Buschmann, I.R.; Unger, T.; Funke-Kaiser, H. Quantitative real-time RT-PCR data analysis: Current concepts and the novel “gene expression’s CT difference” formula. J. Mol. Med. 2006, 84, 901–910. [Google Scholar] [CrossRef]
- Zhou, W.; Lozano-Torres, J.L.; Blilou, I.; Zhang, X.; Zhai, Q.; Smant, G.; Li, C.; Scheres, B. A jasmonate signaling network activates root stem cells and promotes regeneration. Cell 2019, 177, 942–956. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Favero, D.S.; Sakamoto, Y.; Iwase, A.; Coleman, D.; Rymen, B.; Sugimoto, K. Molecular mechanisms of plant regeneration. Annu. Rev. Plant Biol. 2019, 70, 377–406. [Google Scholar] [CrossRef]
- Takeuchi, J.; Fukui, K.; Seto, Y.; Takaoka, Y.; Okamoto, M. Ligand–receptor interactions in plant hormone signaling. Plant J. 2021, 105, 290–3306. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Park, O.S.; Go, J.Y.; Yu, J.; Han, J.H.; Kim, J.; Bae, S.; Jung, Y.J.; Seo, P.J. Arabidopsis ATXR2 represses de novo shoot organogenesis in the transition from callus to shoot formation. Cell Rep. 2021, 37, 109980. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, F.; Chen, L.; Pan, Y.; Sun, L.; Bao, N.; Zhang, T.; Cui, C.; Qiu, Z.; Zhang, Y.; et al. Jasmonate-mediated wound signalling promotes plant regeneration. Nat. Plants 2019, 5, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bo, K.; Pan, Y.; Li, Y.; Yu, D.; Li, C.; Chang, J.; Wu, S.; Wang, Z.; Zhang, X.; et al. Phytochrome-interacting factor PIF3 integrates phytochrome B and UV-B signaling pathways to regulate gibberellin-and auxin-dependent growth in cucumber hypocotyls. J. Exp. Bot. 2023, 74, 4520–4539. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhao, H.; Sun, L.; Wu, W.; Li, C.; Wu, Q. Genome-wide identification of MAPK gene family members in Fagopyrum tataricum and their expression during development and stress responses. BMC Genom. 2022, 23, 96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wu, H.; Su, J.; Wang, H.; Zhu, Q.; Liu, Y.; Xu, J.; Lukowitz, W.; Zhang, S. Maternal control of embryogenesis by MPK6 and its upstream MKK4/MKK5 in Arabidopsis. Plant J. 2017, 92, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Winkelmann, T. Somatic versus zygotic embryogenesis: Learning from seeds. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1359. [Google Scholar]
- Jiang, H.; Chen, Y.; Liu, Y.; Shang, J.; Sun, X.; Du, J. Multifaceted roles of the ERECTA family in plant organ morphogenesis. J. Exp. Bot. 2022, 73, 7208–7218. [Google Scholar] [CrossRef]
- Kazan, K.; Manners, J.M. MYC2: The master in action. Mol. Plant 2013, 6, 686–703. [Google Scholar] [CrossRef] [PubMed]
- López-Ruiz, B.A.; Juárez-González, V.T.; Sandoval-Zapotitla, E.; Dinkova, T.D. Development-related miRNA expression and target regulation during staggered in vitro plant regeneration of Tuxpeño VS-535 maize cultivar. Int. J. Mol. Sci. 2019, 20, 2079. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Yan, Y.; Peng, H.; Long, Y.; Zhang, Y.; Jiang, Z.; Liu, P.; Zou, C.; Peng, H.; et al. Transcriptome sequencing analysis of maize embryonic callus during early redifferentiation. BMC Genom. 2019, 20, 159. [Google Scholar] [CrossRef] [PubMed]
- Change, E.; Guo, W.; Xie, Y.; Jiang, Z.; Dong, Y.; Jia, Z.; Zhao, X.; Liu, J.; Zhang, J. Changes of lignified-callus and wound-induced adventitious rooting in ancient Platycladus orientalis cuttings as affected by tree age. Ind. Crops Prod. 2023, 203, 117183. [Google Scholar] [CrossRef]
- Laukkanen, H.; Rautiainen, L.; Taulavuori, E.; Hohtola, A. Changes in cellular structures and enzymatic activities during browning of Scots pine callus derived from mature buds. Tree Physiol. 2000, 20, 467–475. [Google Scholar] [CrossRef]
- Siddique, A.B.; Islam, S.S. Effect of light and dark on callus induction and regeneration in tobacco (Nicotiana tabacum L.). Bangladesh J. Bot. 2015, 44, 643–651. [Google Scholar] [CrossRef]
- Pan, X.; Cao, P.; Su, X.; Liu, Z.; Li, M. Structural analysis and comparison of light-harvesting complexes I and II. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148038. [Google Scholar] [CrossRef] [PubMed]
- Bricker, T.M.; Frankel, L.K. Auxiliary functions of the PsbO, PsbP and PsbQ proteins of higher plant Photosystem II: A critical analysis. J. Photoch. Photobio. B 2011, 104, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Ifuku, K.; Takabayashi, A.; Shikanai, T.; Endo, T.; Sato, F. Functional dissection of two Arabidopsis PsbO proteins. FEBS J. 2005, 272, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Q.; Wang, J.; Yan, Y.; Zhang, G.; Yan, Y.; Zhang, H.; Wu, J.; Chen, F.; Wang, X.; et al. YR36/WKS1-mediated phosphorylation of PsbO, an extrinsic member of photosystem II, inhibits photosynthesis and confers stripe rust resistance in wheat. Mol. Plant 2019, 12, 1639–1650. [Google Scholar] [CrossRef]
- Wang, L.; Wang, B.; Du, Q.; Chen, J.; Tian, J.; Yang, X.; Zhang, D. Allelic variation in PtoPsbW associated with photosynthesis, growth, and wood properties in Populus tomentosa. Mol. Genet. Genom. 2016, 292, 77–91. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Line | EIR (%) | GCR (%) | PRR (%) |
|---|---|---|---|
| KN5585 (WT) | 96.58 ± 1.23 A 1 | 92.66 ± 1.59 A | 75.25 ± 3.50 A |
| Zmprx19-1 | 80.65 ± 2.70 C | 59.24 ± 2.21 B | 8.78 ± 1.04 B |
| Zmprx19-2 | 85.92 ± 1.63 B | 60.30 ± 2.15 B | 15.08 ± 1.23 B |
| Zmprx19-3 | 83.35 ± 1.52 BC | 64.29 ± 1.76 B | 12.29 ± 1.61 B |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, L.; Li, T. Comparative Transcriptomic Insights into the Mechanisms Underlying Maize (Zea mays L.) Embryogenic Callus Differentiation. Agronomy 2024, 14, 1689. https://doi.org/10.3390/agronomy14081689
Dai L, Li T. Comparative Transcriptomic Insights into the Mechanisms Underlying Maize (Zea mays L.) Embryogenic Callus Differentiation. Agronomy. 2024; 14(8):1689. https://doi.org/10.3390/agronomy14081689
Chicago/Turabian StyleDai, Liqiang, and Tianjiao Li. 2024. "Comparative Transcriptomic Insights into the Mechanisms Underlying Maize (Zea mays L.) Embryogenic Callus Differentiation" Agronomy 14, no. 8: 1689. https://doi.org/10.3390/agronomy14081689