
Focal Adhesion’s Role in Cardiomyocytes Function: From Cardiomyogenesis to Mechanotransduction
 ,
,  , and
, and
Abstract
:1. Focal Adhesions: Dynamic Sites Involved in Cell Adhesion and Function
2. Focal Adhesions and Cardiac Cell Differentiation
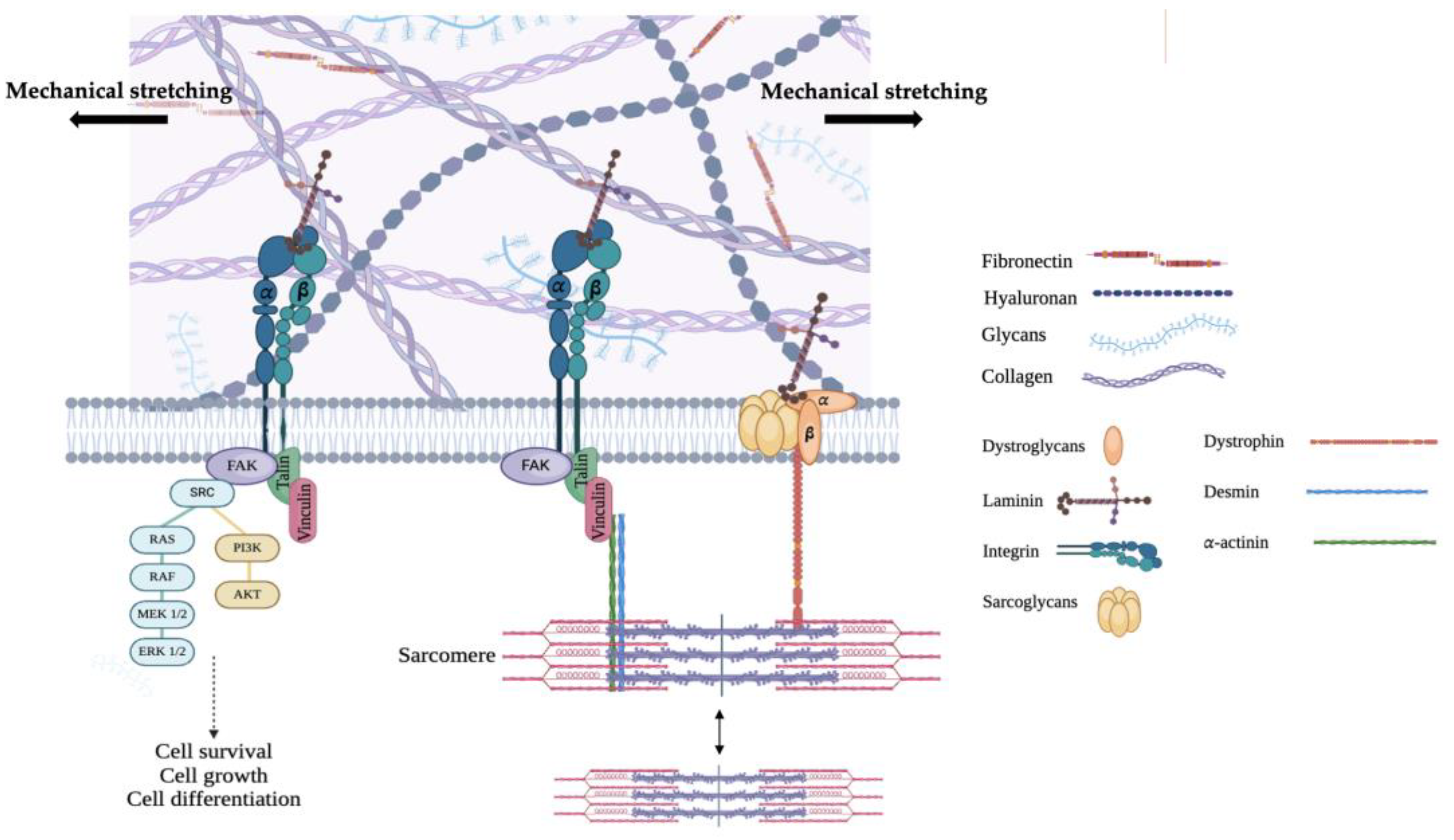
3. Focal Adhesion-Mediated Mechanosensing in Cardiac Muscle
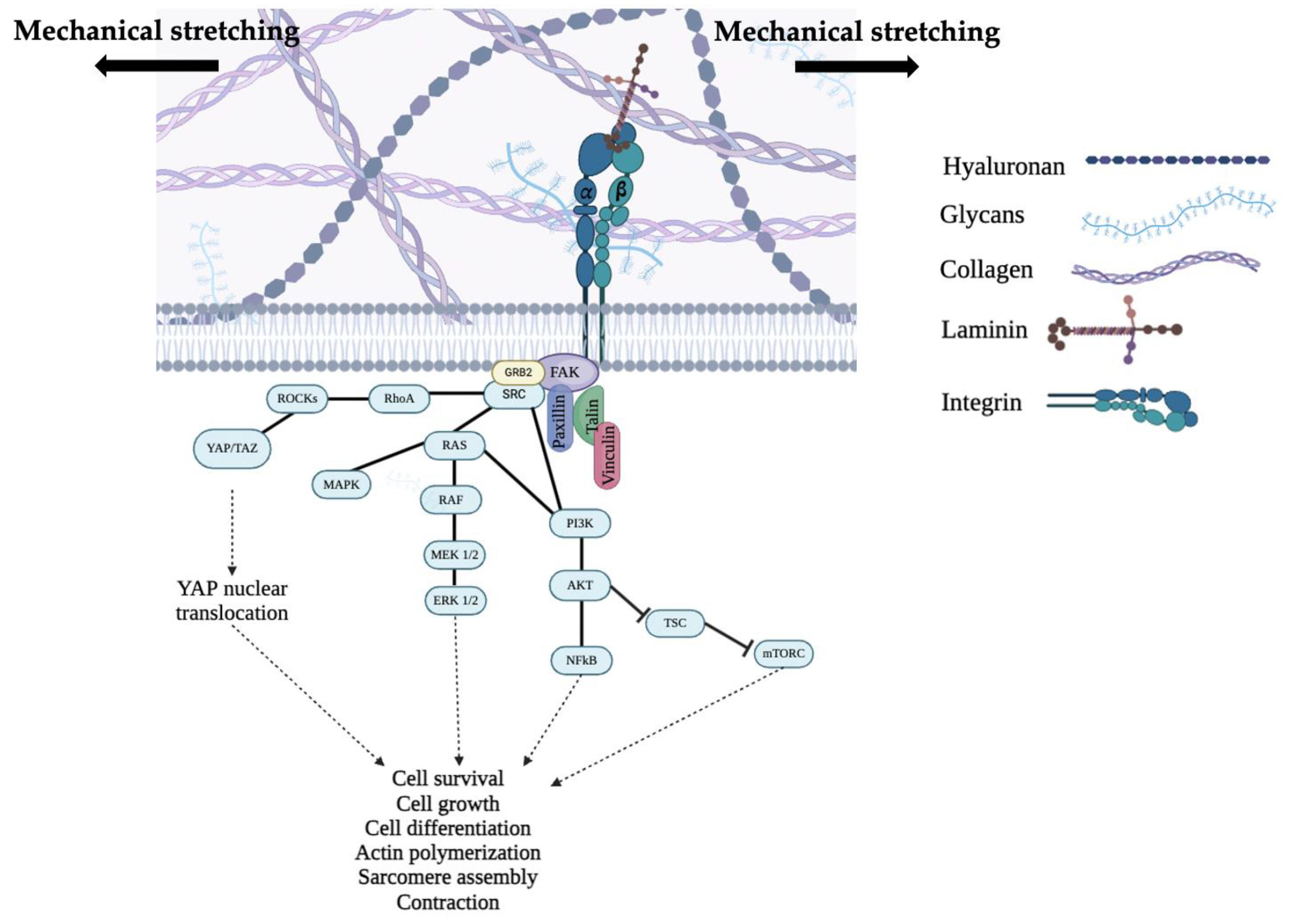
4. Focal Adhesion-Mediated Mechanosignaling in Cardiac Muscle
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zamir, E.; Geiger, B. Molecular complexity and dynamics of cell-matrix adhesions. J. Cell Sci. 2001, 114, 3583–3590. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Fath, K. Focal contacts: Transmembrane links between the extracellular matrix and the cytoskeleton. BioEssays 1989, 10, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Partridge, M.A.; Marcantonio, E.E. Initiation of Attachment and Generation of Mature Focal Adhesions by Integrin-containing Filopodia in Cell Spreading. Mol. Biol. Cell 2006, 17, 4237–4248. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Vinogradova, O.; Plow, E.F. Integrin Bidirectional Signaling: A Molecular View. PLoS Biol. 2004, 2, e169. [Google Scholar] [CrossRef]
- Legerstee, K.; Houtsmuller, A.B. A Layered View on Focal Adhesions. Biology 2021, 10, 1189. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Bershadsky, A. Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 2001, 13, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Legerstee, K.; Geverts, B.; Slotman, J.A.; Houtsmuller, A.B. Dynamics and distribution of paxillin, vinculin, zyxin and VASP depend on focal adhesion location and orientation. Sci. Rep. 2019, 9, 10460. [Google Scholar] [CrossRef]
- Samarel, A.M. Focal adhesion signaling in heart failure. Pflüg. Arch. Eur. J. Physiol. 2014, 466, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.E. Paxillin and focal adhesion signalling. Nat. Cell Biol. 2000, 2, E231–E236. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.C.; Cary, L.A.; Jamieson, J.S.; Cooper, J.A.; Turner, C.E. Src and FAK Kinases Cooperate to Phosphorylate Paxillin Kinase Linker, Stimulate Its Focal Adhesion Localization, and Regulate Cell Spreading and Protrusiveness. Mol. Biol. Cell 2005, 16, 4316–4328. [Google Scholar] [CrossRef]
- Gehmlich, K.; Pinotsis, N.; Hayeß, K.; van der Ven, P.F.M.; Milting, H.; El Banayosy, A.; Körfer, R.; Wilmanns, M.; Ehler, E.; Fürst, D.O. Paxillin and Ponsin Interact in Nascent Costameres of Muscle Cells. J. Mol. Biol. 2007, 369, 665–682. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Zhu, L.; Bromberger, T.; Yang, J.; Yang, Q.; Liu, J.; Plow, E.F.; Moser, M.; Qin, J. Mechanism of integrin activation by talin and its cooperation with kindlin. Nat. Commun. 2022, 13, 2362. [Google Scholar] [CrossRef]
- Sun, Z.; Costell, M.; Fässler, R. Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol. 2019, 21, 25–31. [Google Scholar] [CrossRef]
- Sit, B.; Gutmann, D.; Iskratsch, T. Costameres, dense plaques and podosomes: The cell matrix adhesions in cardiovascular mechanosensing. J. Muscle Res. Cell Motil. 2019, 40, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wu, X.; Sun, S.; Wang, C.; Vangelatos, Z.; Ash-Shakoor, A.; Grigoropoulos, C.P.; Mather, P.T.; Henderson, J.H.; Ma, Z. Profiling the responsiveness of focal adhesions of human cardiomyocytes to extracellular dynamic nano-topography. Bioact. Mater. 2022, 10, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Samarel, A.M. Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am. J. Physiol.-Heart Circ. Physiol. 2005, 289, H2291–H2301. [Google Scholar] [CrossRef]
- Pardo, J.V.; Siliciano, J.D.; Craig, S.W. A vinculin-containing cortical lattice in skeletal muscle: Transverse lattice elements (“costameres”) mark sites of attachment between myofibrils and sarcolemma. Proc. Natl. Acad. Sci. USA 1983, 80, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Münch, J.; Abdelilah-Seyfried, S. Sensing and Responding of Cardiomyocytes to Changes of Tissue Stiffness in the Diseased Heart. Front. Cell Dev. Biol. 2021, 9, 642840. Available online: https://www.frontiersin.org/articles/10.3389/fcell.2021.642840 (accessed on 27 September 2023). [CrossRef]
- Peter, A.K.; Cheng, H.; Ross, R.S.; Knowlton, K.U.; Chen, J. The costamere bridges sarcomeres to the sarcolemma in striated muscle. Prog. Pediatr. Cardiol. 2011, 31, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Zemljic-Harpf, A.; Manso, A.M.; Ross, R.S. Vinculin and Talin: Focus on the Myocardium. J. Investig. Med. 2009, 57, 849–855. [Google Scholar] [CrossRef]
- Dabiri, G.A.; Turnacioglu, K.K.; Sanger, J.M.; Sanger, J.W. Myofibrillogenesis visualized in living embryonic cardiomyocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 9493–9498. [Google Scholar] [CrossRef] [PubMed]
- Sanger, J.W.; Kang, S.; Siebrands, C.C.; Freeman, N.; Du, A.; Wang, J.; Stout, A.L.; Sanger, J.M. How to build a myofibril. J. Muscle Res. Cell Motil. 2005, 26, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.C.; Schöck, F. The initial steps of myofibril assembly: Integrins pave the way. Nat. Rev. Mol. Cell Biol. 2009, 10, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Geach, T.J.; Hirst, E.M.A.; Zimmerman, L.B. Contractile activity is required for Z-disc sarcomere maturation in vivo. Genesis 2015, 53, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Majkut, S.; Idema, T.; Swift, J.; Krieger, C.; Liu, A.; Discher, D.E. Heart-Specific Stiffening in Early Embryos Parallels Matrix and Myosin Expression to Optimize Beating. Curr. Biol. 2013, 23, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Weitkunat, M.; Kaya-Çopur, A.; Grill, S.W.; Schnorrer, F. Tension and Force-Resistant Attachment Are Essential for Myofibrillogenesis in Drosophila Flight Muscle. Curr. Biol. 2014, 24, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Crocini, C.; Gotthardt, M. Cardiac sarcomere mechanics in health and disease. Biophys. Rev. 2021, 13, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.E.; Tokuyama, T.; Anzai, T.; Chanthra, N.; Uosaki, H. Sarcomere maturation: Function acquisition, molecular mechanism, and interplay with other organelles. Philos. Trans. R. Soc. B Biol. Sci. 2022, 377, 20210325. [Google Scholar] [CrossRef] [PubMed]
- Kresh, J.Y.; Chopra, A. Intercellular and extracellular mechanotransduction in cardiac myocytes. Pflüg. Arch. Eur. J. Physiol. 2011, 462, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Chopra, A.; Kutys, M.L.; Zhang, K.; Polacheck, W.J.; Sheng, C.C.; Luu, R.J.; Eyckmans, J.; Hinson, J.T.; Seidman, J.G.; Seidman, C.E.; et al. Force Generation via β-Cardiac Myosin, Titin, and α-Actinin Drives Cardiac Sarcomere Assembly from Cell-Matrix Adhesions. Dev. Cell 2018, 44, 87–96.e5. [Google Scholar] [CrossRef] [PubMed]
- Bennett, P.; Rees, M.; Gautel, M. The Axial Alignment of Titin on the Muscle Thick Filament Supports Its Role as a Molecular Ruler. J. Mol. Biol. 2020, 432, 4815–4829. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.R.; Granzier, H.L. Titin-based tension in the cardiac sarcomere: Molecular origin and physiological adaptations. Prog. Biophys. Mol. Biol. 2012, 110, 204–217. [Google Scholar] [CrossRef]
- Yotti, R.; Seidman, C.E.; Seidman, J.G. Advances in the Genetic Basis and Pathogenesis of Sarcomere Cardiomyopathies. Annu. Rev. Genomics Hum. Genet. 2019, 20, 129–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, Y.; Liu, H.; Tong, C.; Ying, Q.; Sachinidis, A.; Li, L.; Peng, L. Laminin promotes differentiation of rat embryonic stem cells into cardiomyocytes by activating the integrin/FAK/PI3K p85 pathway. J. Cell. Mol. Med. 2019, 23, 3629–3640. [Google Scholar] [CrossRef]
- Wang, D.; Liu, C.; Liu, H.; Meng, Y.; Lin, F.; Gu, Y.; Wang, H.; Shang, M.; Tong, C.; Sachinidis, A.; et al. ERG1 plays an essential role in rat cardiomyocyte fate decision by mediating AKT signaling. Stem Cells 2021, 39, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Tang, Y.; Zhou, Y.; Zhang, J. Deciphering Role of Wnt Signalling in Cardiac Mesoderm and Cardiomyocyte Differentiation from Human iPSCs: Four-dimensional control of Wnt pathway for hiPSC-CMs differentiation. Sci. Rep. 2019, 9, 19389. [Google Scholar] [CrossRef] [PubMed]
- Robert, S.; Flowers, M.; Ogle, B.M. Kinases of the Focal Adhesion Complex Contribute to Cardiomyocyte Specification. Int. J. Mol. Sci. 2021, 22, 10430. [Google Scholar] [CrossRef]
- Doherty, J.T.; Conlon, F.L.; Mack, C.P.; Taylor, J.M. Focal adhesion kinase is essential for cardiac looping and multichamber heart formation. Genesis 2010, 48, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Grabbe, C.; Zervas, C.G.; Hunter, T.; Brown, N.H.; Palmer, R.H. Focal adhesion kinase is not required for integrin function or viability in Drosophila. Development 2004, 131, 5795–5805. [Google Scholar] [CrossRef]
- DiMichele, L.A.; Hakim, Z.S.; Sayers, R.L.; Rojas, M.; Schwartz, R.J.; Mack, C.P.; Taylor, J.M. Transient Expression of FRNK Reveals Stage-Specific Requirement for Focal Adhesion Kinase Activity in Cardiac Growth. Circ. Res. 2009, 104, 1201–1208. [Google Scholar] [CrossRef]
- Peng, X.; Wu, X.; Druso, J.E.; Wei, H.; Park, A.Y.-J.; Kraus, M.S.; Alcaraz, A.; Chen, J.; Chien, S.; Cerione, R.A.; et al. Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6638–6643. [Google Scholar] [CrossRef] [PubMed]
- Hakuno, D.; Takahashi, T.; Lammerding, J.; Lee, R.T. Focal Adhesion Kinase Signaling Regulates Cardiogenesis of Embryonic Stem Cells. J. Biol. Chem. 2005, 280, 39534–39544. [Google Scholar] [CrossRef] [PubMed]
- Männer, J. The anatomy of cardiac looping: A step towards the understanding of the morphogenesis of several forms of congenital cardiac malformations. Clin. Anat. 2009, 22, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Pentassuglia, L.; Sawyer, D.B. ErbB/integrin signaling interactions in regulation of myocardial cell–cell and cell–matrix interactions. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Aponte-Santamaría, C.; Sturm, S.; Bullerjahn, J.T.; Bronowska, A.; Gräter, F. Mechanism of Focal Adhesion Kinase Mechanosensing. PLoS Comput. Biol. 2015, 11, e1004593. [Google Scholar] [CrossRef] [PubMed]
- Clemente, C.F.M.Z.; Xavier-Neto, J.; Dalla Costa, A.P.; Consonni, S.R.; Antunes, J.E.; Rocco, S.A.; Pereira, M.B.; Judice, C.C.; Strauss, B.; Joazeiro, P.P.; et al. Focal adhesion kinase governs cardiac concentric hypertrophic growth by activating the AKT and mTOR pathways. J. Mol. Cell. Cardiol. 2012, 52, 493–501. [Google Scholar] [CrossRef]
- Beuriot, A.; Eichel, C.A.; Dilanian, G.; Louault, F.; Melgari, D.; Doisne, N.; Coulombe, A.; Hatem, S.N.; Balse, E. Distinct calcium/calmodulin-dependent serine protein kinase domains control cardiac sodium channel membrane expression and focal adhesion anchoring. Heart Rhythm. 2020, 17, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Panaviene, Z.; Moncman, C.L. Linker region of nebulin family members plays an important role in targeting these molecules to cellular structures. Cell Tissue Res. 2007, 327, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Hirschy, A.; Schatzmann, F.; Ehler, E.; Perriard, J.-C. Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev. Biol. 2006, 289, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Hagel, M.; George, E.L.; Kim, A.; Tamimi, R.; Opitz, S.L.; Turner, C.E.; Imamoto, A.; Thomas, S.M. The Adaptor Protein Paxillin Is Essential for Normal Development in the Mouse and Is a Critical Transducer of Fibronectin Signaling. Mol. Cell. Biol. 2002, 22, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Atherton, P.; Stutchbury, B.; Jethwa, D.; Ballestrem, C. Mechanosensitive components of integrin adhesions: Role of vinculin. Exp. Cell Res. 2016, 343, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, D.A.; Campbell, I.D.; Critchley, D.R. Talins and kindlins: Partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 2013, 14, 503–517. [Google Scholar] [CrossRef] [PubMed]
- McCleverty, C.J.; Lin, D.C.; Liddington, R.C. Structure of the PTB domain of tensin1 and a model for its recruitment to fibrillar adhesions. Protein Sci. 2007, 16, 1223–1229. [Google Scholar] [CrossRef]
- Belkin, A.M.; Zhidkova, N.I.; Balzac, F.; Altruda, F.; Tomatis, D.; Maier, A.; Tarone, G.; Koteliansky, V.E.; Burridge, K. Beta 1D integrin displaces the beta 1A isoform in striated muscles: Localization at junctional structures and signaling potential in nonmuscle cells. J. Cell Biol. 1996, 132, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Choudhury, D.M.; Craig, S.W. Coincidence of Actin Filaments and Talin Is Required to Activate Vinculin. J. Biol. Chem. 2006, 281, 40389–40398. [Google Scholar] [CrossRef] [PubMed]
- Auernheimer, V.; Lautscham, L.A.; Leidenberger, M.; Friedrich, O.; Kappes, B.; Fabry, B.; Goldmann, W.H. Vinculin phosphorylation at residues Y100 and Y1065 is required for cellular force transmission. J. Cell Sci. 2015, 128, 3435–3443. [Google Scholar] [CrossRef] [PubMed]
- Bogatan, S.; Cevik, D.; Demidov, V.; Vanderploeg, J.; Panchbhaya, A.; Vitkin, A.; Jacobs, J.R. Talin Is Required Continuously for Cardiomyocyte Remodeling during Heart Growth in Drosophila. PLoS ONE 2015, 10, e0131238. [Google Scholar] [CrossRef] [PubMed]
- Martel, V.; Racaud-Sultan, C.; Dupe, S.; Marie, C.; Paulhe, F.; Galmiche, A.; Block, M.R.; Albiges-Rizo, C. Conformation, Localization, and Integrin Binding of Talin Depend on Its Interaction with Phosphoinositides. J. Biol. Chem. 2001, 276, 21217–21227. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, A.P.; Burridge, K. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-bisphosphate. Nature 1996, 381, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Bachir, A.I.; Zareno, J.; Moissoglu, K.; Plow, E.F.; Gratton, E.; Horwitz, A.R. Integrin-Associated Complexes Form Hierarchically with Variable Stoichiometry in Nascent Adhesions. Curr. Biol. 2014, 24, 1845–1853. [Google Scholar] [CrossRef] [PubMed]
- Pasapera, A.M.; Schneider, I.C.; Rericha, E.; Schlaepfer, D.D.; Waterman, C.M. Myosin II activity regulates vinculin recruitment to focal adhesions through FAK-mediated paxillin phosphorylation. J. Cell Biol. 2010, 188, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Case, L.B.; Baird, M.A.; Shtengel, G.; Campbell, S.L.; Hess, H.F.; Davidson, M.W.; Waterman, C.M. Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions. Nat. Cell Biol. 2015, 17, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Ewen, E.P.; Snyder, C.M.; Wilson, M.; Desjardins, D.; Naya, F.J. The Mef2A Transcription Factor Coordinately Regulates a Costamere Gene Program in Cardiac Muscle. J. Biol. Chem. 2011, 286, 29644–29653. [Google Scholar] [CrossRef]
- Moustafa, A.; Hashemi, S.; Brar, G.; Grigull, J.; Ng, S.H.S.; Williams, D.; Schmitt-Ulms, G.; McDermott, J.C. The MEF2A transcription factor interactome in cardiomyocytes. Cell Death Dis. 2023, 14, 240. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Desjardins, C.A.; Cooper, O.; Kontor, A.; Nocco, S.E.; Naya, F.J. EGR1 Functions as a Potent Repressor of MEF2 Transcriptional Activity. PLoS ONE 2015, 10, e0127641. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.A.; Atherton, P.; Ballestrem, C. Mechanotransduction at the cell-matrix interface. Mechanosens. Mol. Tissues 2017, 71, 75–83. [Google Scholar] [CrossRef]
- Rienks, M.; Papageorgiou, A.-P.; Frangogiannis, N.G.; Heymans, S. Myocardial Extracellular Matrix. Circ. Res. 2014, 114, 872–888. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, M.; Wirrig, E.; Phelps, A.; Wessels, A. Extracellular matrix and heart development. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Jacot, J.G.; McCulloch, A.D.; Omens, J.H. Substrate Stiffness Affects the Functional Maturation of Neonatal Rat Ventricular Myocytes. Biophys. J. 2008, 95, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Geisse, N.A.; Sheehy, S.P.; Parker, K.K. Control of myocyte remodeling in vitro with engineered substrates. Vitro Cell. Dev. Biol.—Anim. 2009, 45, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Tallawi, M.; Rai, R.; Boccaccini, A.R.; Aifantis, K.E. Effect of Substrate Mechanics on Cardiomyocyte Maturation and Growth. Tissue Eng. Part B Rev. 2015, 21, 157–165. [Google Scholar] [CrossRef]
- Bildyug, N. Extracellular Matrix in Regulation of Contractile System in Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 5054. [Google Scholar] [CrossRef] [PubMed]
- Baharvand, H.; Azarnia, M.; Parivar, K.; Ashtiani, S.K. The effect of extracellular matrix on embryonic stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2005, 38, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Budina, E.; Stoppel, W.L.; Sullivan, K.E.; Emani, S.; Emani, S.M.; Black, L.D. Cardiac extracellular matrix–fibrin hybrid scaffolds with tunable properties for cardiovascular tissue engineering. Acta Biomater. 2015, 14, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Stoppel, W.L.; Kaplan, D.L.; Black, L.D. Electrical and mechanical stimulation of cardiac cells and tissue constructs. Adv. Drug Deliv. Rev. 2016, 96, 135–155. [Google Scholar] [CrossRef]
- McCain, M.L.; Parker, K.K. Mechanotransduction: The role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function. Pflüg. Arch. Eur. J. Physiol. 2011, 462, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Dobner, S.; Amadi, O.C.; Lee, R.T. Chapter 14—Cardiovascular Mechanotransduction. In Muscle; Hill, J.A., Olson, E.N., Eds.; Academic Press: Boston, MA, USA; Waltham, MA, USA, 2012; pp. 173–186. ISBN 978-0-12-381510-1. [Google Scholar]
- Saucerman, J.J.; Tan, P.M.; Buchholz, K.S.; McCulloch, A.D.; Omens, J.H. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat. Rev. Cardiol. 2019, 16, 361–378. [Google Scholar] [CrossRef]
- Israeli-Rosenberg, S.; Manso, A.M.; Okada, H.; Ross, R.S. Integrins and Integrin-Associated Proteins in the Cardiac Myocyte. Circ. Res. 2014, 114, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Ross, R.S. Molecular and mechanical synergy: Cross-talk between integrins and growth factor receptors. Cardiovasc. Res. 2004, 63, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Gerilechaogetu, F.; Feng, H.; Golden, H.; Nizamutdinov, D.; Dostal, J.; Jacob, J.; Afroze, S.; Foster, D.; Bowman, J.; Ochoa, B. Current concepts in the role of mechanosensing in the regulation of cardiac contractile function. Austin J. Clin. Med. 2014, 1, 11015. [Google Scholar]
- Kwon, M.S.; Park, C.S.; Choi, K.; Park, C.-S.; Ahnn, J.; Kim, J.I.; Eom, S.H.; Kaufman, S.J.; Song, W.K. Calreticulin Couples Calcium Release and Calcium Influx in Integrin-mediated Calcium Signaling. Mol. Biol. Cell 2000, 11, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Gui, P.; Chao, J.-T.; Wu, X.; Yang, Y.; Davis, G.E.; Davis, M.J. Coordinated Regulation of Vascular Ca2+ and K+ Channels by Integrin Signaling. In Integrins and Ion Channels: Molecular Complexes and Signaling; Becchetti, A., Arcangeli, A., Eds.; Springer: New York, NY, USA, 2010; pp. 69–79. ISBN 978-1-4419-6066-5. [Google Scholar]
- Wang, Y.G.; Samarel, A.M.; Lipsius, S.L. Laminin acts via β1 integrin signalling to alter cholinergic regulation of L-type Ca2+ current in cat atrial myocytes. J. Physiol. 2000, 526, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Lai, N.C.; Kawaraguchi, Y.; Liao, P.; Copps, J.; Sugano, Y.; Okada-Maeda, S.; Banerjee, I.; Schilling, J.M.; Gingras, A.R.; et al. Integrins protect cardiomyocytes from ischemia/reperfusion injury. J. Clin. Investig. 2013, 123, 4294–4308. [Google Scholar] [CrossRef]
- Gaetani, R.; Zizzi, E.A.; Deriu, M.A.; Morbiducci, U.; Pesce, M.; Messina, E. When Stiffness Matters: Mechanosensing in Heart Development and Disease. Front. Cell Dev. Biol. 2020, 8, 334. Available online: https://www.frontiersin.org/articles/10.3389/fcell.2020.00334 (accessed on 19 November 2023). [CrossRef] [PubMed]
- Iwata, Y.; Ohtake, H.; Suzuki, O.; Matsuda, J.; Komamura, K.; Wakabayashi, S. Blockade of sarcolemmal TRPV2 accumulation inhibits progression of dilated cardiomyopathy. Cardiovasc. Res. 2013, 99, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Aguettaz, E.; Bois, P.; Cognard, C.; Sebille, S. Stretch-activated TRPV2 channels: Role in mediating cardiopathies. Prog. Biophys. Mol. Biol. 2017, 130, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Costa-Neto, C.M.; Duarte, D.A.; Lima, V.; Maria, A.G.; Prando, É.C.; Rodríguez, D.Y.; Santos, G.A.; Souza, P.P.C.; Parreiras-e-Silva, L.T. Non-canonical signalling and roles of the vasoactive peptides angiotensins and kinins. Clin. Sci. 2014, 126, 753–774. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. β-Arrestins and Cell Signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1213–1226. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Garbincius, J.F.; Michele, D.E. Dystrophin–glycoprotein complex regulates muscle nitric oxide production through mechanoregulation of AMPK signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 13663–13668. [Google Scholar] [CrossRef] [PubMed]
- Stutchbury, B.; Atherton, P.; Tsang, R.; Wang, D.-Y.; Ballestrem, C. Distinct focal adhesion protein modules control different aspects of mechanotransduction. J. Cell Sci. 2017, 130, 1612–1624. [Google Scholar] [CrossRef] [PubMed]
- Vite, A.; Caporizzo, M.A.; Corbin, E.A.; Brandimarto, J.; McAfee, Q.; Livingston, C.E.; Prosser, B.L.; Margulies, K.B. Extracellular stiffness induces contractile dysfunction in adult cardiomyocytes via cell-autonomous and microtubule-dependent mechanisms. Basic. Res. Cardiol. 2022, 117, 41. [Google Scholar] [CrossRef] [PubMed]
- Vejandla, R.M.; Orgil, B.-O.; Alberson, N.R.; Li, N.; Munkhsaikhan, U.; Khuchua, Z.; Martherus, R.; Azeloglu, E.U.; Xu, F.; Lu, L.; et al. Deficiency in nebulin repeats of sarcomeric nebulette is detrimental for cardiomyocyte tolerance to exercise and biomechanical stress. Am. J. Physiol.-Heart Circ. Physiol. 2021, 320, H2130–H2146. [Google Scholar] [CrossRef] [PubMed]
- Boateng, S.Y.; Lateef, S.S.; Mosley, W.; Hartman, T.J.; Hanley, L.; Russell, B. RGD and YIGSR synthetic peptides facilitate cellular adhesion identical to that of laminin and fibronectin but alter the physiology of neonatal cardiac myocytes. Am. J. Physiol.-Cell Physiol. 2005, 288, C30–C38. [Google Scholar] [CrossRef] [PubMed]
- Zemljic-Harpf, A.E.; Ponrartana, S.; Avalos, R.T.; Jordan, M.C.; Roos, K.P.; Dalton, N.D.; Phan, V.Q.; Adamson, E.D.; Ross, R.S. Heterozygous Inactivation of the Vinculin Gene Predisposes to Stress-Induced Cardiomyopathy. Am. J. Pathol. 2004, 165, 1033–1044. [Google Scholar] [CrossRef]
- Wang, X.; Gerdes, A.M. Chronic Pressure Overload Cardiac Hypertrophy and Failure in Guinea Pigs: III. Intercalated Disc Remodeling. J. Mol. Cell. Cardiol. 1999, 31, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Jannie, K.M.; Ellerbroek, S.M.; Zhou, D.W.; Chen, S.; Crompton, D.J.; García, A.J.; DeMali, K.A. Vinculin-dependent actin bundling regulates cell migration and traction forces. Biochem. J. 2015, 465, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Dumbauld, D.W.; Lee, T.T.; Singh, A.; Scrimgeour, J.; Gersbach, C.A.; Zamir, E.A.; Fu, J.; Chen, C.S.; Curtis, J.E.; Craig, S.W.; et al. How vinculin regulates force transmission. Proc. Natl. Acad. Sci. USA 2013, 110, 9788–9793. [Google Scholar] [CrossRef]
- Carton, F.; Casarella, S.; Di Francesco, D.; Zanella, E.; D’urso, A.; Di Nunno, L.; Fusaro, L.; Cotella, D.; Prat, M.; Follenzi, A.; et al. Cardiac Differentiation Promotes Focal Adhesions Assembly through Vinculin Recruitment. Int. J. Mol. Sci. 2023, 24, 2444. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Ichikawa, T.; Matsuyama, D.; Kimura, Y.; Ueda, K.; Craig, S.W.; Harada, I.; Kioka, N. The role of the interaction of the vinculin proline-rich linker region with vinexin α in sensing the stiffness of the extracellular matrix. J. Cell Sci. 2014, 127, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Kioka, N.; Ueda, K.; Amachi, T. Vinexin, CAP/ponsin, ArgBP2: A novel adaptor protein family regulating cytoskeletal organization and signal transduction. Cell Struct. Funct. 2002, 27, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tujague, M.; Thomsen, J.S.; Mizuki, K.; Sadek, C.M.; Gustafsson, J.-Å. The Focal Adhesion Protein Vinexin α Regulates the Phosphorylation and Activity of Estrogen Receptor α. J. Biol. Chem. 2004, 279, 9255–9263. [Google Scholar] [CrossRef] [PubMed]
- Chorev, D.S.; Moscovitz, O.; Geiger, B.; Sharon, M. Regulation of focal adhesion formation by a vinculin-Arp2/3 hybrid complex. Nat. Commun. 2014, 5, 3758. [Google Scholar] [CrossRef] [PubMed]
- DeMali, K.A.; Barlow, C.A.; Burridge, K. Recruitment of the Arp2/3 complex to vinculin: Coupling membrane protrusion to matrix adhesion. J. Cell Biol. 2002, 159, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Torsoni, A.S.; Fonseca, P.M.; Crosara-Alberto, D.P.; Franchini, K.G. Early activation of p160ROCK by pressure overload in rat heart. Am. J. Physiol.-Cell Physiol. 2003, 284, C1411–C1419. [Google Scholar] [CrossRef] [PubMed]
- Tornatore, T.F.; Dalla Costa, A.P.; Clemente, C.F.M.Z.; Judice, C.; Rocco, S.A.; Calegari, V.C.; Cardoso, L.; Cardoso, A.C.; Gonçalves, A.; Franchini, K.G. A role for focal adhesion kinase in cardiac mitochondrial biogenesis induced by mechanical stress. Am. J. Physiol.-Heart Circ. Physiol. 2011, 300, H902–H912. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, F.; Gentile, A.; Fukuda, R.; Tsedeke, A.T.; Jiménez-Amilburu, V.; Ramadass, R.; Iida, A.; Sehara-Fujisawa, A.; Stainier, D.Y.R. Focal adhesions are essential to drive zebrafish heart valve morphogenesis. J. Cell Biol. 2019, 218, 1039–1054. [Google Scholar] [CrossRef] [PubMed]
- van der Stoel, M.M.; Kotini, M.P.; Schoon, R.M.; Affolter, M.; Belting, H.-G.; Huveneers, S. Vinculin strengthens the endothelial barrier during vascular development. Vasc. Biol. 2023, 5, e220012. [Google Scholar] [CrossRef]
- Sussman, M.A.; Welch, S.; Walker, A.; Klevitsky, R.; Hewett, T.E.; Price, R.L.; Schaefer, E.; Yager, K. Altered focal adhesion regulation correlates with cardiomyopathy in mice expressing constitutively active rac1. J. Clin. Investig. 2000, 105, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Gunawan, F.; Ramadass, R.; Beisaw, A.; Konzer, A.; Mullapudi, S.T.; Gentile, A.; Maischein, H.-M.; Graumann, J.; Stainier, D.Y.R. Mechanical Forces Regulate Cardiomyocyte Myofilament Maturation via the VCL-SSH1-CFL Axis. Dev. Cell 2019, 51, 62–77.e5. [Google Scholar] [CrossRef] [PubMed]
- Holle, A.W.; Tang, X.; Vijayraghavan, D.; Vincent, L.G.; Fuhrmann, A.; Choi, Y.S.; Álamo, J.C.; Engler, A.J. In situ mechanotransduction via vinculin regulates stem cell differentiation. Stem Cells 2013, 31, 2467–2477. [Google Scholar] [CrossRef]
- Sarker, M.; Lee, H.T.; Mei, L.; Krokhotin, A.; de los Reyes, S.E.; Yen, L.; Costantini, L.M.; Griffith, J.; Dokholyan, N.V.; Alushin, G.M.; et al. Cardiomyopathy Mutations in Metavinculin Disrupt Regulation of Vinculin-Induced F-Actin Assemblies. J. Mol. Biol. 2019, 431, 1604–1618. [Google Scholar] [CrossRef] [PubMed]
- Zahavich, L.; Akilen, R.; George, K.; Mital, S. Heart Failure with Recovered Ejection Fraction in Patients with Vinculin Loss-of-function Variants. J. Cardiovasc. Transl. Res. 2023, 16, 1303–1309. [Google Scholar] [CrossRef] [PubMed]
- Bang, M.-L. Animal Models of Congenital Cardiomyopathies Associated with Mutations in Z-Line Proteins. J. Cell. Physiol. 2017, 232, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Vasile, V.C.; Ommen, S.R.; Edwards, W.D.; Ackerman, M.J. A missense mutation in a ubiquitously expressed protein, vinculin, confers susceptibility to hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 2006, 345, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Manso, A.M.; Okada, H.; Sakamoto, F.M.; Moreno, E.; Monkley, S.J.; Li, R.; Critchley, D.R.; Ross, R.S. Loss of mouse cardiomyocyte talin-1 and talin-2 leads to β-1 integrin reduction, costameric instability, and dilated cardiomyopathy. Proc. Natl. Acad. Sci. USA 2017, 114, E6250–E6259. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Gramlich, M.; Atherton, J.; McNabb, M.; Trombitás, K.; Sasse-Klaassen, S.; Seidman, J.G.; Seidman, C.; Granzier, H.; Labeit, S.; et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002, 30, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Takahashi, M.; Sakamoto, T.; Hiroe, M.; Marumo, F.; Kimura, A. Structural Analysis of the Titin Gene in Hypertrophic Cardiomyopathy: Identification of a Novel Disease Gene. Biochem. Biophys. Res. Commun. 1999, 262, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Peled, Y.; Gramlich, M.; Yoskovitz, G.; Feinberg, M.S.; Afek, A.; Polak-Charcon, S.; Pras, E.; Sela, B.-A.; Konen, E.; Weissbrod, O.; et al. Titin Mutation in Familial Restrictive Cardiomyopathy. Int. J. Cardiol. 2014, 171, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Larson, A.; Codden, C.J.; Huggins, G.S.; Rastegar, H.; Chen, F.Y.; Maron, B.J.; Rowin, E.J.; Maron, M.S.; Chin, M.T. Altered intercellular communication and extracellular matrix signaling as a potential disease mechanism in human hypertrophic cardiomyopathy. Sci. Rep. 2022, 12, 5211. [Google Scholar] [CrossRef] [PubMed]
- Meagher, P.B.; Lee, X.A.; Lee, J.; Visram, A.; Friedberg, M.K.; Connelly, K.A. Cardiac Fibrosis: Key Role of Integrins in Cardiac Homeostasis and Remodeling. Cells 2021, 10, 770. [Google Scholar] [CrossRef] [PubMed]
- Happe, C.L.; Engler, A.J. Mechanical Forces Reshape Differentiation Cues That Guide Cardiomyogenesis. Circ. Res. 2016, 118, 296–310. [Google Scholar] [CrossRef]
- Simpson, D.G.; Sharp, W.W.; Borg, T.K.; Price, R.L.; Terracio, L.; Samarel, A.M. Mechanical regulation of cardiac myocyte protein turnover and myofibrillar structure. Am. J. Physiol.-Cell Physiol. 1996, 270, C1075–C1087. [Google Scholar] [CrossRef] [PubMed]
- Harston, R.K.; Kuppuswamy, D. Integrins Are the Necessary Links to Hypertrophic Growth in Cardiomyocytes. J. Signal Transduct. 2011, 2011, 521742. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Kamm, R.D.; Lee, R.T. Mechanotransduction in Cardiac Myocytes. Ann. N. Y. Acad. Sci. 2004, 1015, 53–70. [Google Scholar] [CrossRef]
- Sheehy, S.P.; Grosberg, A.; Parker, K.K. The contribution of cellular mechanotransduction to cardiomyocyte form and function. Biomech. Model Mechanobiol. 2012, 11, 1227–1239. [Google Scholar] [CrossRef]
- Koudelková, L.; Brábek, J.; Rosel, D. Src kinase: Key effector in mechanosignalling. Int. J. Biochem. Cell Biol. 2021, 131, 105908. [Google Scholar] [CrossRef] [PubMed]
- Salazar, E.P.; Rozengurt, E. Src Family Kinases Are Required for Integrin-mediated but Not for G Protein-coupled Receptor Stimulation of Focal Adhesion Kinase Autophosphorylation at Tyr-397. J. Biol. Chem. 2001, 276, 17788–17795. [Google Scholar] [CrossRef]
- Torsoni, A.S.; Constancio, S.S.; Nadruz, W.; Hanks, S.K.; Franchini, K.G. Focal Adhesion Kinase Is Activated and Mediates the Early Hypertrophic Response to Stretch in Cardiac Myocytes. Circ. Res. 2003, 93, 140–147. [Google Scholar] [CrossRef] [PubMed]
- De Mets, R.; Wang, I.; Balland, M.; Oddou, C.; Moreau, P.; Fourcade, B.; Albiges-Rizo, C.; Delon, A.; Destaing, O. Cellular tension encodes local Src-dependent differential β1 and β3 integrin mobility. Mol. Biol. Cell 2019, 30, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Martino, F.; Perestrelo, A.R.; Vinarský, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 378185. Available online: https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2018.00824 (accessed on 10 October 2023). [CrossRef] [PubMed]
- Crosara-Alberto, D.P.; Inoue, R.Y.; Costa, C.R.C. FAK signalling mediates NF-κB activation by mechanical stress in cardiac myocytes. Clin. Chim. Acta 2009, 403, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.M.; Turner, K.; Mitchell, C.; Griffin, K.R.; Middlemiss, S.; Lau, L.; Dagg, R.; Taran, E.; Cooper-White, J.; Fabry, B.; et al. The focal adhesion targeting domain of p130Cas confers a mechanosensing function. J. Cell Sci. 2017, 130, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.C.; Pereira, A.H.M.; Ambrosio, A.L.B.; Consonni, S.R.; Rocha de Oliveira, R.; Bajgelman, M.C.; Dias, S.M.G.; Franchini, K.G. FAK Forms a Complex with MEF2 to Couple Biomechanical Signaling to Transcription in Cardiomyocytes. Structure 2016, 24, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, R.; Nagai, T.; Kudoh, S.; Zou, Y.; Tanaka, M.; Tamura, M.; Akazawa, H.; Takano, H.; Nagai, R.; Komuro, I. Integrins Play a Critical Role in Mechanical Stress–Induced p38 MAPK Activation. Hypertension 2002, 39, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Weinheimer, C.; Courtois, M.; Kovacs, A.; Zhang, C.E.; Cheng, A.M.; Wang, Y.; Muslin, A.J. The role of the Grb2–p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J. Clin. Investig. 2003, 111, 833–841. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Highlights | Proteins Involved | Process Sustained | References |
|---|---|---|---|
| Cardiomyocyte Myofibrillar Assembly | NMM II, α-actinin-2 fibers Titin, α- and/or β-cardiac myosins | Pre-myofibril composition. Final myofibril composition. | [22] |
| Sarcomere Remodeling during Heart Development | Myosin, actin, titin (sarcomeric proteins) | Influence the contractile properties of cardiomyocytes and force generation. | [28,30] |
| Titin | Provide elasticity and contribute to the passive stiffness of cardiomyocytes. | [31,32] | |
| β-cardiac myosin | Direct sarcomere assembly by generating the required basal tension. | [31] | |
| Focal Adhesion Signaling and Cardiomyogenesis | Integrin/FAK/PI3K-P85 pathway | FA’s maturation process in cardiac differentiation. | [35,36,37,38] |
| FAK-Tyr397 phosphorylation | Myocardial development, activate survival-promoting pathways. | [45,46,47] | |
| Proteins Involved in FA Maturation | Mint1/Veli/SAP97/CASK complex | Structural and functional support to the sarcomere. | [48] |
| VEGF | Enhance the adhesion of cardiomyocytes to the ECM. | [49] |
| Highlights | Main Players | Process | References |
|---|---|---|---|
| Forces Affecting Cardiomyocytes | Contractions, hemodynamic pressure, ECM-related passive elasticity | Changes in laminin, collagen, matrix protease, and proteoglycan expression. | [68,69] |
| Focal Adhesion proteins involved in mechanotransduction | FAK/Src complex | Cardiac hypertrophic growth and survival signaling. | [15,78] |
| FAK | Adaptive responses via MAPK and AKT/TSC2/mTOR pathways. | [78,137] | |
| Vinculin | Regulation in FA maturation. | [101,107] | |
| Talin, vinculin, tensin1 | Structural module. | [52,53] | |
| FAK, paxillin | Signaling module. | [52,53] | |
| Signaling Pathways in Response to Matrix Rigidity | PI3K/AKT, p38/JNK pathways | Regulation of interactions between adhesion complex and structural proteins. | [14] |
| Wnt/beta-catenin signaling | Cytoskeletal organization, regulation of contractility during cardiomyogenesis. | [14,95] | |
| Non-canonical Hippo pathway through YAP/TAZ | Heart development, cellular mechanics. | [18,135] | |
| AKT/TSC2/mTOR and ERK1/2 pathways | Prevent cardiomyocytes’ apoptosis. | [98,136] | |
| NF-kβ | Cell survival, correct assembling of FAs. | [136] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casarella, S.; Ferla, F.; Di Francesco, D.; Canciani, E.; Rizzi, M.; Boccafoschi, F. Focal Adhesion’s Role in Cardiomyocytes Function: From Cardiomyogenesis to Mechanotransduction. Cells 2024, 13, 664. https://doi.org/10.3390/cells13080664
Casarella S, Ferla F, Di Francesco D, Canciani E, Rizzi M, Boccafoschi F. Focal Adhesion’s Role in Cardiomyocytes Function: From Cardiomyogenesis to Mechanotransduction. Cells. 2024; 13(8):664. https://doi.org/10.3390/cells13080664
Chicago/Turabian StyleCasarella, Simona, Federica Ferla, Dalila Di Francesco, Elena Canciani, Manuela Rizzi, and Francesca Boccafoschi. 2024. "Focal Adhesion’s Role in Cardiomyocytes Function: From Cardiomyogenesis to Mechanotransduction" Cells 13, no. 8: 664. https://doi.org/10.3390/cells13080664